
Abstract
Deregulated proliferation is key to tumor progression. Although unrestricted proliferation of solid tumor cells correlates with the cold-shock protein Y-box (YB)-binding protein-1 accumulation in the nuclei, little is known about its expression and function in hematopoietic malignancies, such as T-cell acute lymphoblastic leukemia (T-ALL). Here we show that YB-1 protein is highly enriched in the nuclei of activated T cells and malignant human T-ALL cell lines but not in resting T cells. YB-1 S102 mutations that either mimic (S102D) or prevent phosphorylation (S102N) led to accumulation of YB-1 in the nucleus of T cells or strictly excluded it, respectively. Inactivation of ribosomal S6 kinase (RSK) was sufficient to abrogate T-cell and T-ALL cell proliferation, suggesting that RSK mediates cell-cycle progression, possibly dependent on YB-1-phosphorylation. Indeed, phosphomimetic YB-1S102D enhanced proliferation implying that S102 phosphorylation is a prerequisite for malignant T-cell proliferation. At initial diagnosis of T-ALL, YB-1 localization was significantly altered in the nuclei of tumor blasts derived from bone marrow or peripheral blood. Our data show deregulated YB-1 in the nucleus as a yet unreported characteristic of T-ALL blasts and may refine strategies to restrict progression of hematopoietic tumors.
Similar content being viewed by others


Main
T-ALLs are aggressive hematological tumors resulting from malignant transformation of lymphoid progenitor cells.1, 2 With current chemotherapy protocols, only about 50% of adults are cured,3 and the outcome of T-ALL patients with primary resistance to chemotherapy or relapse remains poor.4, 5 For the development of aggressive cancer cells, abnormal proliferation is necessary: hyperproliferation when expanding and hypoproliferation when resting until a relapse. Therefore, an advanced understanding of the molecular events underlying deregulated proliferation of leukemic T-cell blasts will help refining therapeutic approaches.
YB-1 has emerged as a potential oncogene promoting tumor cell proliferation in solid cancers when expressed at elevated levels.6, 7 Human YB-1- and its paralog, DbpA, are members of the CSD protein family that regulate the expression of target genes at the level of transcription and translation. YB-1 has a role in the regulation of mRNA packaging and stabilization and controls mRNA translation globally because of its capacity as a major protein of cytoplasmic mRNPs.8, 9, 10, 11 Target mRNAs include IL-2, GM-CSF, CD44, and IFNR2 (ref. 12, 13, 14). YB-1 is a transcription factor promoting the expression of many genes involved in cell growth, including PCNA, EGFRα, and DNA polymerase A15,16,17,18 that was regulated through either direct or indirect YB-1 interaction with the Y-box (inverted CCAAT-box) or other sequences in gene promoters.19 Notably, the role of YB-1 in regulating transcription is influenced through interaction with mRNA and DNA and by co-transcriptional events (splicing, mRNA packaging and stabilization).19 During tumor progression, YB-1 regulates transcription of genes such as MDR1, cyclin A, and cyclin B1 (ref. 20, 21). In fibroblasts and breast cancer cell lines, it has been shown that nuclear localization of YB-1 is mediated by its phosphorylation at Ser102 (ref. 22, 23, 24). In HeLa cells, nuclear YB-1 localization is linked to cell-cycle progression during the G1/S phases.21 Thus, to ensure a healthy balance between tissue maintenance and renewal, YB-1’s expression as well as localization is tightly controlled.
T-ALL can develop from multiple stages of lymphoid progenitors during T-cell development; however, its characteristic malignant signal transduction machinery resembles activated effector T cells.25, 26 Expansion of T cells requires engagement of the TCR/CD3 complex and the co-stimulatory molecule CD28. CD28 co-stimulation has been shown to augment cytokine mRNA levels and G1-kinases.27 Moreover, it increases cellular metabolism and promotes cell survival.28, 29 We asked in the present study how YB-1 expression is regulated in malignant T cells and whether it serves as central switch for malignant T-cell transformation, leading to deregulated cell proliferation.
Results
Elevated YB-1 expression in activated primary human CD4+ T cells and T-ALL cell lines
First, we quantified YB-1 expression in primary T cells versus Jurkat, Molt-16, and RPMI-8402 cell lines derived from peripheral blood of T-ALL patients.30 Western blotting analysis revealed weak expression of YB-1 in subtypes of resting primary CD4+ T cells but much higher YB-1 levels in T-ALL cells (Figure 1a).

YB-1 protein expression in primary and malignant human CD4+ T cells. Primary CD4+ T-cell subsets express YB-1 constitutively at low levels, but it is strongly expressed in malignant T-ALL cell lines. (a) T-ALL cell lines were used as indicated. Effector/memory (CD45−RO+CD4+), naive (CD45-RA+CD4+) and recent thymic emigrant (CD45−RA+CD31+CD4+) T cells were isolated from PBMCs of four different donors. In all, 1 × 107 T-ALL cells and each subset of CD4+ T cells (pooled from four different donors) were subsequently lysed, and 30 μg of protein was subjected to 12% SDS-PAGE, and western blotting analysis was performed using the indicated antibodies. Experiments were performed in triplicates and one representative example is shown. (b) PBMCs of four different donors were stimulated either with 1 μg/ml ConA or 1 μg/ml SEB or 10 ng/ml PMA plus 1 μg/ml Ionomycin for specified time points. For flow cytometry, 1 × 106 PBMCs were fixed and permeabilized and subsequently incubated with primary anti-YB-1 c-terminal antibody and fluorescence-labeled secondary antibody together with direct-labeled anti-CD45 Ab, anti-CD3 Ab and anti-CD4 Ab for identification of CD4+ T cells. Frequencies of YB-1-expressing CD4+ T cells of four different donors are shown in the graph, and the mean value and S.E.M. are indicated. (c) YB-1 mRNA expression is significantly enhanced by CD28 co-stimulation. CD4+ T cells and Jurkat cells were stimulated either with IgG1 isotype or anti-CD3/IgG1 isotype or anti-CD3/anti-CD28-coupled beads for the indicated time. The mRNA expression of YB-1 and DbpA in CD4+ T cells and Jurkat cells were performed by qRT-PCR analysis. Graphs show relative transcript levels of YB-1 mRNA and DbpA mRNA normalized to GAPDH (glyceraldehyde 3-phosphate dehydrogenase) mRNA. Data are summarized from three different experiments. The mean value and S.E.M. are indicated. P-values are shown (analysis of variance) *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (d) Kinetic of YB-1 expression in stimulated T cells. Activation of CD4+ T cells and Jurkat cells was performed using antibody-coated beads as indicated. As negative controls, either T cells before stimulation were used (0 h) and, in addition, resting T cells incubated with isotype-matched antibodies for each time point (control/IgG1 isotype). YB-1 and DbpA protein expression were analyzed as in panel (a). Samples of two time points were always run on one gel as indicated. GAPDH was used for internal normalization and the numbers indicate relative intensity. Experiments were performed four times, and one representative example is shown. (e) 1 × 106 CD4+ T cells isolated from four different donors were stimulated for 96 h using anti-CD3/anti-CD28-coupled beads. Cells were fixed and permeabilized. Total YB-1 and Ki-67 expression were analyzed by flow cytometry. Graph illustrates the average percentages of YB-1 or Ki-67-expressing CD4+ T cells of four different donors with mean value and S.E.M. (Student’s t-test)
As T-ALL cells share characteristics with activated T cells, we assessed YB-1 levels in primary CD4+ T cells activated by Staphylococcal enterotoxin B (SEB), Concanavalin A (ConA), PMA, and Ionomycin (Figure 1b). The percentage of CD4+ T cells with detectable YB-1 expression clearly increased within 24 h of stimulation, yet either hardly reached or remained below the 100% level detected for non-stimulated Jurkat cells (Figure 1b). Next, we analyzed the effect of TCR/CD3 activation with or without CD28 co-stimulation on YB-1 expression. qRT-PCR analysis revealed that, at 6 h, co-stimulation via CD28 led to a seven-fold increase in YB-1 mRNA levels, which returned to baseline at 24 h (Figure 1c). In contrast, transcript numbers for the YB-1 paralog DbpA remained unaltered. In Jurkat cells, mRNA expression for YB-1 and DbpA mRNA remained unaffected by TCR/CD3 triggering even upon co-stimulation (Figure 1c).
At the protein level, YB-1 remained low for >8 h after beginning of the stimulation but was increased 4–5 times at 16 h compared with 0 and 16 h resting T cells (control/IgG isotype) (Figure 1d). Compared with anti-CD3 stimulation, anti-CD3/anti-CD28 engagement led to a doubling of YB-1 amounts by 16 h. YB-1 protein expression in Jurkat cells remained invariable upon stimulation (Figure 1d). Similarly, DbpA did not change significantly over time (Figure 1d). Flow cytometric analysis (Figure 1e) extended the western blotting data to single cells, showing that YB-1-expressing cells appeared around 20 h after the onset of stimulation. Comparison of YB-1 with the proliferation-upregulated molecule Ki-67 demonstrated that YB-1 became upregulated already 20 h before Ki-67 (Figure 1e).
YB-1 localization in the T-cell nuclei is stimulation dependent
In order to analyze the subcellular YB-1 localization during proliferation and differentiation, we stimulated primary and malignant CD4+ T cells either with anti-CD3 or anti-CD3/anti-CD28 and prepared cytoplasmic and nuclear protein extracts. Western blotting analysis revealed that in unstimulated primary T cells YB-1 is weakly expressed in the cytoplasm and nucleus (Figure 2a). Upon stimulation using anti-CD3 or anti-CD3/antiCD28, a significant amount of YB-1 was detectable in the nucleus (Figure 2a). Ser102-phosphorylated YB-1 (pYB-1S102) was found in the cytoplasm and nucleus of primary and Jurkat cells and its distribution between the two compartments appeared unaffected upon stimulation (Figure 2a). Of note, cytoplasmic and nuclear DbpA expression in primary CD4+ T cells and Jurkat cells were differently affected by stimulation showing that the two cold-shock proteins are regulated independently (Figure 2a).

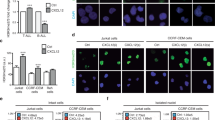
Subcellular localization of YB-1 in activated human CD4+ T cells and Jurkat cells. Activated primary human CD4+ T cells and Jurkat cells show cytoplasmic and nuclear YB-1 localization. (a) CD4+ T cells isolated from four different donors and Jurkat cells were stimulated either with anti-CD3/IgG1 isotype or anti-CD3/anti-CD28-coupled beads for 16 h. Subsequently, subcellular fractionation of cells was performed. CD4+ T-cell lysates of four different donors were pooled. Thirty micrograms of cytoplasmic and nuclear proteins were subjected to 12% SDS-PAGE, and western blotting analysis was performed using the indicated antibodies. GAPDH (glyceraldehyde 3-phosphate dehydrogenase; cytoplasm) and Lamin B (nucleus) were used for internal normalization and relative intensity is shown below each blot. Experiments were performed in triplicates, and one representative example is shown. (b) Establishing YB-1 detection in isolated nuclei of T cells by flow cytometry. CD4+ T cells labeled with carboxyfluorescein succinimidyl ester (CFSE) and isolated nuclei of CD4+ T cells marked with DAPI were mixed 1:1 and analyzed by flow cytometry. Left panels show the different distribution of the non-labeled cells and nuclei population; the right panels display the population of CFSE-labeled CD4+ T cells and DAPI-labeled nuclei. (c) Amounts of YB-1 in isolated nuclei of activated CD4+ T cells and Jurkat cells. T cells were stimulated either with IgG1 isotype or anti-CD3/anti-CD28-coupled beads for the indicated time periods. Nuclei were isolated, fixed, permeabilized, and incubated with primary anti-YB-1 C-terminal antibody and fluorescence-labeled secondary antibody. Numbers shown indicate the mean fluorescence intensity (MFI) of YB-1-positive nuclei of stimulated (number below) or unstimulated T cells (number on top). Experiments were performed in triplicates, and one representative example is shown
To detect the localization of YB-1 in the nuclei, we isolated the nuclei of primary CD4+ T cells and labeled them with 4′,6-diamidino-2-phenylindole (DAPI) and mixed them with CFSE-labeled cells as controls. Using flow cytometry, this allowed us to gate unambiguously on individual nuclei (Figure 2b). When isolated nuclei stained for YB-1 were analyzed in this way, a nuclear accumulation of YB-1 upon stimulation became evident (Figure 2c).
YB-1 translocation is dependent on MAP kinase pathway
Previous work on breast cancer cell lines and fibroblasts showed that nuclear translocation of YB-1 depends on Ser102 phosphorylation.22, 31 Ser102 can be phosphorylated by Akt, PKCα, and RSK by binding to RxRxxS, RR/FRRK/RxS, and R/KxRxxS motifs.22, 32 Therefore, we incubated primary CD4+ T cells during anti-CD3/anti-CD28 stimulation and spontaneously proliferating Jurkat cells with the respective kinase inhibitors. Flow cytometric analysis of nuclear YB-1 protein in CD4+ T cells revealed that the Akt inhibitor II exerted only a minor effect, whereas treatment with the PKC inhibitor Rottlerin caused a 48% reduction of nuclear YB-1 in CD4+ T cells (Figure 3a). RSK inhibitor SL0101 reduced activation-induced nuclear YB-1 accumulation by 60% compared with controls (Figure 3a). In Jurkat cells, Akt or PKC inhibition showed no effect, whereas the RSK inhibitor reduced the nuclear accumulation of YB-1 by half (Figure 3a, lower row).

Nuclear YB-1 translocation in T cells depends on the MAP kinase pathway. Analysis of YB-1 localization in primary and malignant CD4+ T cells in the presence or absence of specific inhibitors for Akt, PKC, and RSK. (a) Flow cytometric analysis of nuclear YB-1 expression in isolated nuclei of CD4+ T cells and Jurkat cells incubated with specific inhibitors for RSK (SL0101: 100 μM), PKC (Rottlerin: 10 μM), and Akt (Akt inhibitor II: 30 μM). In all, 1 × 107 CD4+ T cells and Jurkat cells were preincubated with inhibitors for 20 min. Subsequently, CD4+ T cells were either left inactivated or activated using anti-CD3/anti-CD28 crosslinking for 16 h. Activated CD4+ T cells preincubated with DMSO served as controls. Jurkat cells were employed without additional stimuli. Nuclei were isolated and fixed/permeabilized, and flow cytometric analysis was performed as described in Figure 2c. Numbers represent the mean fluorescence intensity (MFI) of YB-1-positive nuclei isolated from stimulated (number below) or unstimulated CD4+ T cells (number on top) as well as YB-1-positive nuclei isolated from Jurkat cells (number below) and Jurkat cells without primary antibody as control (number on top). One representative example is shown out of three experiments. (b) Fluorescence microscopic assay of mutated YB-1 proteins in primary CD4+ T cells following transduction with GFP, GFP-YB-1S102D, and GFP-YB-1S102N constructs. GFP-YB-1S102D localizes to the cytoplasm and nuclei, while GFP-YB-1S102N is excluded from the nuclei of primary CD4+ T cells. One representative example is shown out of two experiments. (c) Suggestive model of YB-1 translocation into the nucleus in CD4+ T cells. Upon TCR triggering and CD28 costimulation, YB-1 is mainly phosphorylated by RSK kinase and to a minor extent by kinases Akt and PKC. Phosphorylated YB-1 in the cytoplasm controls translation. Additionally, YB-1 translocates from the cytoplasm into the nucleus and regulates gene expression
To confirm that YB-1 phosphorylation was essential for nuclear translocation, Ser102 was mutated to either asparagine or aspartic acid, and the resulting mutants were expressed as GFP fusion proteins (GFP-YB-1S102N and GFP-YB-1S102D). Fluorescence microscopy of lentivirus-transduced primary T cells revealed that the phosphomimetic GFP-YB-1S102D was predominantly found in the nucleus, whereas the phosphorylation-deficient GFP-YB-1S102N version was exclusively localized in the cytoplasm of CD4+ T cells (Figure 3b). We summarized our results in a scheme (Figure 3c), which suggest that YB-1 S102 phosphorylation by RSK and, to a minor extent, by Akt and PKC in primary human CD4+ T cells and, especially in malignant T cells, is a prerequisite for YB-1 translocation into the nucleus, where it controls gene expression (Figure 3c).
Upregulation of YB-1 expression in G1 phase of the cell cycle
To investigate whether YB-1 is enhanced in proliferating T cells, CD4+ T cells were labeled with CFSE and left either unstimulated or stimulated with anti-CD3/anti-CD28-coated beads. On day 5, CFSE/YB-1 co-staining revealed that 37% of stimulated cells express YB-1 in the G0 generation to various extents. Consistently high levels of YB-1, however, were found in proliferative generations (G1–G6), whereas YB-1low/neg cells persisted almost exclusively in the G0 generation (Figure 4a). Additionally, we analyzed Ki-67 in YB-1high- and YB-1low-expressing CD4+ T cells. Therefore, primary CD4+ T cells isolated from 8 to 11 different donors were stimulated using anti-CD3/anti-CD28-coated beads. On days 2, 4, and 6, YB-1/Ki-67 co-staining allowed us to gate on YB-1high CD4+ T-cell populations and identify the Ki-67-positive CD4+ T cells (Figure 4a, right). The results demonstrated that YB-1high CD4+ T cells exhibit approximately 80% Ki-67+ cells at the indicated time points, whereas YB-1low CD4+ T cells had significantly lower frequencies of Ki-67 expression.

A central role for YB-1 in T-cell proliferation. YB-1high is a prerequisite for proliferation of CD4+ T cells. (a) In all, 1 × 106 primary human CD4+ T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and cultured for 5 days in the absence or presence of anti-CD3/anti-CD28-coated beads. Co-staining of YB-1 allows discrimination of YB-1high from YB-1low-proliferating cells. Cells were further gated according to the number of cell divisions (G0–G6), and frequencies of undivided and divided YB-1high and YB-1low cells are indicated in the gates at the top of each generation. Representative data are shown from one out of two independent experiments. YB-1high-expressing T cells exhibit a high expression of the proliferation marker Ki-67. In all, 1 × 106 primary human CD4+ T cells isolated from different donors (n=8–11) were stimulated with anti-CD3 and anti-CD28-coupled beads for specified time. Subsequently, T cells were fixed, permeabilized and co-stained using specific antibodies for YB-1 and Ki-67 followed by flow cytometric analysis. The illustrated graphs show the percentages of Ki-67-positive cells in YB-1high- and YB-1low-expressing CD4+ T cells on days 2, 4, and 6 after activation. The data are summarized using CD4+ T cells from 8 to 11 different donors. The mean value and S.E.M. are indicated, and for statistics, the two-tailed Student’s t-test was applied *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (b) YB-1 is already upregulated in G1 phase of the cell cycle. In all, 1 × 107 CD4+ T cells from four different donors were either left unstimulated or stimulated with anti-CD3/anti-CD28-coupled beads for 24 h in the presence or absence of cell-cycle inhibitors Nocodazole (N; 100 nM) and Paclitaxel (P; 200 nM). Thirty micrograms of total cell lysates (proteins from four different donors were pooled) were prepared, and immunoblotting was performed as described before. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used for internal normalization and relative intensities are shown below the blot. One representative example is shown out of two experiments. (c) Inactivation of YB-1 by specific small interfering RNA (siRNA) abrogates proliferation of CD4+ T cells. Also, 5 × 106 CD4+ T cells were transfected with specific YB-1siRNA, and 72 h later, CD4+ T cells were fixed, permeabilized, stained, and analyzed by flow cytometry as described before. Left diagram shows the YB-1siRNA knockdown efficiency. The dark blue histogram represents CD4+ T cells transfected with YB-1siRNA, and light blue histogram shows CD4+ T cells transfected with control siRNA. Numbers indicate the percentages of YB-1-positive CD4+ T cells. Right panel shows CD4+ T cells labeled with CFSE. After transfections with YB-1siRNA or siRNA control, cells were activated using anti-CD3/anti-CD28 crosslinking and analyzed at the indicated time points by flow cytometry. Numbers on the top indicate frequencies of proliferating cells in each generation (G0–G7). (d) Inactivation of YB-1 in CD4+ T cells by viral transduction using specific YB-1shRNA. CD4+ T cells were transduced using measles virus system. CD4+ T cells were infected with measles virus carrying YB-1shRNA or control shRNA (short hairpin RNA). At specified time points, anti-CD3/anti-CD28-activated CD4+ T cells were fixed, permeabilized, stained, and analyzed by flow cytometry as described before. The top panel shows the knockdown efficiency in the percentage of cells either transduced with control vector or specific YB-1shRNA over specified time. The panel below demonstrates the average amounts of CFSE-positive CD4+ T cells analyzed by flow cytometry that were either transduced with YB-1shRNA or control shRNA, followed by activation using anti-CD3/anti-CD28 crosslinking for the indicated time points. Data summarized from four different experiments using CD4+ T cells. The mean value and S.E.M. are indicated, and for statistics, the two-tailed Student’s t-test was applied. *P<0.05, **P<0.01, ***P<0.001. (e) Viable Jurkat cells after YB-1 inactivation detected by trypan blue staining. Jurkat cells were transduced with lentiviral particle containing YB-1shRNA (MOI 10) or control shRNA and a puromycin-resistance gene. On day 5, puromycin was added for selection. Viable cells were counted at the indicated time points. The data are summarized and normalized from three different experiments. The mean value and S.E.M. are indicated, and for statistics, analysis of variance was applied
As many CD4+ T cells in G0 already expressed YB-1 (Figure 4a), completion of mitosis was obviously dispensable for YB-1 upregulation. To further determine the onset of YB-1 upregulation, CD4+ T cells were preincubated with the cell-cycle inhibitors Nocodazole for a G2/M arrest and paclitaxel for a G1 arrest and either left unstimulated or stimulated for 24 h (Figure 4b). Inhibitor-induced cell-cycle arrest was confirmed by CFSE-based proliferation monitoring as in Figure 4a (data not shown). Immunoblot analysis revealed that neither G2 nor G1 cell-cycle arrest prevented YB-1 upregulation (Figure 4b), implying that enhanced YB-1 expression is initiated in G1.
To analyze the impact of YB-1 on T-cell proliferation, we conducted siRNA- and shRNA-mediated knockdown of YB-1 (Figures 4c and d). Flow cytometric analysis of transfected CD4+ T cells demonstrated a 5–6-fold downregulation of YB-1 protein compared with control-transfected cells (Figure 4c, left panel). Analysis of CFSE dilution revealed that the knockdown of YB-1 strongly impaired cell division (Figure 4c, right panel). In addition, we investigated the impact of YB-1 inactivation in T-cell proliferation using specific shRNA. Resting CD4+ T cells were infected with virus (multiplicity of infection (MOI) 100) containing either control shRNA (pLKO) or YB-1shRNA (pLKO-YB-1shRNA) for 6 days. At the indicated time point, the ‘knockdown’ efficiency was determined by analyzing intracellular YB-1 using flow cytometry (Figure 4d; graphs on the top). In parallel, we analyzed the proliferation capacity of CD4+ T cells, which were labeled with CFSE prior to virus infection and stimulated with anti-CD3/anti-CD28-coupled beads. Flow cytometric analysis on day 6 revealed that CD4+ T cells with the predominant knockdown of YB-1 (Figure 4d; right graph on top) showed significant higher CFSE-positive cells (MFI 8000) compared with control shRNA (MFI 4500) (Figure 4d; lower graph on the right), demonstrating that limiting YB-1 protein resulted in reduction of mitotic events. Strikingly, even highly proliferating Jurkat cells displayed a significant blockade of cell proliferation after shRNA-mediated YB-1 knockdown (Figure 4e).
We next searched to determine the impact of the nuclear entity of YB-1, specifically pYB-1S102, on proliferation. As RSK inhibition strongly interfered with nuclear accumulation of YB-1 (Figures 3a and b), we analyzed its effect on the expansion of primary and malignant T cells. In fact, the RSK inhibitor SL0101 clearly reduced the proliferation of activated CD4+ T cells in a dose-dependent manner (Figure 5a). Moreover, SL0101 at 100 μM strongly diminished the expansion of Jurkat, RPMI-8402, and Molt16 cells (Figures 5a and b). Notably, inhibition of Akt only slightly impaired the proliferation of activated CD4+ T cells and had virtually no effect on the expansion of malignant T cells (Figures 5a and b). To further assess a possible pro-proliferative effect of S102-phosphorylated YB-1, we expressed the phosphomimetic GFP-YB-1S102D variant in preactivated CTV-labeled CD4+ T cells. Flow cytometric analysis on day 5 revealed significantly higher proliferation rates for YB-1S102D-expressing cells compared with control cells expressing GFP or the phosphorylation-deficient GFP-YB-1S102N variant (Figure 5c). These findings strongly support the idea that S102 phosphorylation-dependent nuclear accumulation of YB-1 is the rate-limiting step to the proliferation of T cells.

The impact of Akt, PKC, and RSK kinases on primary and malignant CD4+ T-cell proliferation. (a) CD4+ T cells were labeled with carboxyfluorescein succinimidyl ester CFSE; 1.6 μM) and subsequently stimulated with anti-CD3/anti-CD28-coupled beads. At day 3, Akt inhibitor II was added as described in Figure 3a. The RSK inhibitor was used at increasing concentrations (25–100 μM) as depicted. At day 5, flow cytometric analysis of CFSE-stained CD4+ T cells was used to track multiple mitotic events of single cells. The histograms illustrate the proliferation of anti-CD3/anti-CD28-stimulated CD4+ T cells with or without inhibitors, as indicated. Activated CD4+ T cells preincubated with DMSO served as controls. Numbers indicate the percentage of dividing cells. One representative example is shown out of three experiments. (b) CFSE-labeled T-ALL cell lines were cultured with inhibitors against Akt or RSK for 4 days. Dividing Jurkat, RPMI-8402, and Molt16 cells were analyzed daily using flow cytometry. The histograms illustrate the proliferation of cells from day 1 in the presence or absence of specific inhibitors. DMSO-treated Jurkat, RPMI-8402, and Molt16 cells served as controls. One representative example is shown out of three experiments. (c) Proliferation analysis of GFP-YB-1-S102D- and GFP-YB-1-S102N-expressing primary CD4+ T cells. Constructs were lentivirally transduced in CTV-labeled and, for 48 h, in preactivated CD4+ T cells. At day 5, flow cytometric analyses of CTV-stained CD4+ T cells was used to track cell-cycle progression. The histograms illustrate the proliferation of GFP-YB-1-S102D-, GFP-YB-1-S102N-, and control vector-expressing CD4+ T cells of two different donors. (d) Phosphorylation of YB-1 Ser102 in CD4+ T cells is abrogated in the presence of the inhibitor Fisetin (60 μM) after 48 h of stimulation. Total cell lysates (proteins from four different donors were pooled) were prepared, and immunoblotting was performed. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used for internal normalization and relative intensities are shown below the blot. One representative example is shown out of three experiments. G1-phase cell-cycle arrest mediated by Fisetin incubation in primary human CD4+ T cells. CD4+ T cells isolated from four different donors were stimulated for 48 h either with IgG1 isotype or anti-CD3/anti-CD28-coupled beads in the presence or absence of Fisetin. Total cell lysates (proteins from four different donors were pooled) were prepared, and immunoblotting was performed. Relative band intensities normalized to GAPDH are shown in the graph. The data are summarized from three different experiments. The mean value and S.E.M. are indicated, and for statistics, analysis of variance was applied. ***P<0.001, ****P<0.0001
We also exploited the ability of Fisetin, a plant-derived polyphenol, to inhibit phosphorylation of YB-1 at Ser102 (Figure 5d, left).33 Anti-CD3/anti-CD28-stimulated CD4+ T cells and unstimulated controls were treated with Fisetin for 48 h and analyzed for the expression of G1 kinases cyclin D2 and cdk6 that need to be upregulated to induce cell-cycle progression into S phase. After 48 h of T-cell activation plus Fisetin treatment, no significant effect on cyclin D2 protein expression appeared, but Fisetin caused a drastically reduced cdk6 expression (Figure 5d, right).
Nuclear YB-1 expression in samples of T-ALL patients
According to the aforementioned data, YB-1 is likely a key switch controlling cell-cycle progression. To assess whether YB-1 is deregulated in hematopoietic malignancies, we investigated the nuclear localization of YB-1 in bone marrow cells of 16 T-ALL patients, consisting of adult (n=5) and children (n=11) samples at initial diagnosis showing >80% lymphoblasts. To monitor the proliferation indices of bone marrow cells of T-ALL patients (n=8, at least 80% tumor blasts), we analyzed the expression of Ki-67 in samples of resting and activated primary CD4+ T cells (n=5) from healthy donors and T-ALL cell lines by qRT-PCR. Results showed a significant reduction of Ki-67 transcripts in CD4+ T cells isolated from bone marrow of T-ALL patients compared with resting primary CD4+ T cells and activated CD4+ T cells of healthy donors (Figure 6a). Also, hyperproliferative T-ALL cell lines showed higher numbers of Ki-67 transcripts (Figure 6a). As the proliferation-dependent expression of Ki-67 is low, these data suggest that bone marrow-derived tumor blasts from T-ALL patients exhibit low proliferation (Figure 6a), whereas activated primary T cells and T-ALL cell lines showed a high proliferative profile.

Nuclear YB-1 content in samples isolated from T-ALL patients. (a) Analysis of proliferation marker Ki-67 in primary, malignant CD4+ T cells and T-ALL cell lines. Total RNA was isolated from T cells of bone marrow samples from T-ALL patients (n=8) and from resting (nil) and 24–72 h activated CD4+ T cells (n=5) from healthy individuals, respectively, and analyzed by qRT-PCR. Graph shows relative transcript numbers of Ki-67 mRNA. The means and S.E.M. are indicated. P-values are shown (analysis of variance (ANOVA)). (b) Nuclei of cells isolated from bone marrow samples of T-ALL patients (adult, n=5; children, n=11) and indicated controls such as nuclei isolated from cells of patients in remission or 24 h inactivated/activated CD4+ T cells, or T-ALL cell lines, were isolated, fixed, and permeabilized as described in Figure 2c. The graph demonstrates the mean nuclear YB-1 expression of T-ALL patients and controls. Significances are determined using ANOVA. *P<0.05, **P<0.01, ***P<0.001. (c) Demonstration of the nuclear YB-1 in the isolated nuclei of lymphoblasts isolated from peripheral blood of T-ALL patients compared with inactivated/activated CD4+ T cells isolated from three different donors. Primary CD4+ T cells were stimulated for 24 h. Nuclei were isolated, fixed and permeabilized, and stained for YB-1 as described in Figure 2c. Nuclear co-staining was performed with direct fluorescence-labeled anti-Ki-67 Ab. Numbers shown correspond to the mean fluorescence intensity (MFI) of YB-1- or Ki-67-positive nuclei of lymphoblasts from T-ALL patients (number below), stimulated (number below), or unstimulated T cells (number on top). (d) Nuclear YB-1 in the isolated nuclei of Jurkat cells compared with the nuclei isolated from resting CD4+ T cells. Nuclei were isolated, fixed and permeabilized, and stained for YB-1 as described in Figure 2c. Nuclear co-staining was performed as described before. Numbers shown symbolize the MFI of YB-1- or Ki-67-positive nuclei of Jurkat T cells (number below) or resting T cells (number on top). One representative example is shown out of three experiments
Next, nuclei were isolated from bone marrow cells from 16 patients (see above) and nuclear YB-1 expression was determined by flow cytometry as described in Figures 2b and c. Resting and anti-CD3/CD28-activated T cells of healthy volunteers and bone marrow cells from patients (n=5) without blasts who recently achieved remission from AML served as controls (Table 1). Our analyses demonstrated that nuclear YB-1 derived from bone marrow was similarly low in adults and children with T-ALL. It was highly significantly reduced by 80% in blasts of both adults and children with T-ALL when compared with patients in remission (Figure 6b). Anti-CD3/anti-CD28-activated T cells displayed 12-fold enhanced nuclear YB-1 compared with T-ALL patients (Figure 6b). Notably, at the time point of initial diagnosis of T-ALL (>80% blasts), YB-1 protein expression between patients reaching remission or not upon treatment later on were almost identical (data are shown combined in Figure 6b, samples: T-ALL bone marrow). In contrast to the bone marrow-derived tumor blasts, the aggressively proliferating Jurkat, Molt16, and RPMI-8402 cells exhibited a high MFI of 4284±273 of nuclear YB-1 compared with resting CD4+ T cells (Figure 6b). The data also demonstrated that no matter whether T cells are malignant or ‘normal’, when having a high-proliferative profile determined by Ki-67, they also showed high YB-1 content in the nucleus, and cells with a low-proliferative profile showed low YB-1 expression in the nucleus. In addition, T-ALL blasts and cell lines showed extreme values either lower than resting T cells or as high as activated primary T cells.
As we have found bone marrow cells of T-ALL patients to have low YB-1 expression in the nucleus but cell lines derived from peripheral blood30 to express high levels of nuclear YB-1, we tested whether high nuclear YB-1 is in particular a characteristic for tumor blasts from peripheral blood. Although quite rare to find in the periphery, we chose to investigate tumor blasts from PBMCs of three T-ALL patients from initial diagnosis, including one who eventually experienced fatal relapse within 2 years after diagnosis. Using 24 h activated T cells, T cells of three healthy donors as a reference, the strongly activated and proliferating T cells showed a 2–3-fold enhanced Ki-67 expression (Figure 6c, left panel, anti-CD3/anti-CD28) compared with resting T cells from the same donors (Figure 6c, left panel, unstimulated). In terms of nuclear YB-1 expression of activated primary T cells, it was enhanced 2–3-fold after activation (Figure 6c, left panels). Analyzing patients at initial diagnosis, all T-ALL samples from peripheral blood showed enhanced Ki-67 expression, even higher than 24 h activated primary T cells (Figure 6c, left and right panels). The two patients who responded well to standard therapy protocols (#1 and #2) showed overall equal or even lower YB-1 expression (MFI), just as unstimulated CD4+ T cells from healthy donors (Figure 6c right upper two panels). Looking at individual nuclei, they contained one-third or two-third of a YB-1low fraction, respectively. Indeed, just as in bone marrow, a fraction of the cells expressing very low amounts of YB-1 in the nucleus was detected in all three patients but not in healthy controls. In addition, also a small fraction with very high YB-1 content appeared. Interestingly, only the T-ALL patient who suffered from fatal relapse within 2 years after diagnosis exhibited an overall 1.5-fold enhanced MFI of nuclear YB-1, that is, as high as for strongly activated CD4+ T cells of healthy donors (Figure 6c; lower right versus lower left panel). Besides an enhanced overall MFI, patient #3 showed a YB-1low subfraction and, most importantly, more than half of the nuclei had a very high content of YB-1, comparable to that of strongly activated T cells and even as high as T-ALL lines (Figures 6a and d). Thus, all patients showed deregulated nuclear YB-1 in T-ALL blasts in bone marrow and peripheral blood.
Collectively, our results demonstrate that RSK-dependent nuclear localization of YB-1 is a key limiting step to cell-cycle progression of primary and malignant T cells. Our studies further support an oncogenic role of deregulated nuclear YB-1 in T-ALL.
Discussion
In the present study, we have uncovered YB-1 as a central player driving proliferation of primary and malignant T-cell blasts. We found that YB-1, but not its paralog DbpA, is induced upon T-cell stimulation in primary CD4+ T cells. This observation is reminiscent to previous reports on stimulation-dependent enhancement of YB-1 in non-lymphoid cell types.34, 35, 36, 37 Although the low constitutive YB-1 level in non-stimulated T cells might be important for slow, homeostatic proliferation of resting T cells,38, 39 our data indicate that strong upregulation of YB-1 is obligatory for extensive T-cell expansion. Specifically, this conclusion was drawn from our findings that (i) Ki-67high leukemic blasts express very high levels of YB-1, (ii) only YB-1high T cells extensively proliferate and (iii) YB-1 inactivation abrogates cell proliferation (Figures 4a and d). Consistent with this notion, we found that co-stimulation of T cells with anti-CD3/anti-CD28 as compared with stimulation with anti-CD3 alone significantly induced YB-1 protein expression (Figures 1c and d), suggesting that YB-1 may be involved in mediating pro-proliferative effects of CD28 such as IL-2 mRNA stabilization and upregulation of G1 kinases. In light of a previous study showing that YB-1 stabilizes IL-2 transcripts in Jurkat cells40, 41and the here presented effects of YB-1 on T-cell proliferation (Figures 4a and c), it is conceivable that YB-1 and CD28 signaling pathways are interconnected.27 However, as YB-1 did not regulate cyclin D2, it is not just amplifying CD28 or TCR effects in general.42 Moreover, the herein reported proliferation control by YB-1 was not solely a consequence of enhanced IL-2 production, as YB-1-dependent upregulation of G1 kinase cdk6 occurs independently from IL-2.27
Subcellular localization and regulatory activities of YB-1 are tightly controlled by several mechanisms, including phosphorylation. Several studies suggested that, next to Akt, RSK and isoforms of PKC (α, β, and γ) may regulate YB-1 via Ser102 phosphorylation.22, 24, 43 For PKCα, however, only 19% phosphorylation efficiency was demonstrated for a YB-1 target peptide.24 Additionally, although Akt was originally the main candidate to phosphorylate and activate YB-1 in breast cancer cell lines,22 it was corrected to be less important compared with RSK.24 Distinguishing between conventional T cells and T-cell blasts, RSK inhibition, and not Akt or PKC inhibition, could specifically inhibit YB-1 nuclear localization and proliferation in aggressively expanding T-ALL cell lines. Thus targeting RSK for restraining aggressive expansion of leukemic tumor cells might be an attractive option.
As we find that TCR and CD28 signaling induces not only the expression of YB-1 but also triggers its phosphorylation and translocation into the nucleus, both signals regulate YB-1 in at least two ways, transcriptionally and posttranslationally. In malignant T-cell lines, where YB-1 is highly expressed and found in the nucleus, inhibition of YB-1 expression led to cell-cycle arrest (Figure 5c), presumably owing to YB-1 dilution during mitosis. YB-1 thus emerges as a promising target to halt cancer progression. Resting cancer cells, however, might not be affected by suppression of YB-1, that is, the disease might persist in a quiet but chronic state. On the other hand, as even homeostatic proliferation of T cells might need low amounts of YB-1, quiescent leukemic cells with low-proliferation rates might be eradicated by long-term manipulation of YB-1.40, 41
T-memory cells in a resting state favor the bone marrow as a survival niche and lymphocyte-derived tumor cells might behave similarly.44, 45, 46 Just as our T-ALL tumor blasts, T-memory cells showed reduced proliferation and acquire a resting state, although they may be reactivated rapidly. It is tempting to speculate that tumor cells in these niches exploit mechanisms used by memory T cells, e.g. contact with stroma cells, availability of surviving factors such as IL-7 to keep cells in a low proliferative state, which likely is accompanied by nuclear YB-1 exclusion.46, 47, 48
We have illustrated that a characteristic of malignant T-ALL blasts and cell lines is deregulated YB-1 nuclear localization. Already at diagnosis, localization and amounts of YB-1 expression in tumor blasts of the peripheral blood are likely connected to patient’s outcome (Figure 6). In fact, YB-1 translocation from the cytosol to the nucleus could mark the switch from low to highly aggressive proliferating tumor cells. Thus knowing YB-1 deregulation at the time point of diagnosis could well help to determine the best therapeutic protocol for each individual patient. Of note, enhanced YB-1 expression and translocation was even superior to the common marker Ki-67 in terms of timing. Thus, besides being a potential oncogenic target, the newly identified characteristics of T-ALL cells might offer a diagnostic parameter to predict aggressiveness of cancer progression as early as possible and thus to pick the most appropriate treatment protocol for a favorable long-term outcome in patients.
Materials and methods
Antibodies, inhibitors, and plasmids
Allophycocyanin-coupled anti-CD4 (RPA-T4), FITC-coupled anti-CD45RA (HI100), PE-coupled anti-CD45RO (UCHL1), and PE-coupled anti-CD31 (WM59) antibodies (Ab) were purchased from BD Biosciences (Heidelberg, Germany). PeCy7-coupled anti-Ki-67 (Ki-67) Ab was obtained from BioLegend (San Diego, CA, USA). Primary Ab for YB-1 C-terminal (EP2708Y) was purchased from Abcam (Cambridge, UK). Ab for YB-1 (59-Q), GAPDH (A-3), and Lamin B (C-5) were acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Ab for phospho-YB-1 (Ser102; C34A2), cyclin D2 (D52F9), cdk6 (D4S8S), and p27Kip1 (D37H1) were obtained from Cell Signaling (Danvers, MA, USA). A peptide-based affinity-purified anti-DbpA Ab was generated by Eurogentech (Liege, Belgium). Secondary Abs, such as Brilliant Violet 421 donkey anti-rabbit (Poly4064) and Alexa Fluor 647 donkey anti-rabbit (Poly4064), were obtained from BioLegend. Green or red infrared dye-conjugated secondary Abs IRDye 800CW goat anti-mouse IgG, IRDye 800CW goat anti-rabbit IgG, IRDye 680RD goat anti-mouse IgG, and IRDye 680RD goat anti-rabbit IgG were purchased from LI-COR (Lincoln, NE, USA). Akt (II) and RSK (SL0101) inhibitors were purchased from Merck Millipore (Darmstadt, Germany), and PKC inhibitor Rottlerin from Santa Cruz Biotechnology. Nocodazole and Paclitaxel were obtained from Sigma-Aldrich (St. Louis, MO, USA). Fisetin was purchased from Santa Cruz Biotechnology. The DNA constructs pCG-HΔ24 and pCG-FΔ30 for transduction using measles virus system was kindly provided by F-L Cosset and F Fusil (International Center for Infectiology Research; University of Lyon, Lyon, France). The expression vectors pLKO and pLKO-YB-1shRNA were purchased from Sigma-Aldrich and were further genetically modified.
Samples
PBMCs were obtained from leukocyte reduction filters (Sepacell RZ-2000; Asahi Kasei Medical, Tokyo, Japan) from the Department of Transfusion Medicine and Immunohematology and blood bank at the University Hospital Magdeburg. Material from blood and bone marrow punctures of T-ALL patients were collected at the Division of Hematology and Oncology, University Hospital Magdeburg, Magdeburg, Germany and the Division of General Pediatrics, University Hospital Schleswig Holstein in Kiel, Germany. This study was approved by the local Ethics Committee (MD 115/08) and the institutional review board, and all patients provided informed written consent in accordance with the Declaration of Helsinki.
Human T-cell purification
PBMCs from filters from healthy donors were purified using a Ficoll 400-based (Lymphocyte Separation Medium LSM 1077; PAA Laboratories, Pasching, Austria) density gradient. Total CD4+ T cells (naive, effector, memory) were enriched using CD4-MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and autoMACS-Pro isolation (Miltenyi directly) from purified PBMCs according to the manufacturer’s instructions. CD4+ T cells were maintained at 37 °C in serum-free X-VIVO 20 medium (Lonza, Verviers, Belgium) supplemented with 10 μg/ml streptomycin and 10 U/ml penicillin (Life Technologies GmbH, Darmstadt, Germany).
Cell lines
The T-ALL cell lines Jurkat, Molt-16, and RPMI-8402 were provided by the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Cells were cultured in RPMI-1640 medium containing 10% fetal calf serum (Biochrom AG, Berlin, Germany), 2 mM L-glutamine, 10 μg/ml streptomycin, and 10 U/ml penicillin (Life Technologies GmbH). HEK-293T cells were cultured in DMEM medium containing high glucose, sodium pyruvate, 10% fetal calf serum (Biochrom AG), 10 μg/ml streptomycin and 10 U/ml penicillin (Life Technologies GmbH). All cell cultures were maintained at 37 °C and 5% CO2.
T-cell activation
Cells were incubated either with 1 μg/ml ConA or 1 μg/ml superantigen SEB, or 10 ng/ml PMA and 1 μg/ml Ionomycin (Sigma-Aldrich). For polyclonal stimulation, both 5 × 105 CD4+ T cells and 2 × 105 Jurkat cells were cultured in U-bottom 96-well plates with latex beads (PostNova Analytics, Salt Lake City, UT, USA), which were either coated with anti-CD3 (UCHT1; BD Biosciences) or anti-CD3 and anti-CD28 (CD28.2; BD Biosciences) at a cell-to-bead ratio of 1:1. The beads were prepared by coupling either 2.5 μg/ml IgG1 isotype (MG1-45; BD Biosciences) Ab only or 0.5 μg/ml anti-CD3 and 2 μg/ml IgG1 isotype Abs or 0.5 μg/ml anti-CD3 and 2 μg/ml anti-CD28 Abs.
RNA isolation and qRT-PCR
Extraction of total RNA and performance of qRT-PCR were described before.49 Appropriate primers were obtained from TIB MolBiol (Berlin, Germany) and sequences are described in Supplementary Table S1. Data analyses were carried out using the CFX96 Manager Software (Bio-Rad, Hercules, CA, USA). Fold change in the expression of each gene was normalized to the expression of GAPDH using the 2−ΔΔCT method.50
Subcellular fractionation
To produce cytoplasmic and nuclear extracts, cells were washed and suspended in buffer A (10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 1 mM sodium orthovanadate, complete Mini protease inhibitor cocktail (Roche Diagnostics GmbH; Mannheim, Germany) for 2 min on ice, followed by addition of 0.5% Nonidet P-40 (Sigma-Aldrich) for further 2 min. After centrifugation at 14 000 × g, the supernatant was collected as cytoplasmic extract. The pellet was washed in buffer A and incubated in buffer C (20 mM HEPES (pH 7.9), 420 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM sodium orthovanadate, protease inhibitor cocktail) for at least 2 h with constant shaking at 4 °C. Nuclear extract was collected by centrifugation at 14 000 × g for 20 min. Protein concentrations were determined using BCA protein assay (Thermo Scientific, Waltham, MA, USA).
Nuclei from T cells and samples from blood and bone marrow punctures of T-ALL patients were extracted using sucrose gradient centrifugation. Briefly, 1 × 107 cells were incubated in lysis buffer (320 mM sucrose, 10 mM HEPES, 8 mM MgCl2, EDTA-free protease inhibitor, and 0.5% Triton X-100 (pH 7.6)) for 20 min on ice followed by immediate centrifugation at 600 × g. The pellet was washed twice in wash buffer (320 M sucrose, 10 M HEPES, 8 mM MgCl2, EDTA-free protease inhibitor).
Immunoblotting
Whole-cell lysates were prepared according to Hebel et al.49 The membranes were incubated with Odyssey Blocking Buffer (ODBB) for 1 h at RT and then probed with primary Ab in ODBB/0.1% Tween-20 overnight at 4 °C. Washes (3 ×) with PBS/0.1% Tween-20 (PBS-T) and incubation with secondary Ab for 1 h at RT followed by washes with PBS-T and PBS. For detection, Odyssey infrared dual color imaging system (LI-COR) was used. Quantification of protein expression was determined using appropriate housekeeping protein expression.
Flow cytometric analysis
For intracellular YB-1 staining, cells were fixed with 2% paraformaldehyde, permeabilized with 0.5% saponin (Sigma-Aldrich) in PBS/0.2% BSA, and incubated with primary anti-YB-1 C-terminal Ab and secondary Ab. Cells were washed twice with PBS/0.2% BSA before analysis. For nuclear staining, isolated nuclei were fixed and permeabilized using the Foxp3 Intracellular/Nuclear Staining Kit (eBiosciences, San Diego, CA, USA). Nuclei were stained using Abs as described above and labeled with 0.2 μg/ml DAPI.
For proliferation analysis, cells were labeled with 1.6 μM CFSE or with 5 μM CellTrace Violet reagent (Life Technologies) and quenched with serum-containing medium. Cytometric analysis were performed using FACS Canto II (BD Biosciences) together with the FACS Diva software Version 6.1.2 (BD Biosciences) to collect and compensate the data and the FlowJo software Version 7.6.5 (Tree Star, Ashland, OR, USA) for data analysis.
Plasmid construction
For the generation of YB-1 mutants, the open reading frame of human YB-1 was PCR-amplified from cDNA clone IRATp970G04108D (Source BioScience, Nottingham, UK; Supplementary Table S2) and inserted into the pEGFP-C1 vector. The EGFP-YB-1 DNA was then inserted as a NheI-XbaI-fragment into the lentiviral vector FuGW (Addgene Plasmid no. 14883). Two point mutations were introduced to convert the S102-encoding AGT to GAT or AAT encoding for aspartic acid or asparagine. Briefly, in two PCR runs, point mutation-carrying primers (Supplementary Table S2) generated two overlapping fragments and the adjoining sequence of the FuGW (overlapping by 10–18 bp). PCR products were purified using a NucleoSpin Extract II Extraction Kit (MachereyNagel, Bethlehem, PA, USA). These fragments were combined with the FuGW-EGFP-YB-1wt that was cut before with BspEI, using Cold Fusion (System Biosciences, Mountain View, CA, USA). Briefly, the opened vector and both PCR fragments were mixed with the Cold Fusion reaction mix and H2O and incubated 5 min by RT followed by 10 min on ice. Subsequently, the reaction was used for transformation into E.coli.
For YB-1 knockdown using measles viral transduction, the commercially purchased pLKO and pLKO-YB-1 plasmids were genetically modified by deleting the puromycin-resistance gene and adding the cytoplasmic domain-depleted nerve growth factor receptor gene (ΔNGFR) as selection marker. In brief, pLKO-YB-1shRNA was cut with BamHI and SalI and control pLKO was cut with BamHI and KpnI to remove Puromicin-resistance gene. To add the ΔNGFR gene for selection, PCR was performed using the specific primers (Supplementary Table S2). Resulting fragments were combined with Cold Fusion and transformed as described before. All constructs were sequence verified.
Viral transduction of CD4+ T cells T cells
Lentiviral and measles viral particles were generated in HEK-293T cells with transfer vector (GFP-YB-1S102D or GFP-YB-1S102N for lentiviral transduction and pLKO or pLKOYB-1shRNA for measles viral transduction), packaging plasmid psPAX2, and envelope plasmid pVSVG (lentiviral) or pCG-HΔ24 and pCG-FΔ30 (measles viral) using calcium phosphate precipitation. After 48 and 72 h, supernatants containing lentiviral or measles viral particles were collected. For lentiviral transduction, CD4+ T cells were prestimulated for 48 h with anti-CD3/anti-CD28-coated beads. Culture plates were coated with 18 μg/ml Retronectin (Takara Shuzo Co., Otsu, Japan) overnight and blocked with 2% BSA. After washes with PBS, the concentrated lentiviral or measles viral particles were centrifugation-bound to wells for 2 h at 2000 × g. Prestimulated CD4+ T cells (1 × 105 cells/well) for lentiviral infection were added, whereas for the infection with measles virus resting CD4+ T cells were used. After overnight incubation, supernatants were replaced with media. Transduction efficiencies were assessed by FACS analysis 48 h after infection.
Fluorescence microscopy
GFP-YB-1 localization of CD4+ T cells was analyzed 48 h after transduction with the GFP-tagged YB-1 lentiviral particles. Cells were plated on 35 mm glas bottom dishes (Fluorodish WPI Inc., Sarasota, FL, USA). Nuclei were counterstained with Hoechst 33342 (Life Technologies GmbH) and the localization of the GFP-tagged YB-1 was visualized using a Zeiss Axiovert 200 m equipped with a Neo-Fluor 40 × /0.75 Ph2, an AxioCam MRm, fluorescence filter set Zeiss No. 49, and HQ-FITC (EX BP 480/40, BS 505, EM 535/50) (Chroma Technology Corp., Bellows Falls, VT, USA).
Silencing of YB-1 expression by siRNA and lentiviral shRNA
CD4+ T cells were either transfected with predesigned 1 μM YB-1siRNA (no. 115541; Life Technologies GmbH) or 1 μM All Stars negative siRNA AF488 (Qiagen, Hilden, Germany) using the Human T-cell Nucleofector Kit according to the manufacturer’s instruction (Lonza). After 6–24 h of transfection, CD4+ T cells were stimulated with anti-CD3/anti-CD28-coupled latex beads. Transfection rate was determined after 24–72 h of incubation by FACS and immunoblotting. For lentiviral transduction, Jurkat cells were infected with an MOI of 10 of YB-1shRNA-containing lentiviral particle (Santa Cruz Biotechnology) according to the manufacturer’s protocol. Cells were stained using trypan blue and analyzed by microscopy.
Statistical Analysis
A two-tailed Student’s t-test was applied to perform statistical analysis. For multiparametric experiments, an ANOVA test was performed. Results were considered significant at *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Calculations were performed with GraphPad Prism 6 (San Diego, CA, USA).
References
Pui CH, Robison LL, Look AT . Acute lymphoblastic leukemia. Lancet 2008; 371: 1030–1043.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pilieri SA, Stein H et al. WHO Classification of Tumours of Hematopoetic and Lymphoid Tissue 4th edn. IARC Press, 2008.
Jain P, Kantarjian H, Ravandi F, Thomas D, O'Brien S, Kadia T et al. The combination of hyper-CVAD plus nelarabine as frontline therapy in adult T-cell acute lymphoblastic leukemia and T-lymphoblastic lymphoma: MD Anderson Cancer Center experience. Leukemia 2014; 28: 973–975.
Bhojwani D, Pui CH . Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol 2013; 14: e205–e217.
Vogelstein B, Kinzler KW . The path to cancer-three strikes and you’re out. N Engl J Med 2015; 373: 1895–1898.
To K, Fotovati A, Reipas KM, Law JH, Hu K, Wang J et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res 2010; 70: 2840–2851.
Lasham A, Samuel W, Cao H, Patel R, Mehta R, Stern JL et al. YB-1, the E2F pathway, and regulation of tumor cell growth. J Natl Cancer Inst 2012; 104: 133–146.
Evdokimova VM, Ovchinnikov LP . Translational regulation by Y-box transcription factor: involvement of the major mRNA-associated protein, p50. J Biol Chem 1999; 31: 139–149.
Pisarev AV, Skabkin MA, Thomas AA, Merrick WC, Ovchinnikov LP, Shatsky IN . Positive and negative effects of the major mammalian messenger ribonucleoprotein p50 on binding of 40S ribosomal subunits to the initiation codon of beta-globin mRNA. J Biol Chem 2002; 277: 15445–15451.
Nekrasov MP, Ivshina MP, Chernov KG, Kovrigina EA, Evdokimova VM, Thomas AA et al. The mRNA-binding protein YB-1 (p50) prevents association of the eukaryotic initiation factor eIF4G with mRNA and inhibits protein synthesis at the initiation stage. J Biol Chem 2003; 278: 13936–13943.
Guryanov SG, Filimonov VV, Timchenko AA, Melnik BS, Kihara H, Kutyshenko VP et al. The major mRNP protein YB-1: structural and association properties in solution. Biochim Biophys Acta 2013; 1834: 559–567.
Kohno Y, Matsuki Y, Tanimoto A, Izumi H, Uchiumi T, Kohno K et al. Expression of Y-box-binding protein dbpC/contrin, a potentially new cancer/testis antigen. Br J Cancer 2006; 94 (710–716): 13.
Raffetseder U, Liehn EA, Weber C, Mertens PR . Role of cold shock Y-box protein-1 in inflammation, atherosclerosis and organ transplant rejection. Eur J Cell Biol 2012; 91: 567–575.
Evdokimova V, Ruzanov P, Anglesio MS, Sorokin AV, Ovchinnikov LP, Buckley J et al. Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol Cell Biol 2006; 26: 277–292.
Wolffe AP . Structural and functional properties of the evolutionarily ancient Y-box family of nucleic acid binding proteins. Bioessays 1994; 16: 245–251.
Ladomery M, Sommerville J . A role for Y-box proteins in cell proliferation. Bioessays 1995; 17: 9–11.
Chang YW, Mai RT, Fang WH, Lin CC, Chiu CC, Wu Lee YH . YB-1 disrupts mismatch repair complex formation, interferes with MutSα recruitment on mismatch and inhibits mismatch repair through interacting with PCNA. Oncogene 2014; 33: 5065–5077.
Panupinthu N, Yu S, Zhang D, Zhang F, Gagea M, Lu Y et al. Self-reinforcing loop of amphiregulin and Y-box binding protein-1 contributes to poor outcomes in ovarian cancer. Oncogene 2014; 33: 2846–2856.
Lyabin DN, Eliseeva IA, Ovchinnikov LP . YB-1 protein: functions and regulation. Wiley Interdiscip Rev RNA 2014; 5: 95–110.
Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M . The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays 2003; 25: 691–698.
Jurchott K, Bergmann S, Stein U, Walther W, Janz M, Manni I et al. YB-1 as a cell cycle-regulated transcription factor facilitating cyclin A and cyclin B1 gene expression. J Biol Chem 2003; 278: 27988–27996.
Sutherland BW, Kucab J, Wu J, Lee C, Cheang MC, Yorida E et al. Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 2005; 24: 4281–4292.
Bader AG, Vogt BK . Phosphorylation by Akt disables the anti-oncogenic activity of YB-1. Oncogene 2008; 27: 1179–1182.
Stratford AL, Fry CJ, Desilets C, Davies AH, Cho YY, Li Y et al. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res 2008; 10: R99.
Truitt KE, Nagel T, Suen LF, Imboden JB . Structural requirements for CD28-mediated costimulation of IL-2 production in Jurkat T-cells. J Immunol 1996; 156: 4539–4541.
Ward SG, June CH, Olive D . PI 3-kinase: a pivotal pathway in T-cell activation? Immunol Today 1996; 17: 187–197.
Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP . CTLA-4-mediated inhibition of early events of T-cell proliferation. J Immunol 1999; 162: 5813–5820.
Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB . Regulation of lymphokine messenger RNA stability by a surface-mediated T-cell activation pathway. Science 1989; 244: 339–343.
Fraser JD, Irving BA, Crabtree GR, Weiss A . Regulation of interleukin-2 gene enhancer activity by the T-cell accessory molecule CD28. Science 1991; 251: 313–316.
Burger R, Hansen-Hagge TE, Drexler HG, Gramatzki M . Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: suggestion for classification by immunophenotype and T-cell receptor studies. Leuk Res 1999; 23: 19–27.
Basaki Y, Hosoi F, Oda Y, Fotovati A, Maruyama Y, Oie S et al. Akt-dependent nuclear localization of Y-box-binding protein 1 in acquisition of malignant characteristics by human ovarian cancer cells. Oncogene 2007; 26: 2736–2746.
Hauge C, Frödin M . RSK and MSK in MAP kinase signalling. J Cell Sci 2006; 119: 3021–3023.
Khan MI, Adhami VM, Lall RK, Sechi M, Joshi DC, Haidar OM et al. YB-1 expression promotes epithelial-tomesenchymal transition in prostate cancer that is inhibited by a small molecule fisetin. Oncotarget 2014; 5: 2462–2474.
Ito K, Tsutsumi K, Kuzumaki T, Gomez PF, Otsu K, Ishikawa K . A novel growth-inducible gene that encodes a protein with a conserved cold-shock domain. Nucleic Acids Res 1994; 22: 2036–2041.
Stenina OI, Poptic EJ, DiCorleto PE . Thrombin activates a Y box-binding protein (DNA-binding protein B) in endothelial cells. J Clin Invest 2000; 106: 579–587.
Raffetseder U, Rauen T, Djudjaj S, Kretzler M, En-Nia A, Tacke F et al. Differential regulation of chemokine CCL5 expression in monocytes/macrophages and renal cells by Y-box protein-1. Kidney Int 2009; 75: 185–196.
Keilhoff G, Titze M, Esser T, Langnaese K, Ebmeyer U . Constitutive and functional expression of YB-1 in microglial cells. Neuroscience 2015; 301: 439–453.
Surh CD, Sprent J . Homeostasis of naive and memory T-cells. Immunity 2008; 29: 848–862.
Sprent J, Surh CD . Normal T-cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol 2011; 12: 478–484.
Chen CY, Gherzi R, Andersen JS, Gaietta G, Jürchott K, Royer HD et al. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev 2000; 14: 1236–1248.
Seko Y, Cole S, Kasprzak W, Shapiro BA, Ragheb JA . The role of cytokine mRNA stability in the pathogenesis of autoimmune disease. Autoimmun Rev 2006; 5: 299–305.
Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A . T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science 1999; 283: 680–682.
Nishizuka Y . Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992; 258: 607–614.
Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, Grün JR, Löhning M et al. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity 2009; 30: 721–730.
Hanazawa A, Löhning M, Radbruch A, Tokoyoda K . CD49b/CD69-dependent generation of resting T helper cell memory. Front Immunol 2013; 4: 183.
Okhrimenko A, Grün JR, Westendorf K, Fang Z, Reinke S, von Roth P et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc Natl Acad Sci USA 2014; 111: 9229–9234.
Schirrmacher V, Feuerer M, Beckhove P, Ahlert T, Umansky V . T-cell memory, anergy and immunotherapy in breast cancer. J Mammary Gland Biol Neoplasia 2002; 7: 201–208.
Müller M, Gounari F, Prifti S, Hacker HJ, Schirrmacher V, Khazaie K . EblacZ tumor dormancy in bone marrow and lymph nodes: active control of proliferating tumor cells by CD8+ immune T-cells. Cancer Res 1998; 58: 5439–5446.
Hebel K, Weinert S, Kuropka B, Knolle J, Kosak B, Jorch G et al. CD4+ T-cells from human neonates and infants are poised spontaneously to run a nonclassical IL-4 program. J Immunol 2014; 192: 5160–5170.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001; 25: 402–408.
Acknowledgements
The study was supported by the Sander Foundation (No. 2012.090.1) (to MCB-W), DFG Br1860/8 (to MCB-W), SFB854 TP1 (PRM), A20 (to FHH, TF), B08 (to JH, UT), TP14 (to MCB-W), ME-1365/7-2, ME-1365/9-1 (to PRM), LI-1031/4-1 (to JAL), and the Jose-Carreras Leukemia Foundation (DJCLF SP12/08) (HTM) (to FHH). We thank G Jorch for support. We also thank K Schulze-Osthoff for helpful discussion and B Kramer and K Kramer for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Gieseler-Halbach, S., Meltendorf, S., Pierau, M. et al. RSK-mediated nuclear accumulation of the cold-shock Y-box protein-1 controls proliferation of T cells and T-ALL blasts. Cell Death Differ 24, 371–383 (2017). https://doi.org/10.1038/cdd.2016.141
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.141
This article is cited by
-
Upregulated PARP1 confers breast cancer resistance to CDK4/6 inhibitors via YB-1 phosphorylation
Experimental Hematology & Oncology (2023)
-
YB-1 activating cascades as potential targets in KRAS-mutated tumors
Strahlentherapie und Onkologie (2023)
-
Combination of RSK inhibitor LJH-685 and FLT3 inhibitor FF-10101 promoted apoptosis and proliferation inhibition of AML cell lines
Cellular Oncology (2022)
-
Cold shock proteins: from cellular mechanisms to pathophysiology and disease
Cell Communication and Signaling (2018)
-
Fisetin targets YB-1/RSK axis independent of its effect on ERK signaling: insights from in vitro and in vivo melanoma models
Scientific Reports (2018)
