
Abstract
Background:
This phase Ib study evaluated afatinib plus vinorelbine in patients with advanced solid tumours overexpressing epidermal growth factor receptor (EGFR) and/or human EGFR 2 (HER2).
Methods:
Maximum tolerated doses (MTDs) were determined for afatinib (20, 40 or 50 mg, once daily) combined with standard intravenous vinorelbine (part A; 25 mg m−2 per week) or oral vinorelbine (part B; 60 mg m−2 per week, increased to 80 mg m−2 per week at week 3). Secondary end points for expanded MTD cohorts included assessments of safety, pharmacokinetics, tumour response and progression-free survival (PFS).
Results:
The afatinib MTD was 40 mg with intravenous (MTDA) and oral (MTDB) vinorelbine. The most frequent cycle 1 dose-limiting toxicities were febrile neutropenia and diarrhoea, consistent with individual safety profiles of vinorelbine and afatinib. Common treatment-related adverse events included: diarrhoea (92.7%), asthenia (76.4%), nausea (63.6%), neutropenia (56.4%) and vomiting (54.5%). No notable pharmacokinetic interactions were observed. Best overall tumour response was stable disease in part A (16 out of 28 patients), and partial response in part B (3 out of 27 patients). Median PFS was 14.6 and 15.9 weeks for patients treated at the MTDA and MTDB, including dose-escalation and expansion cohorts.
Conclusions:
Afatinib in combination with intravenous or oral vinorelbine demonstrated a manageable safety profile and antitumour activity at the MTD of 40 mg per day.
Similar content being viewed by others

Main
The ErbB receptor family of tyrosine kinases, comprising epidermal growth factor receptor (EGFR; ErbB1), human EGFR 2 (HER2; ErbB2), HER3 (ErbB3) and HER4 (ErbB4), plays a central role in cell proliferation and survival, and dysregulation of ErbB receptor signalling promotes tumour progression, metastasis and angiogenesis (Salomon et al, 1995; Lurje and Lenz, 2009; Yarden and Pines, 2012). Abnormalities of ErbB family function and expression are common in epithelial tumours such as breast cancer and lung cancer (Burstein, 2005; Hynes and Lane, 2005; Doebele et al, 2010). For instance, ∼20% of breast cancers overexpress HER2, and as many as 75% and 35% of non-small cell lung cancers (NSCLCs) overexpress EGFR and HER2, respectively (Owens et al, 2004; Yaziji et al, 2004; Soria et al, 2012; Wong and Lee, 2012; Garrido-Castro and Felip, 2013). Further, EGFR mutations are found in 10% to 20% of Caucasian and up to 50% of Asian NSCLC patients (Rosell et al, 2009; Shi et al, 2014). In this context, the ErbB receptors have been extensively studied as targets for selective anticancer therapies, particularly in lung and breast cancer, with numerous such agents currently in clinical use or development (Yarden and Pines, 2012).
Afatinib is an irreversible ErbB family blocker that covalently binds to EGFR, HER2 and HER4, and inhibits signalling from all ErbB family homo- and heterodimers (Li et al, 2008; Solca et al, 2012). In two pivotal phase III studies, afatinib significantly improved progression-free survival (PFS) and patient-reported outcomes vs standard platinum-based doublet chemotherapy in treatment-naïve patients with EGFR mutation-positive NSCLC (Sequist et al, 2013; Yang et al, 2013a; Wu et al, 2014; Geater et al, 2015), and improved overall survival (OS) in patients with EGFR Del19 mutation-positive disease (Yang et al, 2015). More recently, afatinib significantly improved PFS, objective response rate and time to treatment failure compared with gefitinib in a phase IIb trial in this setting (Park et al, 2016). Furthermore, in a recent phase III trial in patients with advanced squamous cell carcinoma (SCC) of the lung who had progressed after receiving platinum-based chemotherapy, afatinib significantly improved PFS and OS vs erlotinib (Soria et al, 2015). Based on these data, afatinib is approved for first-line treatment of patients with EGFR mutation-positive NSCLC, as well as second-line treatment of patients with SCC of the lung following first-line chemotherapy.
Afatinib has also demonstrated clinical activity in breast cancer (Lin et al, 2012) and head and neck squamous cell carcinoma (HNSCC) (Seiwert et al, 2014; Machiels et al, 2015). Preclinical antitumour activity with afatinib has been observed in several other cancer types with ErbB network dysfunction, such as pancreatic cancer and colorectal cancer (Modjtahedi et al, 2014). In preclinical studies, afatinib combined with vinorelbine, an antimitotic chemotherapeutic agent registered in Europe for the treatment of metastatic breast cancer and NSCLC (medac GmBH, 2014, 2017; Pierre Fabre Ltd, 2011), demonstrated significantly enhanced antitumour activity compared with either afatinib or vinorelbine alone in a trastuzumab-resistant model (Bahleda et al, 2011). It was thus hypothesised that the addition of vinorelbine may enhance the antitumour activity of afatinib in tumour types exhibiting ErbB family signalling abnormalities. In addition, the combination of afatinib and vinorelbine is of particular interest due to the potential for combined oral administration, and the largely non-overlapping safety profiles of the drugs, including common adverse events (AEs) of rash/acne and diarrhoea for afatinib (European Medicines Agency, 2013; Food and Drug Administration, 2013), and bone marrow depression and gastrointestinal toxicity, most notably including constipation, for vinorelbine (intravenous or oral) (medac GmbH, 2014, 2017).
The phase Ib study reported here evaluated the maximum tolerated doses (MTDs) of daily oral afatinib in combination with either intravenous or oral vinorelbine in patients with advanced, non-resectable and/or metastatic solid tumours, historically known to overexpress EGFR and/or HER2. For both treatment combinations, expansion cohorts were planned at the MTD to further assess pharmacokinetics (PKs) and safety.
Patients and methods
Patient selection
Eligible patients were aged 18 years or older with histologically or cytologically confirmed advanced, non-resectable and/or metastatic solid tumours that were historically known to overexpress EGFR and/or HER2. Further eligibility criteria included an Eastern Cooperative Oncology Group performance status of 0 or 1; adequate liver, kidney, cardiac and bone marrow function; recovery from previous surgery; and recovery from toxicities of previous chemotherapy, hormone therapy, immunotherapy or radiotherapy to National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI CTCAE; version 3.0) grade 1 or below. Key exclusion criteria included untreated or symptomatic brain metastases, pre-existing interstitial lung disease and HER2-directed therapy, chemotherapy, immunotherapy, radiotherapy, biological therapies or investigational therapy during the trial or within 4 weeks before first trial treatment (2 weeks for EGFR-directed therapy and hormone therapy for breast cancer, and 6 weeks for mitomycin C or nitrosoureas).
Study design and dosing regimens
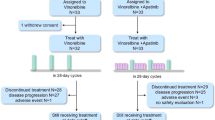
This was a phase Ib, open-label dose-escalation study conducted at two centres in France. A standard 3+3 dose-escalation design was used, wherein patients were sequentially allocated to treatment cohorts of escalating doses of afatinib. Successive cohorts received afatinib 20, 40 or 50 mg once daily, in combination with a fixed dose of vinorelbine 25 mg m−2 intravenously weekly in part A, or vinorelbine 60 mg m−2 oral weekly (increased to 80 mg m−2 oral weekly at week 3) in part B; enrolment to parts A and B was consecutive. The administered doses for vinorelbine in parts A and B were based on recommendations in the summary of product characteristics. Treatment cycles were 28 days in duration, with vinorelbine administered on days 1, 8, 15 and 22 of each cycle, and afatinib administered daily from day 2 of the first cycle onwards, in order to allow for appropriate PK assessment of each agent.
Three patients were initially enrolled to each cohort. In the event of one out of three patients experiencing dose-limiting toxicity (DLT) during the first treatment cycle, another three patients were to be treated at the same dose level. If 0 out of 3 or 1 out of 6 patients experienced DLT, dose escalation could proceed. Dose escalation was stopped if ⩾2 out of 3 or out of 6 patients experienced DLT. The following treatment-related AEs were defined as DLTs: grade 4 neutropenia that was uncomplicated (not associated with fever >38.5 °C) for >7 days; neutropenia of any duration associated with fever >38.5 °C; platelets <25 000/μl or grade 3 thrombocytopenia associated with bleeding requiring transfusion; grade 3 or 4 non-haematologic toxicity (except alopecia, incompletely treated nausea, untreated vomiting or untreated diarrhoea); grade ⩾2 decrease in cardiac left ventricular function; grade ⩾2 worsening of renal function (as measured by serum creatinine, newly developed proteinuria or newly developed decrease in glomerular filtration rate); grade ⩾2 diarrhoea, nausea and/or vomiting persisting for 7 or more days, despite supportive treatment (loperamide or other antidiarrhoeal medication for diarrhoea, and antiemetic treatment for nausea and/or vomiting). Grade 3 neutropenia related to vinorelbine during cycle 1 was also defined as a DLT, and administration of vinorelbine had to be postponed until recovery from the DLT to grade <1. If one of four doses of vinorelbine was missed, this was not considered to be a DLT. If >1 dose of vinorelbine was postponed or missed during cycle 1 for any reason, the event was considered to be a DLT. The MTD was defined as the dose at which ≤1 of six patients experienced DLT during cycle 1. Maximum tolerated dose expansion cohorts for parts A and B included an additional 12 patients to obtain further PK data for the drug combination; safety and antitumour activity was also assessed.
Patients continued therapy at the same dose level until disease progression, DLTs or unacceptable AEs. Afatinib treatment was discontinued or stopped for recovery from the AE and continued at a reduced dose. A maximum of two dose reductions of 10-mg decrements to a minimum dose of 20 mg were allowed. In general, treatment was discontinued if a DLT did not resolve to NCI CTCAE grade ≤1 within 14 days of treatment suspension. Vinorelbine was either dose-reduced or doses were omitted according to the summary of product characteristics until recovery of the AE to NCI CTCAE grade <1. If vinorelbine had to be stopped due to an AE, patients were allowed to continue afatinib monotherapy in case of clinical benefit and absence of progressive disease (PD).
The study was conducted and reported in accordance with the Declaration of Helsinki, the International Conference on Harmonization-Good Clinical Practice guidelines and approved by the relevant regulatory authority (Agence Nationale de Sécurité du Médicament et des Produits de Sante (ANSM), Saint Denis Cedex, France) and an independent ethics committee (Comité de Protection des Personnes Sud Ouest et Outre Mer I, Toulouse, France). All patients provided written, informed consent before study participation.
End points and assessments
The primary trial end point was to determine the MTD of afatinib in combination with intravenous or oral vinorelbine based on the occurrence of DLTs during cycle 1. Secondary end points included safety, PK assessment, best overall tumour response, objective tumour response (OR; including complete response (CR) and partial response (PR)), disease control (combining CR, PR and stable disease (SD)), duration of disease control, best percentage change in tumour size and PFS.
Safety was assessed throughout the study by monitoring AEs, with intensity graded according to the NCI CTCAE version 3.0. Antitumour activity was evaluated according to Response Evaluation Criteria in Solid Tumours version 1.0 (Therasse et al, 2000) every two cycles (8 weeks) during study conduct. A CR or PR had to be confirmed 4 weeks after initial assessment. The duration of an SD was determined to be at least 6 weeks. Disease control was defined as confirmed CR, confirmed PR and/or confirmed SD.
Pharmacokinetic blood sampling (4 ml aliquots of venous blood) was performed immediately before and during the 24 h after the start of administration of vinorelbine (intravenous or oral) on days 1/2, 15/16 and 21/22 of the first treatment cycle. Concentrations of afatinib in plasma and vinorelbine in whole blood were analysed using validated high-performance liquid chromatography tandem mass spectrometry methods. Pharmacokinetic assessments for vinorelbine (day 1/2 and day 15/16) included determination of the area under the plasma concentration–time curve over 24 h (AUC0–24 h), maximum measured plasma concentration (Cmax) and time from dosing to the maximum plasma concentration (tmax). For afatinib, PK assessments (day 15/16 and day 21/22) included area under the plasma–concentration–time curve at steady state over the dosing interval τ (AUCτ,ss), minimum and maximum measured plasma concentration at steady state (Cmax,ss) and time from dosing to the minimum and maximum plasma concentration at steady state (tmax,ss). Pharmacokinetic parameters with and without cotreatment were subjected to intraindividual comparisons to assess the effect of afatinib of the PKs of vinorelbine, and the effect of vinorelbine on afatinib.
Statistical analyses
All efficacy and safety analyses were conducted in the treated set (all patients who received at least one dose of study drug); PK analyses were conducted in all patients who received at least one dose of study drug and provided at least one blood sample following drug administration. Descriptive statistics were applied for all efficacy and safety analyses, and Kaplan–Meier estimates and 95% confidence intervals (CIs) were tabulated for analysis of PFS. For PK assessments, analysis of variance (ANOVA) was used for AUC and Cmax values. No formal statistical testing was planned.
Results
Patients
Between 17 June 2009 and 11 January 2013, 67 patients were screened and 55 were eligible to participate in the trial. Twenty-eight patients were enrolled into part A and 27 into part B (Table 1). Patient baseline characteristics are shown in Table 2. Overall, the median age of patients in the trial was 54 years and 43.6% of patients were male. The most frequent tumour types among the patients were NSCLC (n=14, 25%), HNSCC (n=5, 9%) and breast cancer (n=5, 9%). The majority of patients (85.5%) had been diagnosed with EGFR-positive tumours; 12.7%, 3.6% and 3.6% of patients had HER2 tumour scoring of 1+, 2+ or 3+, respectively. The study population was heavily pre-treated, with the majority of patients (67%) having received more than two lines of chemotherapy before study entry.
MTDs and DLTs
Dose-limiting toxicities for each cohort in parts A and B are displayed in Table 3. In part A, no DLTs were reported in the initial three patients enrolled in either the afatinib 20 or 40 mg cohorts. When one of three patients had DLTs (febrile neutropenia and mucosal inflammation) in the 50 mg cohort, the cohort was expanded to six patients. As all of these additional patients had DLTs, dose escalation was stopped and the 40 mg afatinib cohort was expanded accordingly, with one of six patients experiencing DLT. The MTDA was defined as 40 mg afatinib with intravenous vinorelbine 25 mg m−2 per week.
In part B, no DLT was observed in four patients at afatinib 20 mg (Table 3). One patient had to be replaced due to PD in cycle 1. When one of three patients had DLTs (febrile neutropenia and mucosal inflammation) in the 40 mg cohort, the cohort was expanded without additional DLTs observed, and afatinib was subsequently escalated to 50 mg. After one of three patients had DLTs (febrile neutropenia and stomatitis) at afatinib 50 mg, followed by two additional patients with DLTs (diarrhoea), the MTDB was defined as 40 mg afatinib with oral vinorelbine (60 mg m−2 per week, increased to 80 mg m−2 per week at week 3).
The MTDA and MTDB cohorts were expanded with 13 and 12 patients, respectively, for further PK analysis, particularly assessment of drug–drug interactions. An additional seven patients in the expanded MTDA expansion cohort and two patients in the MTDB expansion cohort had AEs qualifying as DLTs during cycle 1 (Table 3). Observed AEs were consistent with the known safety profiles of afatinib and vinorelbine, the most frequent events being febrile neutropenia (n=8, 14.5%) and diarrhoea (n=7, 12.7%) (Supplementary Table 1), and were generally manageable with afatinib or vinorelbine dose reduction or omission of vinorelbine doses. As shown in Table 1, DLTs led to permanent treatment discontinuation in five patients in the MTDA expansion cohort (grade 3 asthenia, grade 3 febrile neutropenia and grade 4 lipase increase (n=1); grade 3 cholestasis and grade 4 hepatic cytolysis (n=1); grade 3 dyspnoea (n=1); grade 3 oral mucositis (n=1); grade 3 febrile neutropenia and grade 3 leucopoenia (n=1)). The patient with grade 3 cholestasis and grade 4 hepatic cytolysis was rechallenged with vinorelbine alone (as standard medical treatment) following recovery from the DLT and hepatic cytolysis recurred, suggesting this event was related to vinorelbine. In the MTDB expansion cohort, one patient experienced DLT leading to permanent treatment discontinuation (grade 4 febrile neutropenia). In both the dose escalation and expansion cohorts of parts A and B, a further five patients experienced DLT during cycle 2 or later, with one patient in part A discontinuing due to grade 3 asthenia occurring 156 days after first intake of the trial drugs.
Overall safety
All 55 patients had treatment-related AEs during the course of the study (Table 4). Diarrhoea (n=51, 92.7%), asthenia (n=42, 76.4%) nausea (n=35, 63.6%), neutropenia (n=31, 56.4%) and vomiting (n=30, 54.5%) were most frequently reported. Grade 3 and 4 AEs were reported in 28 (50.9%) and 14 (25.5%) patients, respectively, with neutropenia (n=14, 25.5%) and diarrhoea (n=13, 23.6%) being the most frequently observed grade 3 AEs, and neutropenia the most frequent grade 4 AE (n=8; 14.5%).
Overall, 32 patients (58.2%) had at least one serious AE (SAE; 16 patients each in parts A and B). The most frequently reported SAEs, which occurred in at least 5% of patients, were diarrhoea (n=7, 12.7%), febrile neutropenia (n=5, 9.1%), dyspnoea (n=4, 7.3%), vomiting (n=4, 7.3%), neutropenia (n=3, 5.5%), dehydration (n=3, 5.5%) and general physical health deterioration (n=3, 5.5%). Five patients died during the trial due to general physical health deterioration (n=3), pneumonia (n=1) and acute respiratory failure (n=1). All five deaths were deemed related to disease progression and occurred during the treatment period. None of the deaths was considered to be related to study drug. Adverse events were the reason for permanent discontinuation of afatinib in 19 patients (34.5%; 10 in part A and 9 in part B). Twenty-six patients (47.3%; 12 in part A and 14 in part B) permanently discontinued vinorelbine due to AEs. Asthenia, neutropenia, diarrhoea, nausea, vomiting and febrile neutropenia were the most frequent reasons for drug discontinuation.
Pharmacokinetics
No notable differences in key PK parameters for afatinib and vinorelbine were observed in the presence or absence of the other agent (Table 5). In part A, afatinib exposure in terms of gMean values was slightly higher in the presence of intravenous vinorelbine based on AUCτ,ss and Cmax,ss, compared with afatinib exposure as monotherapy. In contrast, vinorelbine exposure was slightly lower in the presence of afatinib in terms of AUC0–24 h and Cmax, compared with vinorelbine exposure in the absence of afatinib (Table 5). In part B, exposure to both oral afatinib and vinorelbine was slightly lower in the presence of the other drug, compared with their respective monotherapy exposures. The variability of the PK parameters was moderate to high for both afatinib and vinorelbine (intravenous or oral). Results of the exploratory statistical analysis by ANOVA are shown in Supplementary Table 2. In all cases, the 90% CIs for the AUC and Cmax ratios were not located within the defined bioequivalence range of 80–125% but were overlapping, and intraindividual variability was moderate to high. Overall, these PK analyses suggested no relevant interaction between afatinib and vinorelbine in the treatment schedules evaluated.
Antitumour activity
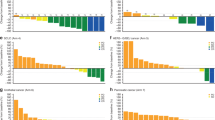
Objective response and duration of therapy for individual patients are shown in Figure 1. The best overall response observed in part A was SD, achieved by 16 of 28 patients (57%). Of these 16 patients, 4 had unconfirmed CR or PR. Median (range) duration of disease control (CR+PR+SD) was 167 (94–351) days among patients treated at the 40 mg MTDA and 168 (81–202) days in the 50 mg cohort. Only one patient exhibited disease control for 110 days in the 20 mg cohort. Median PFS of patients treated at the MTDA was 14.6 weeks (95% CI: 7.1–31.9).

Duration of therapy in individual patients by dose, objective response and tumour type. HNSCC=head and neck squamous cell carcinoma; i.v.=intravenous; NE=not estimable; NSCLC=non-small cell lung cancer; PD=progressive disease; p.o.=oral; PR=partial response; SD=stable disease.
In part B, the best overall tumour response observed was PR in 3 of 27 patients (11%; two patients with breast cancer and one with NSCLC). The median (range) duration of OR was 114.0 (113–151) days, with a median (range) time to response of 55.0 (55–343) days. The disease control rate was 52% (14 of 27 patients). Median (range) duration of disease control was 162 (50–493) days among patients treated at the 40 mg MTDB and 120 (54–230) days in the 50 mg cohort. All patients in the 20 mg cohort had PD at the first tumour assessment. Patients treated at the MTDB had a median PFS of 15.9 weeks (95% CI: 7.6–23.1).
Discussion
In this phase I study of heavily pre-treated patients with advanced solid tumours, historically known to overexpress EGFR and/or HER2, the MTD of afatinib in combination with standard dose intravenous (part A) or oral (part B) vinorelbine was 40 mg per day. The most frequently occurring DLTs in parts A and B were febrile neutropenia and diarrhoea, consistent with the established safety profiles of single-agent vinorelbine and afatinib, respectively (European Medicines Agency, 2013; Food and Drug Administration, 2013; medac GmbH, 2014, 2017). Prophylactic treatment with granulocyte-colony-stimulating factors (G-CSFs), such as filgrastim (including approved biosimilars), lenograstim or pegfilgrastim is known to reduce the risk of chemotherapy-induced neutropenia, but its use varies widely in clinical practice (Aapro et al, 2011). The overall high incidence of treatment-related febrile neutropenia observed in this study (16.4%; with all occurrences of grade 3 severity, apart from one that was grade 4) may reflect the fact that prophylactic G-CSF supportive therapy (Aapro et al, 2011) was not used. Diarrhoea is a recognised but manageable side effect of afatinib, usually occurring during the first 2 weeks of therapy (European Medicines Agency, 2013; Food and Drug Administration, 2013; Yang et al, 2013b). Although management of diarrhoea in the current study was according to standard procedures for afatinib, the incidence of grade 3 diarrhoea observed with the combination (23.6%) was higher than observed for afatinib alone (Sequist et al, 2013; Wu et al, 2014).
Overall, the AE profile observed in the current study was consistent with the types of AEs previously reported for afatinib and vinorelbine, with no new or unexpected events reported. Although AEs led to a number of dose interruptions, the majority of patients remained on study, indicating that effective management of AEs allowed for treatment continuation. In addition, PK assessments indicated no clinically relevant drug–drug interactions between afatinib and either intravenous or oral vinorelbine. These findings are consistent with those from a previously reported phase I study in Japanese patients with advanced solid tumours (Mukai et al, 2015). Although this study was not designed to evaluate antitumour activity, three ORs were observed and ∼50% of patients in parts A and B achieved disease control, with a median duration ranging from 110.0 to 168.0 days. These findings are noteworthy both in terms of disease control and long-term tolerability of the combination.
To date, phase I clinical findings have suggested that the combination of afatinib and vinorelbine is feasible, with a manageable safety profile, and may provide clinical benefit in patients with advanced cancers (Mukai et al, 2015). However, further evaluation of this combination in phase II/III trials, including two studies in patients with advanced breast cancer, have yielded less favourable results. In the phase II LUX-Breast 3 trial in patients with HER2-overexpressing breast cancer and progressive brain metastases following trastuzumab and/or lapatinib-based therapy, afatinib in combination with vinorelbine did not provide improved efficacy over investigator’s choice of therapy and appeared to be less well tolerated (Cortes et al, 2015). Further, the afatinib plus vinorelbine combination compared unfavourably with trastuzumab plus vinorelbine in the LUX-Breast 1 trial, a pivotal phase III trial in patients with HER2-positive metastatic breast cancer whose disease progressed on/after one prior trastuzumab-based treatment, which was stopped early (Harbeck et al, 2016). The combination of afatinib plus vinorelbine was also evaluated in the LUX-Breast 2 trial in patients with HER2-overexpressing breast cancer who failed HER2-targeted treatment in the neoadjuvant or adjuvant setting, as well as in Study 1200.89 in HER2-overexpressing inflammatory breast cancer (Goh et al, 2016). However, recruitment to all trials of this combination was halted following the results of LUX-Breast 1. Although there are little clinical data available for other EGFR tyrosine kinase inhibitors in combination with vinorelbine, conventional and metronomic dosing schedules of oral vinorelbine plus once-daily erlotinib were recently explored in a phase I study in patients with advanced NSCLC (Sutiman et al, 2016). Both dosing schedules were reported to be feasible and well tolerated, and demonstrated preliminary antitumour activity, leading the investigators to conclude that further investigations are warranted in this setting. In an earlier phase II study in Chinese patients with lung adenocarcinoma, once-daily gefitinib in combination with a lower dose of intravenous vinorelbine (15 mg m−2 every 2 weeks) demonstrated an improved 1-year PFS rate (P=0.008) compared with gefitinib alone. Three of six evaluable patients in the Chinese study were reported an exon 19 deletion, although few patients had qualified samples for mutation analysis (Chen et al, 2007). The combination of afatinib and vinorelbine has not yet been further explored in other distinct indications, such as NSCLC; however, the manageable safety profile and antitumour activity consistently observed in phase I trials of this combination may lend support for future studies.
In summary, this phase I trial established the MTD of afatinib as 40 mg per day in combination with standard doses of intravenous or oral vinorelbine. In this study, both combination regimens demonstrated generally manageable safety profiles consistent with those known for single-agent afatinib and vinorelbine. The treatment combination showed signs of clinical activity in heavily pre-treated patients with advanced solid tumours. Pharmacokinetic analyses suggest no clinically relevant drug–drug interactions between afatinib and vinorelbine in the evaluated treatment schedules.
Change history
06 February 2018
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aapro MS, Bohlius J, Cameron DA, Dal Lago L, Donnelly JP, Kearney N, Lyman GH, Pettengell R, Tjan-Heijnen VC, Walewski J, Weber DC, Zielinski C Cancer EOfRaTo (2011) 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 47 (1): 8–32.
Bahleda R, Soria J, Berge Y, Massard C, Wind S, Uttenreuther-Fischer MM, Fleischer F, De-Montserrat H, Solca F, Tschoepe I, Delord J (2011) Phase I trial assessing safety and pharmacokinetics of afatinib (BIBW 2992) with intravenous weekly vinorelbine in advanced solid tumors. J Clin Oncol 29 (Suppl. 15): abstract 2585.
Burstein HJ (2005) The distinctive nature of HER2-positive breast cancers. N Engl J Med 353 (16): 1652–1654.
Chen YM, Liu JM, Chou TY, Perng RP, Tsai CM, Whang-Peng J (2007) Phase II randomized study of daily gefitinib treatment alone or with vinorelbine every 2 weeks in patients with adenocarcinoma of the lung who failed at least 2 regimens of chemotherapy. Cancer 109 (9): 1821–1828.
Cortes J, Dieras V, Ro J, Barriere J, Bachelot T, Hurvitz S, Le Rhun E, Espie M, Kim SB, Schneeweiss A, Sohn JH, Nabholtz JM, Kellokumpu-Lehtinen PL, Taguchi J, Piacentini F, Ciruelos E, Bono P, Ould-Kaci M, Roux F, Joensuu H (2015) Afatinib alone or afatinib plus vinorelbine versus investigator's choice of treatment for HER2-positive breast cancer with progressive brain metastases after trastuzumab, lapatinib, or both (LUX-Breast 3): a randomised, open-label, multicentre, phase 2 trial. Lancet Oncol 16 (16): 1700–1710.
Doebele RC, Oton AB, Peled N, Camidge DR, Bunn Jr PA (2010) New strategies to overcome limitations of reversible EGFR tyrosine kinase inhibitor therapy in non-small cell lung cancer. Lung Cancer 69 (1): 1–12.
European Medicines Agency (2013) Giotrif European Public Assessment Report (summary). Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002280/WC500152392.pdf.
Food and Drug Administration (2013) Gilotrif prescribing information. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf.
Garrido-Castro AC, Felip E (2013) HER2 driven non-small cell lung cancer (NSCLC): potential therapeutic approaches. Transl Lung Cancer Res 2 (2): 122–127.
Geater SL, Xu CR, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Palmer M, Shi Y, Lungershausen J, Wu YL (2015) Symptom and quality of life improvement in LUX-Lung 6: an open-label phase III study of afatinib versus cisplatin/gemcitabine in Asian patients With EGFR mutation-positive advanced non-small-cell lung cancer. J Thorac Oncol 10 (6): 883–889.
Goh G, Schmid R, Guiver K, Arpornwirat W, Chitapanarux I, Ganju V, Im SA, Kim SB, Dechaphunkul A, Maneechavakajorn J, Spector N, Yau T, Afrit M, Ahmed SB, Johnston SR, Gibson N, Uttenreuther-Fischer M, Herrero J, Swanton C (2016) Clonal evolutionary analysis during HER2 blockade in HER2-positive inflammatory breast cancer: a phase II open-label clinical trial of afatinib +/- vinorelbine. PLoS Med 13 (12): e1002136.
Harbeck N, Huang CS, Hurvitz S, Yeh DC, Shao Z, Im SA, Jung KH, Shen K, Ro J, Jassem J, Zhang Q, Im YH, Wojtukiewicz M, Sun Q, Chen SC, Goeldner RG, Uttenreuther-Fischer M, Xu B, Piccart-Gebhart M group LU-Bs (2016) Afatinib plus vinorelbine versus trastuzumab plus vinorelbine in patients with HER2-overexpressing metastatic breast cancer who had progressed on one previous trastuzumab treatment (LUX-Breast 1): an open-label, randomised, phase 3 trial. Lancet Oncol 17 (3): 357–366.
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5 (5): 341–354.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK (2008) BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27 (34): 4702–4711.
Lin NU, Winer EP, Wheatley D, Carey LA, Houston S, Mendelson D, Munster P, Frakes L, Kelly S, Garcia AA, Cleator S, Uttenreuther-Fischer M, Jones H, Wind S, Vinisko R, Hickish T (2012) A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res Treat 133 (3): 1057–1065.
Lurje G, Lenz HJ (2009) EGFR signaling and drug discovery. Oncology 77 (6): 400–410.
Machiels JP, Haddad RI, Fayette J, Licitra LF, Tahara M, Vermorken JB, Clement PM, Gauler T, Cupissol D, Grau JJ, Guigay J, Caponigro F, de CG Jr, de SV, Keilholz U, Del Campo JM, Cong XJ, Ehrnrooth E, Cohen EE (2015) Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncol 16 (5): 583–594.
medac GmbH (2014) Vinorelbine 10 mg/ml concentrate for solution for infusion – summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/19453/SPC/.
medac GmbH (2017) Navelbine 30 mg soft capsule – summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/1604.
Modjtahedi H, Cho BC, Michel MC, Solca F (2014) A comprehensive review of the preclinical efficacy profile of the ErbB family blocker afatinib in cancer. Naunyn Schmiedebergs Arch Pharmacol 387 (6): 505–521.
Mukai H, Masuda N, Ishiguro H, Mitsuma A, Shibata T, Yamamura J, Toi M, Watabe A, Sarashina A, Uttenreuther-Fischer M, Ando Y (2015) Phase I trial of afatinib plus vinorelbine in Japanese patients with advanced solid tumors, including breast cancer. Cancer Chemother Pharmacol 76 (4): 739–750.
Owens MA, Horten BC, Da Silva MM (2004) HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin Breast Cancer 5 (1): 63–69.
Park K, Tan EH, O'Byrne K, Zhang L, Boyer M, Mok T, Hirsh V, Yang JC, Lee KH, Lu S, Shi Y, Kim SW, Laskin J, Kim DW, Arvis CD, Kolbeck K, Laurie SA, Tsai CM, Shahidi M, Kim M, Massey D, Zazulina V, Paz-Ares L (2016) Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol 17 (5): 577–589.
Pierre Fabre Ltd (2011) Navelbine 20 mg soft capsule: summary of product characteristics. Available at: http://www.medicines.org.uk/emc/medicine/16015/SPC/.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, Paz-Ares L, Bover I, Garcia-Campelo R, Moreno MA, Catot S, Rolfo C, Reguart N, Palmero R, Sanchez JM, Bastus R, Mayo C, Bertran-Alamillo J, Molina MA, Sanchez JJ, Taron M (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361 (10): 958–967.
Salomon DS, Brandt R, Ciardiello F, Normanno N (1995) Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 19 (3): 183–232.
Seiwert TY, Fayette J, Cupissol D, Del Campo JM, Clement PM, Hitt R, Degardin M, Zhang W, Blackman A, Ehrnrooth E, Cohen EE (2014) A randomized, phase 2 study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann Oncol 25 (9): 1813–1820.
Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, Massey D, Zazulina V, Shahidi M, Schuler M (2013) Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31 (27): 3327–3334.
Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, Heeroma K, Itoh Y, Cornelio G, Yang PC (2014) A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol 9 (2): 154–162.
Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR (2012) Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 343 (2): 342–350.
Soria JC, Felip E, Cobo M, Lu S, Syrigos K, Lee KH, Goker E, Georgoulias V, Li W, Isla D, Guclu SZ, Morabito A, Min YJ, Ardizzoni A, Gadgeel SM, Wang B, Chand VK, Goss GD (2015) Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol 16 (8): 897–907.
Soria JC, Mok TS, Cappuzzo F, Janne PA (2012) EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev 38 (5): 416–430.
Sutiman N, Zhang Z, Tan EH, Ang MK, Tan SW, Toh CK, Ng QS, Chowbay B, Lim WT (2016) Phase I study of oral vinorelbine in combination with erlotinib in advanced non-small cell lung cancer (NSCLC) using two different schedules. PLoS One 11 (5): e0154316.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216.
Wong AL, Lee SC (2012) Mechanisms of resistance to trastuzumab and novel therapeutic strategies in HER2-positive breast cancer. Int J Breast Cancer 2012: 415170.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, Shi Y, Geater SL (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15 (2): 213–222.
Yang JC, Hirsh V, Schuler M, Yamamoto N, O'Byrne KJ, Mok TS, Zazulina V, Shahidi M, Lungershausen J, Massey D, Palmer M, Sequist LV (2013a) Symptom control and quality of life in LUX-Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol 31 (27): 3342–3350.
Yang JC, Reguart N, Barinoff J, Kohler J, Uttenreuther-Fischer M, Stammberger U, O'Brien D, Wolf J, Cohen EE (2013b) Diarrhea associated with afatinib: an oral ErbB family blocker. Expert Rev Anticancer Ther 13 (6): 729–736.
Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O'Byrne K, Feng J, Lu S, Huang Y, Geater SL, Lee KY, Tsai CM, Gorbunova V, Hirsh V, Bennouna J, Orlov S, Mok T, Boyer M, Su WC, Lee KH, Kato T, Massey D, Shahidi M, Zazulina V, Sequist LV (2015) Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16 (2): 141–151.
Yarden Y, Pines G (2012) The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 12 (8): 553–563.
Yaziji H, Goldstein LC, Barry TS, Werling R, Hwang H, Ellis GK, Gralow JR, Livingston RB, Gown AM (2004) HER-2 testing in breast cancer using parallel tissue-based methods. JAMA 291 (16): 1972–1977.
Acknowledgements
Medical writing assistance provided by Helen Kitchen of GeoMed, an Ashfield company, part of UDG Healthcare plc, was supported financially by Boehringer Ingelheim during the preparation of this manuscript. We are fully responsible for all content and editorial decisions and are involved at all stages of manuscript development and have approved the final version. This study was funded by the sponsor, Boehringer Ingelheim.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
J-CS received consulting fees from Boehringer Ingelheim. IT was an employee of Boehringer Ingelheim until March 2016, and DS and MU-F are current employees of Boehrineger Ingelheim. The other authors declare no conflict of interest.
Additional information
This work was presented, in part, at the European Society for Medical Oncology 38th Congress—ECCO 17, ESMO 38, ESTRO 32, 27 September–01 October 2014, Amsterdam, The Netherlands; and the 48th Annual Meeting of the American Society of Clinical Oncology, 1–5 June 2012, Chicago, IL, USA.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Bahleda, R., Varga, A., Bergé, Y. et al. Phase I open-label study of afatinib plus vinorelbine in patients with solid tumours overexpressing EGFR and/or HER2. Br J Cancer 118, 344–352 (2018). https://doi.org/10.1038/bjc.2017.436
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.436
