
Abstract
Background:
Bactericidal/Permeability-increasing-fold-containing family B member 1 (BPIFB1, previously termed LPLUNC1) is highly expressed in the nasopharynx, significantly downregulated in nasopharyngeal carcinoma (NPC), and associated with prognosis in NPC patients. Because metastasis represents the primary cause of NPC-related death, we explored the role of BPIFB1 in NPC migration and invasion.
Methods:
The role of BPIFB1 in NPC metastasis was investigated in vitro and in vivo. A co-immunoprecipitation assay coupled with mass spectrometry was used to identify BPIFB1-binding proteins. Additionally, western blotting, immunofluorescence, and immunohistochemistry allowed assessment of the molecular mechanisms associated with BPIFB1-specific metastatic inhibition via vitronectin (VTN) and vimentin (VIM) interactions.
Results:
Our results showed that BPIFB1 expression markedly inhibited NPC cell migration, invasion, and lung-metastatic abilities. Additionally, identification of two BPIFB1-interacting proteins, VTN and VIM, showed that BPIFB1 reduced VTN expression and the formation of a VTN-integrin αV complex in NPC cells, leading to inhibition of the FAK/Src/ERK signalling pathway. Moreover, BPIFB1 attenuated NPC cell migration and invasion by inhibiting VTN- or VIM-induced epithelial–mesenchymal transition.
Conclusions:
This study represents the first demonstration of BPIFB1 function in NPC migration, invasion, and lung metastasis. Our findings indicate that re-expression of BPIFB1 might represent a useful strategy for preventing and treating NPC.
Similar content being viewed by others


Main
Nasopharyngeal carcinoma (NPC) constitutes a squamous cell carcinoma of the nasopharynx that exhibits significant ethnic and regional differences and is often found to occur in Southeast Asia and southern China (Lo et al, 2004; Chua et al, 2016; Song et al, 2016; Tu et al, 2017). Previous studies showed that NPC tumourigenesis comprises a complex, multi-step, and multi-factorial process involving abnormal activation of certain oncogenes, as well as the inactivation of several tumour-suppressor genes (Xiong et al, 2004; Zeng et al, 2011, 2014, 2015; Gong et al, 2016; Tang et al, 2017). NPC shows a tendency towards early lymphatic spread and a relatively high incidence of lymph node metastasis among head and neck cancers (Lu et al, 2013; Bo et al, 2015; He et al, 2016). Radiotherapy is the primary effective method utilised for NPC treatment and has significantly improved its local regional control (Razak et al, 2010). However, radiation resistance, local recurrence, and distant metastasis of NPC comprise the major reasons for treatment failure (Tian et al, 2013). Therefore, a better understanding of the metastatic mechanisms associated with NPC would allow the development of more effective anticancer therapies.
Bactericidal/Permeability-increasing (BPI)-fold-containing family B member 1 (BPIFB1), also known as long-palate lung and nasal epithelium clone 1 (LPLUNC1), belongs to the BPI-fold-containing family (Weston et al, 1999; Bingle and Craven, 2003; Bingle et al, 2011). Our previous studies showed that BPIFB1 is tissue-specifically expressed in nasopharyngeal epithelia and downregulated in NPC tissues (Zhang et al, 2003). Its expression also negatively correlates with the clinical stages of NPC, suggesting that the reduction in BPIFB1 expression constitutes a novel adverse prognostic factor in NPC (Liao et al, 2013). BPIFB1 inhibits NPC growth by downregulating mitogen-activated protein (MAP) kinases and the cyclin D1/E2F pathways (Yang et al, 2013) and can also significantly suppress interleukin (IL)-6-induced inflammation and NPC cell proliferation by inhibiting signal transducer and activator of transcription 3 (STAT3) activity (Liao et al, 2013). However, its role in NPC metastasis has not been clarified.
In this study, BPIFB1 was re-expressed in NPC cell lines to demonstrate its function in NPC migration and invasion in vitro and in vivo. Additionally, a co-immunoprecipitation (co-IP) assay combined with mass spectrometry (MS) analysis was performed to identify BPIFB1-interacting proteins. Among the potential interacting proteins, roles of the cell adhesion molecule vitronectin (VTN) and the intermediate filament protein vimentin (VIM) were confirmed. Our results suggest that BPIFB1 might reduce NPC migration and invasion by interacting with VTN or VIM and their associated signalling pathways.
Materials and methods
Cell lines and transfection constructs
NPC cell lines 5-8F, HNE2, and HONE1 were maintained in our laboratory and grown in RPMI-1640 medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (FBS; Life Technologies) and 1% penicillin-streptomycin (Life Technologies). Cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.
To generate the vector overexpressing BPIFB1, the full-length BPIFB1-coding sequence was amplified, tagged with Flag, cloned into the pIRESneo3 plasmid (Life Technologies), and termed as pIRESneo3-BPIFB1/Flag (BPIFB1-Flag). The full-length VTN- and VIM-coding sequences were amplified and cloned into the pcDNA6-His vector (Life Technologies), generating pcDNA6-His/VTN (VTN-His) and pcDNA6-His/VIM (VIM-His), respectively. Transfection was conducted using Lipofectamine 3000 reagent (Life Technologies) according to the manufacturer’s protocol.
Three-dimensional (3D) invasion assay
3D cell culture was conducted as described previously (Sokol et al, 2017). Briefly, Matrigel (BD Biosciences, San Jose, CA, USA) was fully melted on ice, and 100 μl was added to a 24-well plate until it completely covered the bottom of the plate. The Matrigel was then allowed to solidify for 45 min at 37 °C in a humidified atmosphere with 5% CO2. A 200 μl suspension containing 500 cells was added dropwise to the top of the Matrigel and allowed to settle for 1 h. The cultures were supplemented with 200 μl of media containing 10% Matrigel. After 4 days, 400 μl of media was exchanged, and images depicting cell morphology were obtained for 10 consecutive days. The invasive degree of spheroids was classified into two types: noninvasive spheroids exhibiting smooth edges with no visible cellular protrusions or only occasional scattered protrusions; or invasive spheroids exhibiting fully scattered protrusions.
Clinical NPC samples
A total of 20 NPC samples and 11 paired, adjacent, non-tumour nasopharyngeal epithelium (NPE) tissues were collected from newly diagnosed patients with NPC at the Second Xiangya Hospital of Central South University (Changsha, China). The study was approved by the Joint Ethics Committee of the Central South University Health Authority and informed consent was obtained from each participant. The diagnoses of all specimens were confirmed by histopathological examination.
RNA extraction and quantitative real-time polymerase chain reaction (qPCR)
Total RNAs were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The cDNA was prepared from total RNA using 5 × All-In-One RT master mix (Applied Biologic Materials, Richmond, Canada), after which real-time qPCR was conducted using a Mini Option system (Bio-Rad, Hercules, CA, USA) with SYBR Green (Applied Biologic Materials). The amount of each target gene was quantified by the comparative CT method using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the normalisation control. The following primers were synthesised by Life Technologies: BPIFB1 forward primer (5′-ATC GGA TCC AGC TGA TGA AC-3′) and reverse primer (5′-AGG AGG CTG GAG TAA GCA CA-3′); GAPDH forward primer (5′-CAA CGG ATT TGG TCG TAT TGG-3′) and reverse primer (5′-TGA CGG TGC CAT GGA ATT T-3′).
Western blotting
Whole-cell lysates were extracted using radioimmunoprecipitation assay buffer, and protein concentration was determined using the BCA protein assay kit (Pierce, Grand Island, NY, USA). Equal levels of protein from the samples were separated by 10% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS–PAGE), and the separated proteins were transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA) and blocked with Tris-buffered saline-Tween-20 with 5% dry skimmed milk for 1 h at room temperature. To assess protein expression, the blots were incubated with the following primary antibodies at 4 °C overnight: rabbit antibodies against His-tag, E-cadherin, ZO-1, β-catenin, N-cadherin, VIM, Snail, and Slug (Cell Signaling Technology, Danvers, MA, USA); and mouse antibodies against Flag (Sigma-Aldrich, St Louis, MO, USA), BPIFB1 (Abnova, Taipei, Taiwan); VTN and integrin αV (BD Biosciences). After washing, the blots were incubated with horseradish peroxidase-labelled secondary antibodies (Cell Signaling Technology) and detected by enhanced chemiluminescence (EMD-Millipore). GAPDH (Cell Signaling Technology) served as an endogenous control for equal loading.
Migration and invasion assays
For wound healing assay, cells were seeded in six-well culture plates and grown to a near-confluent monolayer after transfection. Wounds were made using a 10-μl pipette tip, and cellular debris was removed by washing with phosphate-buffered saline. Images of the scratched area were taken at time points of 0, 24, and 48 h under a microscope. Wound width was scored and evaluated to ensure that all wounds were the same width at the beginning of each experiment. Each sample was assayed in triplicate, and a minimum of three independent experiments were performed. For the transwell assay, invasive capacity was detected using Transwell cell culture inserts (8-μm pores; Corning, NY, USA) in 24-well plates. Cells were resuspended in serum-free medium after transfection and added to chambers coated with Matrigel (BD Biosciences). The bottom well contained growth medium with 20% FBS. Following a 24-h incubation, cells that invaded through the filter pores were fixed with methanol and stained with 5% crystal violet. The number of invasive tumour cells was counted from three randomly selected fields for each experiment and averaged.
Lung metastasis in nude mice
To confirm the role of BPIFB1 in metastasis in vivo, we used a nude mouse model with lung metastasis initiated via tail vein injection. Briefly, 4-week-old female nude mice were obtained from the Laboratory Animal Center of Central South University (Changsha, China) and maintained under specific pathogen-free conditions. We injected 1 × 106 5-8F cells transfected with the BPIFB1-overexpression plasmid (5-8F/BPIFB1) or negative control (empty vector; 5-8F/NC) into each of two groups of nude mice intravenously through the tail vein (n=8/group). Mice were killed 8-weeks post-injection, and the lung tissues were removed from each mouse, weighed, photographed, embedded in 10% paraffin, and subjected to sectioning. Haematoxylin and eosin staining was performed for histological examination and evaluation of metastasis, and the number of visible lung-surface metastases in each mouse was recorded. The study was approved by the Joint Ethics Committee of the Central South University Health Authority.
IP assays
5-8F cells transfected with only BPIFB1-Flag or co-transfected with BPIFB1-Flag and VTN-His (or VIM-His) vectors were seeded in 100-mm dishes. The cells were harvested using IP lysis buffer (Beyotime, Shanghai, China) supplemented with an EDTA-free protease-inhibitor cocktail (Roche, Manheim, Germany). Protein lysates (1 mg) were incubated on a rotator with 5 μg of primary antibodies overnight at 4 °C, followed by addition of 40 μl IP beads and incubation for another 3 h on a rotator at 4 °C. The beads and immune complexes were washed with lysis buffer, boiled with sample-loading buffer, and detected by western blotting.
Liquid chromatography tandem MS (LC-MS/MS)
The immunoprecipitated proteins were resolved by SDS–PAGE, stained with Coomassie Brilliant Blue, and the gels washed extensively with ddH2O overnight. The stained bands representing only the BPIFB1 IP fraction compared with the control were excised and cut into small (<1 mm3) pieces and kept in clean tubes. The slices were subjected to in-gel reduction, alkylation, and trypsinised overnight at 37 °C. Digested peptides were dried and resuspended in MS-compatible buffer, and the mixture was analysed by nano-LC-MS/MS using an LTQ Velos-orbitrap MS (Thermo Fisher Scientific, Waltham, MA, USA) coupled with an Ultimate RSLC nano-LC system (Dionex, Sunnyvale, CA, USA). Proteins were identified using Proteome Discoverer 1.4 software (Thermo Fisher Scientific), and the resulting raw files were imported and used to conduct a search of the UniProt KB/Swiss-Prot database. For database searches, mass tolerances were set to 10 ppm and 0.8 Da for precursor and fragmentations, respectively. Peptides identified with false discovery rates <1% (q-value<0.01) were discarded.
Immunofluorescence
5-8F cells transfected or co-transfected with the Flag-BPIFB1 and His-VTN (or -VIM) expression vectors were seeded on coverslips in a six-well plate. After a 24-h culture, cells were fixed in 4% paraformaldehyde for 20 min, subjected to membrane permeabilisation with 0.25% Triton X-100 for 40 min, and blocked in 5% bovine serum albumin for 1 h at room temperature. The cells were then incubated with mouse antibodies against Flag-BPIFB1, ITGAV, VIM and rabbit antibodies against His-VTN/VIM, Src, and N-cadherin antibodies at 4 °C overnight, followed by incubation with secondary fluorochrome-labelled antibodies (Alexa Fluor 488 donkey anti-rabbit IgG (H+L) antibody and Alexa Fluor 568 goat anti-mouse IgG (H+L) antibody; Life Technologies) for 40 min at 37 °C. After incubation with DAPI for 10 min at room temperature to stain the nuclei, cells were imaged using a confocal laser scanning microscope (UltraView Vox; Perkin-Elmer, Waltham, MA, USA). The scatter analysis of channels 561 and 488, as well as the mean relative fluorescence intensity (not total fluorescence intensity), were measured using the image processing and analysis software Velocity (Perkin-Elmer).
Immunohistochemistry
Immunohistochemistry was characterised using a streptavidin-peroxidase-complex method. Briefly, tissue slices were de-waxed and rehydrated, antigen retrieval was performed by pressure cooking the slides, and blocker was used to block the endogenous peroxidase. Tissue sections were incubated with rabbit anti-BPIFB1 (Abcam, Cambridge, MA, UK), anti-VTN (Abcam), anti-ITGAV (Abcam), anti-FAK and anti-Src (Cell Signaling Technology) at 4 °C overnight. Normal rabbit immunoglobulin G was used as the isotype control. After washing, the sections were incubated with biotin-labelled secondary antibodies (Maixin, Fuzhou, China) for 40 min at room temperature, followed by treatment with Streptomyces anti-biotin peroxidase solution for 10 min. Colour reaction using 3-3′-diaminobenzidine (Maixin) and counterstaining with haematoxylin was used to detect immunohistochemistry signals. The intensity of staining was scored from 0–2, according to the standards of 0 (no staining), 1 (weak staining), and 2 (strong staining).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 (GraphPad, La Jolla, CA, USA). Student’s t tests were performed to evaluate significant differences between any two groups of data. All data were represented as the mean±standard deviation (s.d.) unless otherwise stated. A P<0.05 was considered to indicate statistical significance.
Results
BPIFB1 inhibits cell migration and invasion in NPC cell lines
To explore the role of BPIFB1 in NPC migration and invasion, BPIFB1 was re-expressed in three NPC cell lines (5-8F, HNE2, and HONE1; Figure 1A). Cell migration and invasion abilities were measured by wound healing and Matrigel invasion assays. The results revealed that BPIFB1 overexpression led to reduced migratory (Figure 1B) and invasive (Figure 1C) properties in the three NPC cell lines. To simulate the extracellular microenvironment for NPC cell growth and reproduce the process of cell adhesion, invasion, and metastasis, we established a 3D cell culture model, using Matrigel, to explore the role of BPIFB1 in cell invasion. The results showed that 5-8F cells could form spheroids capable of invading 3D Matrigel; however, re-expression of BPIFB1 almost completely blocked the invasive ability of the spheroids (Figure 1D). Therefore, these data suggested that BPIFB1 was capable of inhibiting NPC cell migration and invasion in both monolayers and 3D cell-culture systems.

BPIFB1 inhibits NPC cell migration and invasion in vitro. (A) BPIFB1 expression in 5-8F, HNE2, and HONE1 cells transfected with the BPIFB1-Flag vector was measured by qPCR and western blot using the anti-Flag antibody. (B) Wound healing assays were performed to measure the cell migration ability of 5-8F, HNE2, and HONE1 cells transfected with the BPIFB1-Flag vector or empty vector. The picture shows the scratch width at 0, 24, and 48 h. (C) A transwell assay was performed to detect the BPIFB1-specific invasion ability of 5-8F, HNE2, and HONE1 cells. Cells were transfected with BPIFB1-Flag or empty vector. The graph summarises data from three independent experiments. *P<0.05; **P<0.01; ***P<0.001. Scale bars=200 μm. (D) BPIFB1 re-expression inhibited the invasive progression of spheroids in a 3D cell culture model. Images of spheroids formed by 5-8F cells in Matrigel (left). The black arrows indicate the scattered protrusions formed on the spheroid surface. Scale bars=50 μm. Quantification of two spheroid types in NC and BPIFB1-overexpressing cells (right).
BPIFB1 reduces NPC lung metastasis in nude mice
A lung-metastasis model was generated by tail vein injection of nude mice with the NPC cell line 5-8F transfected with the BPIFB1-overexpression vector or empty vector. Each group was assigned eight mice, and at 8-weeks post-injection, the number of metastatic tumour nodules in each mouse lung was assessed. The results showed that BPIFB1 significantly reduced the size and number of metastatic tumour foci (Figure 2A and B). A lower number of lung metastatic nodules was also observed in the BPIFB1 group (n=8; 6/8 mice; 3.1±0.9 nodules per mouse) as compared with that in the control group (n=8, 8/8 mice; 9.4±2.6 nodules per mouse; Figure 2C). Haematoxylin and eosin staining of paraffin-embedded lung tissues also showed that the size and number of metastatic nodules in the BPIFB1 group were significantly reduced compared with those in the control group (Figure 2D). Therefore, our data further demonstrated that BPIFB1 significantly reduced NPC lung metastasis in vivo.

BPIFB1 inhibits NPC-cell lung metastasis in vivo. (A) Bright-field images of mouse lungs taken at 8-weeks post-injection of 1 × 106 5-8F cells transfected with BPIFB1-Flag or empty vector into the tail vein. (B) Representative images of visible nodules on the lung surface. Arrows indicate clusters of tumour cells that have colonised in the lung. (C) The number of lung-metastasis nodules on each lung surface was counted, and the data represent the mean±s.d. (each data point represents a different mouse; n=8 mice per group). *P<0.05. (D) Representative images of lung metastasis according to H&E staining. Rectangular boxes indicate clusters of micro-metastatic cells in the lung. Scale bars=200 and 50 μm.
Identification of potential BPIFB1-interacting partners by IP-MS
To explore BPIFB1-specific mechanisms in NPC cell migration and invasion, a co-IP assay coupled with MS was performed to identify potential BPIFB1-binding proteins. 5-8F cells transfected with the BPIFB1-expression vector tagged with Flag (BPIFB1-Flag) or empty vector (NC) were lysed and precipitated using an anti-Flag antibody. Following SDS–PAGE, several specific bands in the BPIFB1 lane were detected (Figure 3A) and subjected to LC-MS/MS analysis, from which we identified 108 candidate BPIFB1-interacting proteins. Among these, Gene Ontology molecular-function analysis indicated that several proteins were associated with cytoskeleton, cell motility, cell cycle, or apoptosis (Supplementary Table S1). We then focused on two proteins, VTN and VIM, which had higher scores or numbers of associated areas and are closely related to cell motility. Figure 3B shows examples of the peptide precursor m/z values and charge states of these two proteins identified from the corresponding MS/MS spectra. Interactions between BPIFB1 and exogenous (Figure 3C and D) or endogenous (Figure 3F) VTN or VIM were confirmed using co-IP experiments in 5-8F cells. Notably, exogenous and endogenous VTN or VIM were each detected in the precipitation complex containing an anti-Flag antibody (Figure 3D and F), whereas BPIFB1 was detected in a reciprocal experiment precipitated by the anti-His (Figure 3E) or anti-VTN and -VIM antibodies (Figure 3G), indicating that BPIFB1 directly interacts with VTN and VIM in 5-8F cells.

Identification of BPIFB1-interacting proteins via IP-MS. (A) Co-IP was performed in 5-8F cells transfected with the BPIFB1-Flag plasmid using an anti-Flag antibody. The precipitated complex was subjected to SDS–PAGE and Coomassie blue staining. IgG heavy chain (IgG-H) and light chain (IgG-L) are 55 and 25 kDa, respectively. The molecular weight (MW) marker is shown on the left. (B) Representative image and charge states are shown according to the corresponding MS/MS spectrum. LC-MS/MS spectra of precursor ions at m/z 824.42 correspond to VTN residues 198 through 212 (DVWGIEGPIDAAFTR), and m/z 585.36 corresponds to VIM residues 130 through 139 (ILLAELEQLK). (C) BPIFB1 and VTN (or VIM) expression was examined at the protein level by western blot using 5-8F cells co-transfected with BPIFB1-Flag and His-VTN (or -VIM) vectors. (D and E) Co-IP was performed to detect interactions between BPIFB1 and exogenous VTN (or VIM) in 5-8F cells co-transfected with BPIFB1-Flag and His-VTN (or -VIM) using anti-Flag (BPIFB1) and anti-His (VTN or VIM) antibodies. (F and G) Co-IP was performed to detect interactions between BPIFB1 and endogenous VTN (or VIM) in 5-8F cells transfected with only BPIFB1-Flag vector using anti-Flag (BPIFB1) and anti-VTN or VIM antibodies.
BPIFB1 inhibits VTN-enhanced NPC cell migration and invasion
VTN, which was identified as a BPIFB1-binding protein by IP-MS, constitutes a major component of the extracellular matrix (ECM). Among ECM proteins, VTN is a multifunctional glycoprotein involved in several pivotal processes in multiple tumours, including cell adhesion, movement, and proliferation (Pirazzoli et al, 2013). To determine whether BPIFB1 and VTN colocalise in cells, we performed immunofluorescence staining, with the results revealing that the BPIFB1 and VTN proteins clearly colocalised in 5-8F cells (Figure 4A). To further investigate whether BPIFB1 can inhibit NPC cell migration and invasion by interacting with VTN, BPIFB1 was re-expressed in 5-8F, HNE2, and HONE1 cell lines, resulting in a significant downregulation of VTN expression (Figure 4B). Wound-healing and transwell assays showed that BPIFB1 inhibited the migration and invasion of the three cell lines, whereas VTN expression led to an enhancement in the migratory (Figure 4C and D and Supplementary Figure S1A) and invasive (Figure 4E and Supplementary Figure S1B) properties of 5-8F, HNE2, and HONE1 cells. Notably, BPIFB1 reversed the enhancing functions of VTN when BPIFB1 and VTN were co-expressed in NPC cells. These results suggested that BPIFB1 was capable of inhibiting NPC migration and invasion by interacting with and inhibiting VTN.

BPIFB1 inhibits NPC-cell migration and invasion by downregulating VTN expression. (A) BPIFB1 and VTN colocalisation in 5-8F cells. DAPI-stained nuclei: blue; anti-Flag-BPIFB1: red; anti-His-VTN: green; merged image represents the overlay of DAPI, Flag, and His signals; BPIFB1 and VTN colocalisation: yellow. Scatter analysis shows the signals of Channel 561 (BPIFB1) and 488 (VTN). Pearson’s correlation coefficient of colocalisation is indicated in the top right corner of the plot. Scale bar=29 μm. (B) BPIFB1 downregulates VTN expression in 5-8F, HNE2, and HONE1 cells transfected with the BPIFB1-Flag overexpression vector as compared with the control group. (C) BPIFB1 and VTN protein expression was confirmed by western blot in 5-8F cells transfected or co-transfected with BPIFB1-Flag and VTN-His vectors using anti-Flag and anti-His primary antibodies. (D and E) Representative images of migration and transwell Matrigel invasion assays on 5-8F cells transfected or co-transfected with BPIFB1-Flag and VTN-His vectors. The graph summarises the data from three independent experiments. **P<0.01; ***P<0.001; n.s., no significance. Scale bars=200 μm.
BPIFB1 suppresses the FAK-signalling pathway by disassociating the VTN/ITGAV complex
VTN is involved in cell adhesion mainly through direct binding to its receptor ITGAV and activating downstream signalling pathways (Ruoslahti et al, 1987). The expression of several integrins, including ITGAV, was high in NPC samples according to two published NPC gene-expression databases (GSE12452 and GSE64634; Supplementary Figure S2). Experiments involving co-transfection of BPIFB1- and VTN-expression vectors, followed by IP indicated that the abundance of His-tagged VTN-associated ITGAV decreased notably following BPIFB1 re-expression in 5-8F cells (Figure 5A), indicating that BPIFB1 overexpression reduced VTN/ITGAV-complex formation. BPIFB1 overexpression in 5-8F cells not only resulted in VTN downregulation (Figure 4B), but also altered ITGAV-expression levels. Conversely, VTN overexpression recruited additional ITGAV; however, this positive effect was attenuated by BPIFB1 re-expression in NPC cells (Figure 5B).


BPIFB1 inhibits VTN-ITGAV-complex formation and downstream activation of the FAK signalling pathway. (A) The ITGAV protein was immunoprecipitated by the VTN antibody in 5-8F cells. (B) Expression of ITGAV and some proteins associated with the FAK-signalling pathway, including FAK, p-FAK, Src, p-Src, MEK, and ERK, was detected by western blot in 5-8F cells transfected or co-transfected with BPIFB1-Flag and VTN-His vectors. (C) BPIFB1, VTN, ITGAV, and Src expression was detected by immunofluorescence in 5-8F cells transfected or co-transfected with BPIFB1-Flag and VTN-His vectors. Scale bar=29 μm. Five randomly selected areas were scanned, and the data are shown as the mean±s.d. **P<0.01; ***P<0.001; n.s., no significance. (D) BPIFB1, VTN, ITGAV, FAK, and Src expression as determined in 20 NPC and 11 NPE tissues by immunohistochemistry. H&E staining was also performed on these tissues. Data shown are representative images of the respective molecular expression analyses. Normal rabbit immunoglobulin G was used as the isotype control. Magnification= × 200, scale bars=50 μm; Magnification= × 400, scale bars=20 μm. (E) Administration of the FAK inhibitor PND1186 decreased p-FAK expression. (F and G) PND1186 suppresses NPC cell migration and invasion in 5-8F cells induced by BPIFB1 loss or VTN overexpression. The graph summarises data from three independent experiments. Scale bars=200 μm.
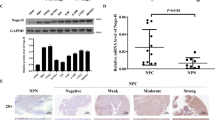
Integrins can activate FAK through binding to its ligand VTN, which recruits Src family kinases to focal adhesions. Src recruitment and activation is required for RAS-MAP kinase (MEK)-extracellular-signal-regulated kinase (ERK)-pathway activation (Hood and Cheresh, 2002). Therefore, we determined whether BPIFB1 affected the activation of the FAK-induced signalling pathway by reducing VTN/ITGAV-complex formation. As shown in Figure 5B, BPIFB1 overexpression downregulated the expression levels of proteins downstream of FAK signalling, including FAK, p-FAK, Src, p-Src, MEK, and ERK. By contrast, VTN overexpression resulted in significant activation of the FAK-signalling pathway, which was dramatically weakened upon BPIFB1 co-expression, leading to reduced signalling-protein expression. The expression of molecules of BPIFB1, VTN, ITGAV and Src was verified by immunofluorescence (Figure 5C). We utilised 20 NPC and 11 non-tumour NPE tissues to assess potential clinical relationships between BPIFB1 and VTN, ITGAV, FAK, and Src proteins (Figure 5D). Immunohistochemical staining showed that BPIFB1 was mainly expressed in the cytoplasm of NPE cells and especially in normal ciliated columnar epithelium cells with apocrine secretion, whereas BPIFB1 was only expressed in 25% (5/20) of NPC samples vs 72.7% (8/11) of NPE samples. Conversely, VTN was highly expressed in NPC tissues as compared with NPE tissues (high expression rate: NPC, 85% (17/20) vs NPE, 18.2% (2/11)). A similar pattern of high expression in NPC tissues and low expression in NPE tissues was exhibited for ITGAV, FAK, and Src proteins (Supplementary Figure S3). These data suggested that BPIFB1 was mainly expressed in NPE tissues, but exhibited low or no expression in NPC tissues, and that BPIFB1 expression was negatively correlated with VTN, ITGAV, FAK, and Src expression in NPC tissues.
We then used the FAK inhibitor PND1186 (1 μ M) to efficiently reduce FAK phosphorylation (Figure 5E), with results indicating that PND1186 markedly suppressed NPC cell migration and invasion induced by BPIFB1 loss or VTN overexpression in vitro (Figure 5F and G). This finding provided important insight into the potential use of BPIFB1 for managing NPC metastasis. The above data demonstrated that BPIFB1 overexpression inhibited VTN/ITGAV-complex formation and activation of the FAK-signalling pathway, resulting in the attenuation of NPC migration and invasion.
BPIFB1 inhibits VIM-induced NPC migration and invasion
VIM represents an additional BPIFP1-binding protein. To confirm the binding between BPIFB1 and VIM, immunofluorescence was used to confirm colocalisation of the two proteins. BPIFB1 and VIM exhibited clear colocalisation in 5-8F cells, which was consistent with co-IP results (Figure 6A). Additionally, these two proteins were primarily colocalised at the cell membrane, based on colocalisation with a well-known cell membrane protein (N-cadherin) (Supplementary Figure S4). VIM overexpression in 5-8F, HNE2, and HONE1 cells resulted in a marked increase in NPC cell migration (Figure 6B and C and Supplementary Figure S5A) and invasion (Figure 6D and Supplementary Figure S5B), whereas BPIFB1 overexpression attenuated VIM-induced NPC cell migration and invasion. These data suggested that BPIFB1 inhibited NPC cell migration and invasion by inhibiting VIM.

BPIFB1 inhibits VIM-induced migration and invasion of NPC cells. (A) BPIFB1 and VIM colocalisation in 5-8F cells. DAPI-stained nuclei: blue; anti-Flag-BPIFB1: red; anti-His-VIM: green; merged image represents the overlay of DAPI, Flag, and His signals; BPIFB1 and VIM colocalisation: yellow. Scatter analysis shows the signals of channel 561 (BPIFB1) and 488 (VIM). Pearson’s correlation coefficient of colocalisation is indicated in the top right corner of the plot. Scale bar=29 μm. (B) BPIFB1 and VIM expression was confirmed by western blot in 5-8F cells transfected or co-transfected with BPIFB1-Flag and VIM-His vectors using anti-Flag and anti-His primary antibodies. (C and D) Representative images of migration and transwell Matrigel invasion assays of 5-8F cells transfected or co-transfected with BPIFB1-Flag and VIM-His vectors. Data represent the mean±s.d. and are representative of three independent experiments. *P<0.05; **P<0.01; ***P<0.001; n.s., no significance.
BPIFB1 overexpression inhibits the epithelial–mesenchymal transition process in NPC through interaction with VIM and VTN
VIM is a canonical marker of epithelial-mesenchymal transition (EMT) (Kalluri and Weinberg, 2009). Because BPIFB1 can inhibit VIM-induced NPC cell migration and invasion, we hypothesised that BPIFB1 participates in the EMT process in NPC. Therefore, we examined EMT markers, including E-cadherin, ZO-1, N-cadherin, β-catenin, VIM, and two EMT-associated transcription factors, Slug and Snail, following BPIFB1 overexpression. Re-expression of BPIFB1 in NPC cells resulted in a significant upregulation of E-cadherin and ZO-1 and downregulation of N-cadherin, VIM, β-catenin, Slug, and Snail expression (Figure 7A), indicating that BPIFB1 was capable of inhibiting the EMT process in NPC cells by directly decreasing the expression of its binding protein, VIM.

BPIFB1 inhibits the EMT process through VTN and VIM in NPC cells. Expression of typical EMT markers, including E-cadherin, N-cadherin, BPIFB1, VIM, β-catenin, Slug, and Snail, according to western blot proteins from of 5-8F, HNE2, and HONE1 cells transfected with the (A) BPIFB1-Flag overexpression vector or (B) co-transfected with the BPIFB1-Flag and VTN-His vectors. GAPDH was used as an internal control. (C) Schematic model illustrating the role of BPIFB1 in regulating NPC migration and invasion by binding and interacting with VTN and VIM.
Several integrins, including ITGAV, ITGB3, and ITGB6, promote the EMT process (Galliher and Schiemann, 2006; Peng et al, 2016); therefore, we further investigated whether BPIFB1 affects the EMT process in NPC via VTN and integrins. vitronectin overexpression in 5-8F cells decreased E-cadherin and ZO-1 expression and significantly increased N-cadherin, VIM, β-catenin, Slug, and Snail levels, whereas this effect was reversed by BPIFB1 (Figure 7B). These results showed that VTN plays a crucial role in the EMT process in NPC, and that BPIFB1 can inhibit the migration and invasion of NPC by binding to VTN and attenuating the VTN-induced EMT process.
Discussion
In our previous study, we identified BPIFB1 as a relatively specific nasopharyngeal gene by suppression-subtractive hybridisation and cDNA microarray hybridisation of NPC biopsies and non-tumour NPE tissues (Zhang et al, 2003). The genes encoding BPI-fold containing family, also known as the PLUNC protein family, are located on human chromosome 20 and include BPI-fold-containing family A, member 1 (BPIFA1) and BPIFB1 (Wei et al, 2014; Zhang et al, 2014; Zhou et al, 2016). This family occupies the ‘epithelial frontier’ owing to their host-defence and innate immune properties (LeClair, 2003). BPIFA1 contains a single BPI structural domain, and BPIFB1 contains two BPI domains (Canny and Levy, 2008), both of which are mainly expressed in human nasopharynx, trachea, lung, and salivary glands (Zhou et al, 2005). We studied the function of BPIFA1 and BPIFB1 proteins in NPC carcinogenesis and found that BPIFA1 acts as a tumour suppressor in the formation of EBV-driven NPC by inhibiting Epstein–Barr virus (EBV) infection and EBV-encoded latent membrane protein 1 (LMP1) and BZLF1 (Zhou et al, 2007; Ou et al, 2015). BPIFA1 can also regulate NPC-cell progression and apoptosis through the microRNA-141-PTEN/p27 pathway, which is negatively regulated by LMP1 (Chen et al, 2013), whereas BPIFB1 delays NPC-cell growth via downregulation of the MAP kinase and cyclin D1/E2F pathways (Yang et al, 2013). Furthermore, BPIFB1 suppresses IL-6-induced NPC-cell proliferation by inhibiting STAT3 activation, suggesting that BPIFB1 can regulate NPC progression by inhibiting inflammation (Liao et al, 2013). Additionally, BPIFB1 expression is positively correlated with the survival rate of patients with NPC, but negatively correlated with the clinical staging of NPC, suggesting that BPIFB1 might affect NPC migration and invasion.
In this study, we found that BPIFB1 inhibited NPC cell migration, invasion, and lung metastasis in vitro and in vivo through its re-expression. A knockdown approach was not performed, because BPIFB1 exhibits very low endogenous expression in NPC cells. We also focused on identifying BPIFB1-interacting proteins and determining the underlying mechanisms of BPIFB1-related NPC migration and invasion. MS approaches represent good methods for investigating potential binding partners of target proteins (Liang et al, 2016). Using co-IP-MS methods, we obtained numerous candidate BPIFB1-binding proteins predicted to participate in several cellular processes, including the cell junction, DNA repair, transcription regulation, protein synthesis, and energy metabolism. The ECM protein VTN and the EMT marker VIM, which are associated with cell adhesion and metastasis, were selected for further study (Bissell and Radisky, 2001; Liotta and Kohn, 2001).
VTN is a component of the ECM, as well as a cell-adhesion glycoprotein (Barnes et al, 1980; Wang et al, 2017). It primarily localises in the ECM, which provides the facility to bind with the secreted protein BPIFB1. vitronectin is generally abundantly in contact with tumour cells once they breach through to the basement membrane during cancer cell invasion. Additionally, VTN can affect tumour cell adhesion, motility, and invasion (Pola et al, 2013), as well as protect tumour cells from apoptosis-related cell death (Uhm et al, 1999). However, the function of VTN in NPC remains unclear. In this study, we found that VTN overexpression dramatically increased NPC-cell migration and invasion, although this positive effect could be attenuated when BPIFB1 and VTN were co-expressed in NPC cells. Therefore, we concluded that BPIFB1 was capable of inhibiting the VTN-induced NPC migration and invasion by downregulating VTN expression. In our previous study, we found that BPIFB1 inhibits the activation of the nuclear factor-κB (NF-κB) and STAT3 pathways, and that they undergo collaboration and cross-talk to promote the development and progression of multiple cancers. Moreover, the maintenance of NF-κB activity in tumours requires STAT3, and a previous study reported that the human VTN promoter contains a Rel/NF-κB transcription factor-binding site (Reuning, 2011). Therefore, BPIFB1 might inhibit STAT3 and NF-κB activation, thereby suppressing VTN-promoter activity and ultimately regulating VTN expression levels.
VTN binding to its receptor integrins on the cell surface can induce integrin clustering and activation of their downstream signalling pathways (Meyer et al, 1998; Hood and Cheresh, 2002). Integrins, which comprise 18-α and 8-β subunits, are a family of glycoproteins that form heterodimeric receptors for ECM molecules (Desgrosellier and Cheresh, 2010; Siret et al, 2015). Several integrins, including ITGAV, ITGA1, ITGA6, ITGB1, and ITGB6, are highly expressed in NPC tissues according to NPC GEO databases GSE12452 and GSE64634. The two primary integrins, αVβ3 and αVβ5, can bind with VTN and induce integrin clustering and activate FAK autophosphorylation at tyrosine 397. Activated FAK protein recruits adaptor proteins to mediate the activation of small GTPases and subsequent downstream effector molecules for tumour metastasis in a complex microenvironment (Hood and Cheresh, 2002; Cabodi et al, 2010). In the present study, we found that BPIFB1 inhibited the formation of the VTN/ITGAV complex. We used co-IP experiments to investigate whether BPIFB1 and VTN could competitively bind integrin αV, finding that BPIFB1 was unable to bind ITGAV. Therefore, we hypothesised that BPIFB1 inhibited the formation of the VTN/ITGAV complex by downregulating VTN and ITGAV expression, followed by suppression of integrin clustering and FAK activation in NPC. The expression of BPIFB1 was also negatively correlated with the expression of VTN, ITGAV, and some molecules associated with the FAK-signalling pathway in human NPC samples. Because activated ERK can directly regulate the enzymes needed for cell migration or indirectly participate in the transcription of many signalling-pathway genes (Roberts and Der, 2007), we concluded that BPIFB1 might suppress NPC migration and invasion by inhibiting VTN/ITGAV-complex formation and activation of the downstream FAK/Src/ERK signalling pathway.
VIM, another BPIFB1-binding protein identified by IP-MS, is mainly expressed in cells of mesenchymal origin and often used as an EMT marker (Franke et al, 1982). VIM acts as a primary regulator of cytoskeleton-interacting proteins and cellular-adhesion molecules, participating in several critical processes of tumour endothelial cells, such as cell adhesion, migration, invasion, and related signal transduction mechanisms (Mendez et al, 2010; Yan et al, 2015). In this study, we found that VIM colocalised with BPIFB1 on the cell membrane of NPC cells. Notably, VIM significantly promoted NPC cell migration and invasion, whereas BPIFB1 reversed this activity. Consistent with this finding, the acquisition of mesenchymal markers, such as N-cadherin and VIM, accompanies the process of EMT, a key step in distant tumour metastasis, along with loss of epithelial markers, such as E-cadherin (Kalluri and Weinberg, 2009; Horikawa et al, 2011). During EMT, epithelial cells lose apical–basal polarity and cell–cell contact, followed by acquisition of an increased ability to move and connect with the ECM (Thiery et al, 2009). In the present study, we found that BPIFB1 reduced the expression of several EMT molecules, including N-cadherin, β-catenin, VIM, Snail, and Slug, and induced E-cadherin and ZO-1 expression, indicating that BPIFB1 was capable of inhibiting EMT in NPC. Activated STAT3 promotes VIM expression and mediates EMT progression in multiple cancers (Satelli and Li, 2011). Our previous work revealed that BPIFB1 can inhibit IL-6-induced Janus-related kinase-2/STAT3 activation in NPC cells. Therefore, BPIFB1 might suppress VIM expression by inhibiting activation of the STAT3 pathway. Some integrins, such as ITGAV and ITGB3, cooperate with tumour growth factor (TGF)-β or activate ERK signalling to induce EMT of mammary epithelial cells. Therefore, we hypothesised that VTN, as an important integrin ligand, might influence the EMT process in NPC. Accordingly, VTN overexpression promoted the NPC EMT process, which contributed to NPC cell invasion and metastasis. However, BPIFB1 overexpression suppressed this process.
vitronectin also interacts with VIM through a high-affinity binding site located at the VIM N-terminus (Podor et al, 2001). Therefore, we concluded that VTN and VIM exert a synergistic effect on NPC cell migration and invasion, whereas BPIFB1 can inhibit this effect by binding with VTN and VIM. Additionally, VIM plays a key role in cell adhesion, likely via regulation of several integrins, including ITGAV, ITGA6, and ITGB3 (Homan et al, 1998). There also appears to be a functional requirement for VIM in regulating FAK expression, activation, and focal adhesion organisation (Dave et al, 2013). In turn, αvβ3 integrin can alter TGF-β signalling in mammary breast epithelial cells via Src, which significantly enhances the ability of TGF-β to induce EMT and invasion (Galliher and Schiemann, 2006). Moreover, VIM can regulate numerous EMT markers, thereby influencing tumour-cell metastasis and invasion (Desgrosellier and Cheresh, 2010). BPIFB1 can bind and inhibit VTN and VIM, and the two downstream signalling pathways are not independent of each other, but rather can form complicated signal-transduction pathways. In addition to VTN and VIM, we also obtained several other putative BPIFB1-binding proteins associated with cell junctions, cytoskeleton, and motility, indicating that BPIFB1 might also mediate or interact with these proteins to regulate NPC metastasis.
The role of BPIFB1 in NPC metastasis and invasion via its interaction with VTN and VIM is summarised in Figure 7C. Specifically, BPIFB1 can bind with the ECM protein VTN and the EMT marker VIM on the cell surface. BPIFB1 reduces formation of the VTN-ITGAV complex and activation of its downstream FAK/Src/ERK pathway and also suppresses the EMT process mediated by VIM and VTN, ultimately inhibiting NPC migration and invasion. To our knowledge, this represents the first study screening for BPIFB1-binding proteins and demonstrating BPIFB1 function in NPC migration and invasion. The importance of VTN or VIM interaction with BPIFB1 in mediating NPC carcinogenesis indicates that they might represent good biomarkers and potential therapeutic targets for the diagnosis and prognosis of NPC and suggest that targeting the interaction between BPIFB1, VTN, and VIM might constitute a new therapeutic strategy for NPC.
References
Barnes D, Wolfe R, Serrero G, McClure D, Sato G (1980) Effects of a serum spreading factor on growth and morphology of cells in serum-free medium. J Supramol Struct 14: 47–63.
Bingle CD, Craven CJ (2003) Comparative analysis of the PLUNC (palate, lung and nasal epithelium clone) protein families. Biochem Soc Trans 31: 806–809.
Bingle CD, Seal RL, Craven CJ (2011) Systematic nomenclature for the PLUNC/PSP/BSP30/SMGB proteins as a subfamily of the BPI fold-containing superfamily. Biochem Soc Trans 39: 977–983.
Bissell MJ, Radisky D (2001) Putting tumours in context. Nat Rev Cancer 1: 46–54.
Bo H, Gong Z, Zhang W, Li X, Zeng Y, Liao Q, Chen P, Shi L, Lian Y, Jing Y, Tang K, Li Z, Zhou Y, Zhou M, Xiang B, Li X, Yang J, Xiong W, Li G, Zeng Z (2015) Upregulated long non-coding RNA AFAP1-AS1 expression is associated with progression and poor prognosis of nasopharyngeal carcinoma. Oncotarget 6: 20404–20418.
Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P (2010) Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer 10: 858–870.
Canny G, Levy O (2008) Bactericidal/permeability-increasing protein (BPI) and BPI homologs at mucosal sites. Trends Immunol 29: 541–547.
Chen P, Guo X, Zhou H, Zhang W, Zeng Z, Liao Q, Li X, Xiang B, Yang J, Ma J, Zhou M, Peng S, Xiang J, Li X, Le CW, Xiong W, McCarthy JB, Li G (2013) SPLUNC1 regulates cell progression and apoptosis through the miR-141-PTEN/p27 pathway, but is hindered by LMP1. PLoS One 8: e56929.
Chua ML, Wee JT, Hui EP, Chan AT, Lo KW, Huang DP (2016) Nasopharyngeal carcinoma. Lancet 387: 1012–1024.
Dave JM, Kang H, Abbey CA, Maxwell SA, Bayless KJ (2013) Proteomic profiling of endothelial invasion revealed receptor for activated C kinase 1 (RACK1) complexed with vimentin to regulate focal adhesion kinase (FAK). J Biol Chem 288: 30720–30733.
Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10: 9–22.
Franke WW, Grund C, Kuhn C, Jackson BW, Illmensee K (1982) Formation of cytoskeletal elements during mouse embryogenesis. III. Primary mesenchymal cells and the first appearance of vimentin filaments. Differentiation 23: 43–59.
Galliher AJ, Schiemann WP (2006) Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial–mesenchymal transition in mammary epithelial cells. Breast Cancer Res 8: R42.
Gong Z, Yang Q, Zeng Z, Zhang W, Li X, Zu X, Deng H, Chen P, Liao Q, Xiang B, Zhou M, Li X, Li Y, Xiong W, Li G (2016) An integrative transcriptomic analysis reveals p53 regulated miRNA, mRNA, and lncRNA networks in nasopharyngeal carcinoma. Tumour Biol 37: 3683–3695.
He B, Li W, Wu Y, Wei F, Gong Z, Bo H, Wang Y, Li X, Xiang B, Guo C, Liao Q, Chen P, Zu X, Zhou M, Ma J, Li X, Li Y, Li G, Xiong W, Zeng Z (2016) Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis 7: e2353.
Homan SM, Mercurio AM, LaFlamme SE (1998) Endothelial cells assemble two distinct alpha6beta4-containing vimentin-associated structures: roles for ligand binding and the beta4 cytoplasmic tail. J Cell Sci 111: 2717–2728.
Hood JD, Cheresh DA (2002) Role of integrins in cell invasion and migration. Nat Rev Cancer 2: 91–100.
Horikawa T, Yoshizaki T, Kondo S, Furukawa M, Kaizaki Y, Pagano JS (2011) Epstein–Barr Virus latent membrane protein 1 induces Snail and epithelial–mesenchymal transition in metastatic nasopharyngeal carcinoma. Br J Cancer 104: 1160–1167.
Kalluri R, Weinberg RA (2009) The basics of epithelial–mesenchymal transition. J Clin Invest 119: 1420–1428.
LeClair EE (2003) Four reasons to consider a novel class of innate immune molecules in the oral epithelium. J Dent Res 82: 944–950.
Liang F, Li Q, Li X, Li Z, Gong Z, Deng H, Xiang B, Zhou M, Li X, Li G, Zeng Z, Xiong W (2016) TSC22D2 interacts with PKM2 and inhibits cell growth in colorectal cancer. Int J Oncol 49: 1046–1056.
Liao Q, Zeng Z, Guo X, Li X, Wei F, Zhang W, Li X, Chen P, Liang F, Xiang B, Ma J, Wu M, Tang H, Deng M, Zeng X, Tang K, Xiong W, Li G (2013) LPLUNC1 suppresses IL-6-induced nasopharyngeal carcinoma cell proliferation via inhibiting the Stat3 activation. Oncogene 33: 2098–2109.
Liotta LA, Kohn EC (2001) The microenvironment of the tumour–host interface. Nature 411: 375–379.
Lo KW, To KF, Huang DP (2004) Focus on nasopharyngeal carcinoma. Cancer Cell 5: 423–428.
Lu J, Xu X, Liu X, Peng Y, Zhang B, Wang L, Luo H, Peng X, Li G, Tian W, He M, Li X (2013) Predictive value of miR-9 as a potential biomarker for nasopharyngeal carcinoma metastasis. Br J Cancer 110: 392–398.
Mendez MG, Kojima SI, Goldman RD (2010) Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J 24: 1838–1851.
Meyer T, Marshall JF, Hart IR (1998) Expression of alphav integrins and vitronectin receptor identity in breast cancer cells. Br J Cancer 77: 530–536.
Ou C, Sun Z, Zhang HAN, Xiong WEI, Ma J, Zhou M, Lu J, Zeng Z, Bo X, Chen PAN, Li G, Li X, Li X (2015) SPLUNC1 reduces the inflammatory response of nasopharyngeal carcinoma cells infected with the EB virus by inhibiting the TLR9/NF-κB pathway. Oncol Rep 33: 2779–2788.
Peng C, Li Z, Niu Z, Niu W, Xu Z, Gao H, Niu W, Wang J, He Z, Gao C, Lin P, Agrez M, Zhang Z, Niu J (2016) Norcantharidin suppresses colon cancer cell epithelial-mesenchymal transition by inhibiting the αvβ6-ERK-Ets1 signaling pathway. Sci Rep 6: 20500.
Pirazzoli V, Ferraris GM, Sidenius N (2013) Direct evidence of the importance of vitronectin and its interaction with the urokinase receptor in tumor growth. Blood 121: 2316–2323.
Podor TJ, Singh D, Chindemi P, Foulon DM, McKelvie R, Weitz JI, Austin R, Boudreau G, Davies R (2001) Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J Biol Chem 277: 7529–7539.
Pola C, Formenti SC, Schneider RJ (2013) Vitronectin-alphavbeta3 integrin engagement directs hypoxia-resistant mTOR activity and sustained protein synthesis linked to invasion by breast cancer cells. Cancer Res 73: 4571–4578.
Razak ARA, Siu LL, Liu FF, Ito E, O’Sullivan B, Chan K (2010) Nasopharyngeal carcinoma: the next challenges. Eur J Cancer 46: 1967–1978.
Reuning U (2011) Integrin αvβ3 promotes vitronectin gene expression in human ovarian cancer cells by implicating rel transcription factors. J Cell Biochem 112: 1909–1919.
Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26: 3291–3310.
Ruoslahti E, Pierschbacher MD, Liotta LA, Kohn EC, Barnes D, Wolfe R, Serrero G, McClure D, Sato G (1987) New perspectives in cell adhesion: RGD and integrins. Science 238: 491–497.
Satelli A, Li S (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 68: 3033–3046.
Siret C, Terciolo C, Dobric A, Habib MC, Germain S, Bonnier R, Lombardo D, Rigot V, Andre F (2015) Interplay between cadherins and alpha2beta1 integrin differentially regulates melanoma cell invasion. Br J Cancer 113: 1445–1453.
Sokol ES, Feng Y-X, Jin DX, Tizabi MD, Miller DH, Cohen MA, Sanduja S, Reinhardt F, Pandey J, Superville DA, Jaenisch R, Gupta PB (2017) SMARCE1 is required for the invasive progression of in situ cancers. Proc Natl Acad Sci USA 114: 4153–4158.
Song Y, Li X, Zeng Z, Li Q, Gong Z, Liao Q, Li X, Chen P, Xiang B, Zhang W, Xiong F, Zhou Y, Zhou M, Ma J, Li Y, Chen X, Li G, Xiong W (2016) Epstein–Barr virus encoded miR-BART11 promotes inflammation-induced carcinogenesis by targeting FOXP1. Oncotarget 7: 36783–36799.
Tang Y, He Y, Shi L, Yang L, Wang J, Lian Y, Fan C, Zhang P, Guo C, Zhang S, Gong Z, Li X, Xiong F, Li X, Li Y, Li G, Xiong W, Zeng Z (2017) Co-expression of AFAP1-AS1 and PD-1 predicts poor prognosis in nasopharyngeal carcinoma. Oncotarget 8: 39001–39011.
Thiery JP, Acloque H, Huang RYJ, Nieto MA (2009) Epithelial–mesenchymal transitions in development and disease. Cell 139: 871–890.
Tian YM, Tian YH, Zeng L, Liu S, Guan Y, Lu TX, Han F (2013) Prognostic model for survival of local recurrent nasopharyngeal carcinoma with intensity-modulated radiotherapy. Br J Cancer 110: 297–303.
Tu C, Zeng Z, Qi P, Li X, Yu Z, Guo C, Xiong F, Xiang B, Zhou M, Gong Z, Liao Q, Yu J, He Y, Zhang W, Li X, Li Y, Li G, Xiong W, Sandri-Goldin RM (2017) Genome-wide analysis of 18 Epstein–Barr viruses isolated from primary nasopharyngeal carcinoma biopsy specimens. J Virol 91: e00301–e00317.
Uhm JH, Dooley NP, Kyritsis AP, Rao JS, Gladson CL (1999) Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin Cancer Res 5: 1587–1594.
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C (2017) Role of tumor microenvironment in tumorigenesis. J Cancer 8: 761–773.
Wei F, Li XL, Li XY, Zhang WL, Liao QJ, Zeng Y, Gong ZJ, Zhou M, Ma J, Xiong W, Shen SR, Zeng Z (2014) The effect and mechanism of PLUNC protein family against inflammation and carcinogenesis of nasopharyngeal carcinoma. Prog Biochem Biophys 41: 24–31.
Weston WM, LeClair EE, Trzyna W, McHugh KM, Nugent P, Lafferty CM, Ma L, Tuan RS, Greene RM (1999) Differential display identification of plunc, a novel gene expressed in embryonic palate, nasal epithelium, and adult lung. J Biol Chem 274: 13698–13703.
Xiong W, Zeng ZY, Xia JH, Xia K, Shen SR, Li XL, Hu DX, Tan C, Xiang JJ, Zhou J, Deng H, Fan SQ, Li WF, Wang R, Zhou M, Zhu SG, Lu HB, Qian J, Zhang BC, Wang JR, Ma J, Xiao BY, Huang H, Zhang QH, Zhou YH, Luo XM, Zhou HD, Yang YX, Dai HP, Feng GY, Pan Q, Wu LQ, He L, Li GY, Chua ML, Wee JT, Hui EP, Chan AT, Lo KW, Huang DP, Zeng Z, Huang H, Zhang W, Xiang B, Zhou M, Zhou Y, Ma J, Yi M, Li X, Li X, Xiong W, Li G, Lo KW, To KF, Huang DP (2004) A susceptibility locus at chromosome 3p21 linked to familial nasopharyngeal carcinoma Nasopharyngeal carcinoma. Cancer Res 64: 1972–1974.
Yan QJ, Zeng ZY, Gong ZJ, Zhang WL, Li XY, He BY, Song YL, Li Q, Zeng Y, Liao Q, Chen P, Shi L, Fan S, Xiang B, Ma J, Zhou M, Li X, Yang J, Xiong W, Li G (2015) EBV-miR-BART10-3p facilitates epithelial–mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget 6: 41766–41782.
Yang Y, Liao Q, Wei F, Li X, Zhang W, Fan S, Shi L, Li X, Gong Z, Ma J, Zhou M, Xiang J, Peng S, Xiang B, Deng H, Yang Y, Li Y, Xiong W, Zeng Z, Li G (2013) LPLUNC1 inhibits nasopharyngeal carcinoma cell growth via down-regulation of the MAP kinase and cyclin D1/E2F pathways. PLoS One 8: e62869.
Zeng Z, Fan S, Zhang X, Li S, Zhou M, Xiong W, Tan M, Zhang W, Li G (2015) Epstein–Barr virus-encoded small RNA 1 (EBER-1) could predict good prognosis in nasopharyngeal carcinoma. Clin Transl Oncol 18: 206–211.
Zeng Z, Huang H, Huang L, Sun M, Yan Q, Song Y, Wei F, Bo H, Gong Z, Zeng Y, Li Q, Zhang W, Li X, Xiang B, Li X, Li Y, Xiong W, Li G (2014) Regulation network and expression profiles of Epstein–Barr virus-encoded microRNAs and their potential target host genes in nasopharyngeal carcinomas. Sci China Life Sci 57: 315–326.
Zeng Z, Huang H, Zhang W, Xiang B, Zhou M, Zhou Y, Ma J, Yi M, Li X, Li X, Xiong W, Li G (2011) Nasopharyngeal carcinoma: advances in genomics and molecular genetics. Sci China Life Sci 54: 966–975.
Zhang B, Nie X, Xiao B, Xiang J, Shen S, Gong J, Zhou M, Zhu S, Zhou J, Qian J, Lu H, He X, Li X, Hu G, Li G (2003) Identification of tissue-specific genes in nasopharyngeal epithelial tissue and differentially expressed genes in nasopharyngeal carcinoma by suppression subtractive hybridization and cDNA microarray. Genes Chromosomes Cancer 38: 80–90.
Zhang W, Zeng Z, Wei F, Chen P, Schmitt DC, Fan S, Guo X, Liang F, Shi L, Liu Z, Zhang Z, Xiang B, Zhou M, Huang D, Tang K, Li X, Xiong W, Tan M, Li G, Li X (2014) SPLUNC1 is associated with nasopharyngeal carcinoma prognosis and plays an important role in all-trans-retinoic acid-induced growth inhibition and differentiation in nasopharyngeal cancer cells. FEBS J 281: 4815–4829.
Zhou HD, Fan SQ, Zhao J, Huang DH, Zhou M, Liu HY, Zeng ZY, Yang YX, Huang H, Li XL, Shen SR, Li G-Y (2005) Tissue distribution of the secretory protein, SPLUNC1, in the human fetus. Histochem Cell Biol 125: 315–324.
Zhou HD, Li XL, Li GY, Zhou M, Liu HY, Yang YX, Deng T, Ma J, Sheng SR (2007) Effect of SPLUNC1 protein on the pseudomonas aeruginosa and Epstein–Barr virus. Mol Cell Biochem 309: 191–197.
Zhou Y, Liao Q, Li X, Wang H, Wei F, Chen J, Yang J, Zeng Z, Guo X, Chen P, Zhang W, Tang K, Li X, Xiong W, Li G (2016) HYOU1, regulated by LPLUNC1, is up-regulated in nasopharyngeal carcinoma and associated with poor prognosis. J Cancer 7: 367–376.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81372907, 81472531, 81472595, 81672683, 81672688, and 81772928), the Natural Science Foundation of Hunan Province (2015JJ1022 and 2016JC2035), and the Fundamental Research Funds for the Central South University (2014zzts066).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wei, F., Wu, Y., Tang, L. et al. BPIFB1 (LPLUNC1) inhibits migration and invasion of nasopharyngeal carcinoma by interacting with VTN and VIM. Br J Cancer 118, 233–247 (2018). https://doi.org/10.1038/bjc.2017.385
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.385
Keywords
This article is cited by
-
CircCDYL2 bolsters radiotherapy resistance in nasopharyngeal carcinoma by promoting RAD51 translation initiation for enhanced homologous recombination repair
Journal of Experimental & Clinical Cancer Research (2024)
-
Single-cell RNA sequencing of cervical exfoliated cells reveals potential biomarkers and cellular pathogenesis in cervical carcinogenesis
Cell Death & Disease (2024)
-
BPIFB1, Serving as a Downstream Effector of EBV-miR-BART4, Blocks Immune Escape of Nasopharyngeal Carcinoma via Inhibiting PD-L1 Expression
Biochemical Genetics (2024)
-
Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation
Journal of Hematology & Oncology (2022)
-
BPIFB1 inhibits vasculogenic mimicry via downregulation of GLUT1-mediated H3K27 acetylation in nasopharyngeal carcinoma
Oncogene (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}