
Abstract
Aim:
To examine the therapeutic effects and underlying mechanisms of DZ2002, a reversible S-adenosyl-L-homocysteine hydrolase (SAHH) inhibitor, on lupus-prone female NZB×NZW F1 (NZB/W F1) mice.
Methods:
Female NZB/W F1 mice were treated orally with DZ2002 (0.5 mg·kg−1·d−1) for 11 weeks, and the proteinuria level and body weight were monitored. After the mice ware euthanized, serum biochemical parameters and renal damage were determined. Splenocytes of NZB/W F1 mice were isolated for ex vivo study. Toll-like receptor (TLR)-stimulated human peripheral blood mononuclear cells (PBMCs) or murine bone marrow-derived dendritic cells (BMDCs) were used for in vitro study.
Results:
Treatment of the mice with DZ2002 significantly attenuated the progression of glomerulonephritis and improved the overall health. The improvement was accompanied by decreased levels of nephritogenic anti-dsDNA IgG2a and IgG3 antibodies, serum IL-17, IL-23p19 and TGF-β. In ex vivo studies, treatment of the mice with DZ2002 suppressed the development of pathogenic Th17 cells, significantly decreased IL-17, TGF-β, IL-6, and IL-23p19 production and impeded activation of the STAT3 protein and JNK/NF-κB signaling in splenocytes. DZ2002 (500 μmol/L) significantly suppressed TLR agonists-stimulated up-regulation in IL-6, IL-12p40, TNF-α, and IgG and IgM secretion as well as in HLA-DR and CD40 expression of dendritic cells among human PBMCs in vitro. DZ2002 (100 μmol/L) also significantly suppressed TLR agonists-stimulated up-regulation in IL-6 and IL-23p19 production in murine BMDCs, and prevented Th17 differentiation and suppressed IL-17 secretion by the T cells in a BMDC-T cell co-culture system.
Conclusion:
DZ2002 effectively ameliorates lupus syndrome in NZB/W F1 mice by regulating TLR signaling-mediated antigen presenting cell (APC) responses.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterized by the aberrant function of the immune system and the release of self-antigens, along with autoantibody production1,2. The female NZB/W F1 strain is a classic model of spontaneous lupus; these mice develop severe lupus-like manifestations comparable to that of lupus patients, including elevated serum antinuclear autoantibody levels and immune complex-mediated glomerulonephritis3,4.
Toll-like receptors (TLRs) compose a key family of pattern recognition receptors (PRRs) involved in driving autoimmune inflammation. Christensen has demonstrated the opposing inflammatory and regulatory roles of TLR7 and TLR9 in a murine model of lupus, confirming the significance of TLR7 signaling in promoting specific autoantibody production and increasing disease severity in SLE5. Moreover, Hackl et al reported that a TLR7 ligand, rather than a TLR4 ligand, could reduce de novo regulatory T cell (Treg) generation from naïve T cells. Consistently, the researchers also observed a higher level of IL-17 in a splenic DC-T cell co-culture stimulated with a TLR7 ligand, correlating with a reduced number of Tregs6. Moreover, inappropriate TLR4 activation might lead to chronic inflammation, even with subsequent autoimmune complications7. Additionally, repeated injection of a low dose of the TLR4 ligand LPS into female MRL/n, BXSB, and NZW mice accelerates the development of lupus, with increased autoantibody production and exacerbation of renal disease8. These findings suggested that the selective inhibition of TLR signaling pathways and the further activation of pathogenic T cells could have a therapeutic benefit in SLE.
Aberrations in the innate and adaptive arms of the immune system play an important role in the pathogenesis of lupus. DCs, poised at the interface between innate and adaptive immunity, function as the most potent antigen-presenting cells (APCs), with a crucial role in the activation and differentiation of naïve T cells9,10,11. IL-6 and IL-23, regarded as IL-17-inducing APC-derived factors, actively participate in pathogenic Th17 cell differentiation and subsequent responses in various chronic inflammatory and autoimmune diseases, including rheumatoid arthritis (RA)12, inflammatory bowel disease13, systemic sclerosis14 and SLE15,16. DCs are considered a cellular source of IL-6 and IL-23. TLR ligation on a human DC induces high expression of IL-23p19 and IL-6, which is positively correlated with the elevated IL-17A levels among co-cultured T cells17,18.
The immunosuppressive properties of S-adenosyl-L-homocysteine hydrolase (SAHH) inhibitors have been well known for years. Irreversible type I SAHH inhibitors have been demonstrated to have preventive and therapeutic effects in T-cell-mediated experimental animal models19,20,21. Nevertheless, the mechanisms of SAHH inhibitors remain elusive. The reversible type III SAHH inhibitor DZ2002 [methyl-(adenin-9-yl)-2-hydroxybutanoate] has been found to have an immunomodulatory function and to alleviate disease in several inflammatory and autoimmune animal models, with greatly reduced cytotoxicity22,23,24,25. Recently, Tardif et al reported that treatment with DZ2002 prevented lupus-like disease from developing in both BXSB and MRL-Faslpr mouse models and that methylation inhibition might be an approach for the treatment of autoimmune diseases26.
Here, we demonstrated that DZ2002 exerted a therapeutic effect on lupus-prone female NZB/W F1 mice with established nephritis. The mechanism involves the modulation of T cell development in lupus by interfering with TLR-triggered APC function.
Materials and methods
Animals
Female NZB/W F1 mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Female C57BL/6 and BALB/c mice were obtained from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences. All mice were housed under specific pathogen-free conditions. All experiments were performed according to the institutional ethical guidelines on animal care and approved by the Institute Animal Care and Use Committee at the Shanghai Institute of Materia Medica.
Therapeutic regimens
In this study, 23-week-old female NZB/W F1 mice were orally treated with DZ2002 for 11 weeks. At the initiation of treatment, the mice were randomly divided according to body weight and proteinuria level and were orally treated for 6 consecutive days every week. The mice were divided into three groups (n=8 per group): a vehicle-control group (saline), a positive-control group [prednisolone (PNS) 2 mg·kg−1·d−1] and a DZ2002-treated group (0.5 mg·kg−1·d−1), and proteinuria levels and body weight were monitored weekly. During the treatment, individuals were euthanized, followed by serum and kidney collection if their urine protein concentration was >10 mg/mL for 2 consecutive weeks, accompanied by weight loss >20%. In this case, the last known values for urinary protein and body weight were carried forward27.
Biochemical parameters, renal histopathology and immunofluorescence examination
Urinary protein concentrations were determined with a Coomassie blue G dye-binding assay. Blood urea nitrogen (BUN) and creatinine (CRE) concentrations were determined using a HITACHI-7080 automatic biochemical analyzer (Hitachi High-Technologies Corporation, Tokyo, Japan). Formalin-fixed kidney sections were embedded in paraffin and stained with hematoxylin and eosin (H&E). The sections were then graded by two independent renal pathologists for glomerular, interstitial, and vascular lesions according to a grading scheme that was a modification of the method described by Tao and colleagues28. Briefly, renal damage indices included the following: glomerulonephritis, interstitial lymphocytic infiltration, tubular dilatation and perivascular lymphocytic infiltration (0- to 4-point scale, where 0=no proliferation or mononuclear cell infiltration between/around tubules and 4=extensive sclerosis/crescents and mononuclear cell infiltration of the surrounding parenchyma/extensive tubule atrophy/vasculitis). Snap-frozen kidneys were used for immunofluorescence examination of IgG deposits, and glomerular staining was analyzed by Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).
Flow cytometric analysis
Surface marker and intracellular transcription factor staining was conducted and analyzed using our previously reported methods29. Briefly, for surface marker and intracellular transcription factors, cells were collected and stained with FITC-, PE-, PerCP-Cy5.5-, or APC-conjugated monoclonal antibodies (mAbs) for membrane molecules or intracellular staining after being blocked with anti-mCD16/CD32 (Fc Receptor Block, eBioscience, San Diego, CA, USA). For intracellular staining of cytokines, cells were stimulated with PMA (50 ng/mL) and ionomycin (750 ng/mL) in the presence of GolgiStop (10 μg/mL) for 4 h and then collected for the following staining. The immunofluorescent Abs used in this study were all from BD Biosciences (Franklin Lakes, NJ, USA). The FoxP3 Staining Buffer Set was from eBioscience (San Diego, CA, USA). Flow cytometric analysis was performed on a 2-laser/4-color FACSCalibur analytical cytometer or a 4-laser/13-color BD LSR II analytical cytometer (BD Biosciences), and the data were analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
Generation of bone marrow-derived DCs (BMDCs)
BMDCs were prepared by culturing BM cells from normal C57BL/6 mice in RPMI-1640 containing 10% fetal bovine serum (FBS) supplemented with 10 ng/mL GM-CSF (PeproTech, Rocky Hill, NJ, USA). On d 8, loosely adherent cells were collected, washed, and used in assays. The purity of the resulting cells was determined by flow cytometric analysis, and the percentage of CD11c+ cells was consistently >95%.
Purification of naïve splenic CD4+ T cells
CD4+ T cell purification was conducted using our previously reported methods29. Briefly, mAb cocktails were added to deplete CD8+ cells, B220+ cells, NK1.1+ cells, and I-A+ APCs from splenocytes, and an anti-CD44 mAb (KM201) was added to obtain naïve CD4+ T cells (CD4+CD44−CD62L+). The remaining cells were then incubated with L3T4 MicroBeads (Miltenyi Biotec GmbH, German) and positively selected. The purity of the resulting cells was determined by flow cytometric analysis and was consistently >98%.
In vitro T cell differentiation
In vitro induction of T cell differentiation was conducted as described previously29. Briefly, naive CD4+ T cells were activated with an anti-CD3 mAb (5 μg/mL) and an anti-CD28 mAb (2 μg/mL) and were induced to differentiate. For Th1 cell differentiation, mIL-12 (10 ng/mL; PeproTech, Rocky Hill, NJ, USA) and anti-mIL-4 (10 μg/mL; BD Pharmingen) were added to the cultures. For inducible Treg differentiation, human transforming growth factor-β (10 ng/mL; PeproTech, Rocky Hill, NJ, USA), mIL-2 (100 units/mL; BD Pharmingen, San Diego, CA, USA) and anti-mIFN-γ (10 μg/mL) were added to the cultures. For Th17 cell differentiation, we used a cocktail containing mIL-6 (25 ng/mL), hTGF-β (10 ng/mL), mIL-23 (20 ng/mL; eBioscience, San Diego, CA, USA), anti-mIFN-γ (10 μg/mL) and anti-mIL-4 (10 μg/mL). Cells stimulated under neutral conditions (anti-mIL-4 plus anti-mIFN-γ but no additional cytokines) were considered naive cells. DZ2002 (50 μmol/L) was added simultaneously.
Ex vivo stimulation of splenocytes
Splenocytes from NZB/W F1 mice were cultured in 24-well plates (4×106 cells/well) coated with an anti-CD3 mAb (145-2C11, 5 μg/mL) or stimulated with LPS (10 μg/mL) for 48 h. After incubation, supernatants were collected, and the cytokine concentration was determined by ELISA.
In vitro stimulation of peripheral blood mononuclear cells (PBMCs)
PBMCs were isolated from leukocytes acquired from healthy donors by centrifugation over Ficoll-Paque Plus (Amersham Biosciences, Piscataway, NJ, USA) gradients using standard procedures. PBMCs were cultured (1.5×106 cells/well in a 24-well plate or 3×105 cells/well in a 96-well plate) with medium alone, the TLR4 ligand LPS (5 μg/mL; Sigma-Aldrich, St Louis, MO, USA), the TLR7 ligand R837 (imiquimod, 3 μg/mL; Invivogen, San diego, CA, USA) or the TLR9 ligand ODN 2336 (1 μmol/L; Invivogen, San diego, CA, USA) and with or without DZ2002 (500 μmol/L, without cytotoxicity to PBMCs) were added simultaneously. After incubation, for 24-well plates, the supernatants were collected to determine cytokine and Ab concentrations by ELISA, and the cultured cells were analyzed by flow cytometry. For 96-well flat-bottom plates, the cultures were pulsed with 0.5 μCi [3H]thymidine per well for 12 h before harvesting and were assessed for [3H]thymidine incorporation at 72 h.
In vitro stimulation of BMDCs
Immature BMDCs were cultured in 24-well plates (8×105 cells/well) with medium alone, the TLR4 ligand LPS (10 μg/mL; Sigma-Aldrich, St Louis, MO, USA), the TLR7 ligand R837 (imiquimod, 5 μg/mL; Invivogen, San diego, CA, USA) or the TLR9 ligand ODN 1668 (5 μmol/L; Invivogen) and with or without DZ2002 (100 μmol/L, without significant cytotoxicity to BMDCs) were added simultaneously. After incubation, supernatants were collected to determine cytokine concentrations by ELISA.
DC-T cell co-culture
TLR ligand-primed BMDCs (2×105 cells/well) with or without DZ2002 (100 μmol/L) treatment were co-cultured with purified naïve CD4+ T cells (8×105 cells/well) in 24 well-plates pre-coated with an anti-CD3 mAb. After 4 d of culture, the cells were harvested and re-stimulated with PMA (50 ng/mL) plus ionomycin (750 ng/mL) for 4 h in the presence of a protein transport inhibitor (BD GolgiStop™, 10 μg/mL) for intracellular staining.
ELISA for Ab detection
Serum anti-dsDNA Abs were detected using our previously reported methods29. Briefly, 96-well high-binding microplates were coated with calf thymus dsDNA (Sigma-Aldrich, St Louis, MO, USA) and incubated at 4 °C overnight. Each diluted serum sample was added and incubated. HRP-conjugated goat anti-mouse IgG (H+L), IgG1, IgG2a, and IgG3 Abs (Invitrogen, San diego, CA, USA) were then added and incubated. Serum from female NZB/W F1 mice at 10 months of age was used as the standard. Abs in culture supernatants were assayed using a human IgG and IgM ELISA set (Abcam, Cambridge, Massachusetts, USA).
ELISA for cytokine detection
Cytokines in sera or culture supernatants were assayed using mouse IL-17A, TGF-β, IL-12p40, IL-23p19, IL-6, IL-1β, IFN-γ, and IL-10 ELISA kits and human IL-6, IL-12p40, and TNF-α ELISA kits (all from BD Pharmingen) according to the manufacturer's instructions.
Western blot analysis
Splenocytes from different groups of NZB/W F1 mice or BMDC cultures were directly lysed in sodium dodecyl sulfate (SDS) sample buffer and boiled for 10 min at 100 °C. Proteins were resolved by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech) for immunoblotting. Anti-phospho-IκBα, anti-phospho-JNK, anti-STAT3, anti-phospho-STAT3, and anti-GAPDH were from Cell Signaling Technology (Beverly, Massachusetts, USA). Proteins were visualized using HRP-conjugated anti-rabbit or anti-mouse IgG and the ECL system (Amersham Biosciences).
Statistical analysis
Proteinuria and body weight data were analyzed using one-way analysis of variance (ANOVA) for repeated measures, corrected with the Bonferroni posttest. Nonparametric data (histological scores) were analyzed using a Mann-Whitney U test. Additionally, data were analyzed using Student's t-test and one-way ANOVA. P<0.05 was considered statistically significant. For statistical analyses, GraphPad Prism 5.0 software was used.
Results
Effects of DZ2002 on proteinuria and renal function in female NZB/W F1 mice
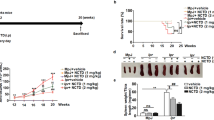
DZ2002 treatment significantly inhibited the progression and aggravation of proteinuria and maintained the body weight of lupus-afflicted mice (Figures 1A and 1B). Additionally, DZ2002 prevented the worsening of renal function, represented by a significant decrease in BUN and CRE levels compared with the vehicle group (Figure 1C).

DZ2002 treatment retarded disease progression in NZB/W F1 mice. Twenty-three weeks old female NZB/W F1 mice with established lupus were orally treated with vehicle (saline), PNS (2 mg/kg) or DZ2002 (0.5 mg/kg). Proteinuria (A) and bodyweight (B) were measured every week. (C) Serum level of BUN and CRE were detected at the end of the 11-week treatment. (D) Representative kidney sections stained with H&E (left-panel, magnification×100) and frozen sections stained for IgG (right-panel, magnification×100) from mice. (E) Up-panel: Glomerular, interstitial and vascular disease was scored from (0) to (4+) as described in materials and methods. Bottom-panel: Score for glomeruli immune deposits was analyzed by image analysis software. Mean±SEM. n=8. bP<0.05; cP<0.01 vs vehicle.
Effects of DZ2002 on the kidney pathology of female NZB/W F1 mice
To evaluate the effects of DZ2002 on renal injury and immune-complex deposition, kidneys were examined by histopathology and an immunofluorescence assay. We found that kidney sections from vehicle-treated mice exhibited severe renal injury, characterized by glomerulosclerosis, tubular cast/cyst formation, an increased mesangial matrix and diffuse perivascular and interstitial mononuclear cell infiltration. Treatment with DZ2002 exerted significant protection against renal injury, with amelioration in glomerular and perivascular injuries and interstitial inflammatory cell infiltration (Figure 1D, left panel, and Figure 1E, upper panel).
Renal cryostat sections were stained to detect IgG deposition. Pronounced scattered granular IgG deposits were observed in kidneys from vehicle-treated mice. In contrast, mice treated with DZ2002 exhibited significantly reduced IgG deposition (Figure 1D, right panel, and Figure 1E, bottom panel).
Effects of DZ2002 on the levels of Abs and serum cytokines in female NZB/W F1 mice
Anti-dsDNA IgG Abs, and especially nephritogenic IgG2a and IgG3 isotypes, appeared to be predominant in the progression of glomerulonephritis in SLE30,31. Both DZ2002 and PNS treatment remarkably decreased the level of serum total anti-dsDNA IgG Abs, the IgG2a, and IgG3 isotypes (Figure 2A).

DZ2002 treatment modulated serum anti-dsDNA autoantibodies and cytokines in NZB/W F1 mice. At the termination of treatment, serum of female NZB/W F1 mice were collected and examined for anti-dsDNA autoantibodies and cytokines. (A) Serum levels of anti-dsDNA total IgG, IgG1, IgG2a, and IgG3 antibodies. (B) Serum levels of IL-17A, TGF-β, IL-23p19, and IL-12p40. Mean±SEM. n=8 for each group, each symbol represented an individual mouse. bP<0.05, cP<0.01 vs vehicle.
DZ2002 treatment significantly decreased the serum levels of IL-17A and IL-23p19, and IL-12p40 was slightly affected (Figure 2B). In addition to its role as a differentiation factor, up-regulated TGF-β has an alternative function in glomerular injury, accelerating nephritis by facilitating excessive matrix deposition31,32,33. The reduced serum level of TGF-β might partially contribute to the protective function of DZ2002 against renal injury.
Effects of DZ2002 on T cell development and APC responses
To confirm the effects of DZ2002 on the development of pathogenic helper T cells, a flow cytometric analysis was conducted on murine splenocytes. The results showed that DZ2002 treatment significantly reduced the accumulation of IL-17-producing cells and enhanced the accumulation of FoxP3+ cells in both CD4+ and CD3+T cells in the spleen. However, the proportion of IFN-γ-producing cells was slightly changed, without altering the percentage of CD4+ T cells, CD8+ T cells, or CD19+B cells in the spleen (Figure 3A, Figure S1 and Figure S2).

DZ2002 treatment suppressed Th17 cell development and inhibited IL-6 and IL-23p19 production from splenocytes while suppressing the activation of STAT3 and JNK/NF-κB signaling. At the termination of treatment, female NZB/W F1 mice in each treated groups were randomly divided into 3 sub-groups. Spleens of each sub-group were pooled together. (A) Up-left: Representative staining of intracellular IL-17 and IFN-γ in CD4+ T cell. Bottom-left: Representative result of FoxP3+CD25+ percentage in CD4+ T cells. Right: Frequency of IL-17+, IFN-γ+, and FoxP3+ populations in CD4+ T cells. (B) Cytokine productions in splenocytes stimulated with anti-CD3 mAb for 48 h. (C) Cytokine productions in splenocytes stimulated with LPS (10 μg/mL) for 48 h. Western blot analysis of total STAT3, phosphorylated STAT3 (D), I-κBα, and JNK (E) proteins in splenocytes of NZB/W F1 mice from vehicle-treated group (F1), PNS-treated group (PNS). and DZ2002-treated group (DZ). Age-matched naïve C57BL/6 mice (B6) were used as normal control. Mean±SEM. n=3 independent experiments. bP<0.05, cP<0.01 vs vehicle.
For splenocytes in the DZ2002 group, upon ex vivo stimulation with an anti-CD3 mAb, the levels of IL-17A, TGF-β, and IFN-γ were much lower, whereas IL-10 level was significantly increased (Figure 3B). Furthermore, the APC responses of splenocytes conditioned with the synthetic TLR4 ligand LPS were examined. The levels of IL-23p19 and IL-6, the two critical boosters of the STAT3-Th17 axis, were significantly decreased in the cultures of splenocytes from DZ2002-treated mice compared with those from vehicle-treated mice. In contrast, the IL-12p40 level in the splenocyte cultures did not vary between DZ2002- and vehicle-treated mice (Figure 3C).
To investigate whether DZ2002 has a direct influence on helper T cell differentiation, naive CD4+ T cells were isolated from naive C57BL/6 mice and stimulated with differentiation-inducing cytokine cocktails. DZ2002 exerted few effects on the generation of a Th1, Th17, or Treg population in vitro (data not shown). Given that DZ2002 treatment could modulate the expression of APC-derived cytokines, which contribute substantially to T cell activation and differentiation by presenting antigens, supplying a co-stimulatory signal and altering the local cytokine milieu, we speculated that the decrease in the Th17 cell population in DZ2002-treated lupus mice could be indirectly mediated by the treatment's effect on APCs.
The STAT3 protein, which is activated by IL-6 and IL-23, is both necessary and sufficient for inflammatory Th17 cell differentiation, thus functioning as an essential regulator of autoimmune processes34. DZ2002 treatment significantly suppressed the phosphorylation of STAT3 in splenocytes, which was consistent with the serum IL-17 level (Figure 3D).
The JNK/NF-κB pathway greatly contributes to DC control of Th cell polarization and to inflammatory responses35,36. We thus evaluated the effect of DZ2002 treatment on JNK/NF-κB signaling proteins. As expected, the results showed that DZ2002 treatment abrogated the phosphorylation of IκBα, an NF-κB inhibitory protein, and the phosphorylation of JNK, a molecule upstream of NF-κB (Figure 3E). These data indicated that DZ2002 treatment significantly abated the transduction of this canonical inflammatory signaling pathway in lupus-afflicted NZB/W F1 mice.
DZ2002 inhibited DC and B cell activation among PBMCs upon TLR ligation
To clarify the definite effects of DZ2002 on the function of APCs, PBMCs from healthy donors were used to assess the correlates of APCs following lupus-related TLR engagement. We examined the expression of HLA-DR and CD40 on the surface of DCs following stimulation with a synthetic ligand of TLR4 (LPS), TLR7 (R837), or TLR9 (ODN2336). The results demonstrated that DZ2002 markedly decreased HLA-DR and CD40 expression on CD11c+ DCs upon stimulation with TLR agonists (Figure 4A).

DZ2002 inhibited DC and B cell activation of PBMCs upon TLR ligation. PBMCs from healthy donor were treated without (w/o) or with TLR agonists in the presence or absence of DZ2002 (500 μmol/L). (A) HLA-DR and CD40 expression of CD11c+ DCs in PBMCs were analyzed after 48-h culture by flow cytometry. Shaded area depicted unstimulated cells, full line represented staining after stimulation with TLR-Ls, and dotted line showed staining after DZ2002 incubation. Data shown represent results of 3 independent experiments with 3 different donors. (B) Cytokine productions were measured from PBMCs supernatants after 48-h culture by ELISA. (C) Proliferation of PBMCs after 72-h incubation. (D) IgG and IgM secretion measured from PBMC supernatants after 6-d incubation by ELISA. Mean±SEM. n=3 independent experiments with 3 different donors. bP<0.05, cP<0.01 versus corresponding medium with similar stimulatory condition.
TLR ligands induce the production of IL-6, IL-12p40, and TNF-α, primarily from DCs. As shown in Figure 4B, DZ2002 significantly suppressed IL-6, IL-12p40, and TNF-α production by PBMCs induced by TLR agonists.
TLR signaling can trigger B cells to proliferate and to secrete IgG and IgM37. We next assessed whether the magnitude of the B cell response to TLR ligands among DZ2002-treated PBMCs was altered. The results showed that the proliferative responses of PBMCs triggered by TLR4-L, TLR7-L, or TLR9-L were significantly suppressed by DZ2002 (Figure 4C). Moreover, the IgG and IgM production by PBMCs induced by TLR7 or TLR9 agonist was correspondingly decreased after treatment with DZ2002 (Figure 4D).
DZ2002 modulated T cell differentiation via altering the cytokine expression pattern of BMDCs induced by TLR ligation
To clarify the definite effects of DZ2002 on the function of APCs, BMDCs from naïve C57BL/6 mice were used to assess the correlates of APCs following lupus-related TLR engagement. We examined the cytokine expression of BMDCs following stimulation with a synthetic ligand of TLR4 (LPS), TLR7 (R837), or TLR9 (ODN1668). The results demonstrated that DZ2002 significantly inhibited IL-23p19 secretion from BMDCs induced by TLR7-L or TLR9-L. Additionally, DZ2002 impaired IL-6 production by BMDCs induced by TLR4-L or TLR7-L (Figure 5A).

DZ2002 altered the cytokine-secreting pattern of BMDCs to suppress Th17 cell differentiation in vitro. (A) BMDCs from normal female C57BL/6 mice were treated without (w/o) or with TLR agonists in the presence or absence of DZ2002 (100 μmol/L) for 24 h. Cytokine productions were measured by ELISA. (B) Naïve CD4+ T cells stimulated with anti-CD3 mAb were co-cultured with BMDCs, which were primed without (w/o) and with TLR agonists in the presence (DZ-DC) or absence (DC) of DZ2002 (100 μmol/L), for 4 d. Cytokine productions were measured from the co-culture supernatants by ELISA. (C) Co-cultured T cells were analyzed by flow cytometer. Left: Intracellular staining of IL-17 and IFN-γ in cultured CD4+ T cells. Middle: The percentage of FoxP3+ Tregs in cultured CD4+ T cells was measured. Data shown represent results of 3 independent experiments. Right: Statistical analysis of 3 independent experiments. Mean±SEM. n=3 independent experiments. bP<0.05, cP<0.01 vs corresponding medium with similar stimulatory condition. fP<0.01 vs the culture contained BMDCs without TLR agonist prime.
Having identified that DZ2002 could significantly inhibit the production of BMDC-derived pro-inflammatory cytokines induced by TLR agonists, we investigated the impact of DZ2002 on T cell differentiation in vitro in the context of BMDCs. Naïve CD4+ T cells were stimulated with an anti-CD3 mAb and co-cultured with BMDCs, which were primed via TLRs and with or without DZ2002, to model the physiological interaction between DCs and T cells. T cells activated by primed BMDCs pretreated with DZ2002 secreted much lower amounts of IL-17A, whereas the amounts of IFN-γ were little changed (Figure 5B). Consistent with the reduction of IL-17 levels in the co-culture, a significantly lower frequency of IL-17+ T cells was observed after incubation with either TLR7 or TLR9 ligand-primed BMDCs pretreated with DZ2002, whereas the percentage of IFN-γ+ T cells was slightly changed (Figure 5C, left and right panels). Moreover, IL-10 levels were much lower in the TLR7-L-primed BMDC-T cell co-culture, regardless of DZ2002 pretreatment (Figure 5B). Accordingly, the expression of FoxP3 was slightly up-regulated in T cells co-cultured with DZ2002-pretreated TLR7-L- or TLR9-L-primed BMDCs (Figure 5C, middle and right panels). It suggested that the dampened Th17 response induced by DZ2002 was associated with the altered TLR-mediated BMDC functions.
Discussion
In this study, we explored the therapeutic effects and mechanisms of DZ2002 in autoimmune syndrome in lupus-prone female NZB/W F1 mice. The results demonstrated that DZ2002 treatment could significantly reverse the imbalance of nephritogenic and inhibitory Ab levels, suppress pathogenic Th17 development and the activation of inflammatory signaling pathways, reduce immune-complex deposition and ameliorate renal injury. Remarkably, the therapeutic effects of DZ2002 were closely linked to its modulation of TLR signaling-mediated APC responses.
TLRs, which play a key role in innate responses to infections, were recently implicated in acute and chronic inflammatory processes induced by endogenous ligands. Numerous studies have established that the activation of particular TLRs, such as TLR4, TLR7, and TLR9, leads to the activation of immune cells, most notably DCs and B cells, and to the inappropriate production of autoantibodies and/or cytokines known to be directly involved in the pathogenesis of SLE5,38,39.
IL-17 has been implicated in many chronic autoimmune diseases, including psoriasis, multiple sclerosis and systemic sclerosis, and in SLE15,40. IL-23-dependent Th17 cells can invade the target organ and promote the development of organ-specific autoimmune inflammation41,42. The current study demonstrated that DZ2002 treatment markedly reduced the Th17 population in NZB/W F1 mice, accompanied by a comprehensive suppression of IL-17 production. Intriguingly, DZ2002 only mildly affects CD4+ T cell differentiation under polarizing cytokine conditions in vitro. According to the findings that we obtained from analyzing the activation of proteins in TLR-related JNK/NF-κB signaling and the levels of APC-derived cytokines in DZ2002-treated NZB/W F1 mice, we examined the effects of DZ2002 on APCs. These APCs included DCs, the APCs that most potently prime naïve T cells and initiate an immune response, and B cells, the major source of pathogenic autoantibodies. Using TLR ligands as activating agents, the data presented here demonstrate that DZ2002 effectively inhibited the activation of co-stimulatory and antigen-presenting molecules and the secretion of pro-inflammatory cytokines from human DCs in response to TLR agonists.
In line with this observation, we further questioned whether the altered cytokine-secreting pattern of DCs could lead to the subsequent variations in T cell differentiation. We then used a primed murine BMDC-T cell co-culture system to mimic in vivo DC-T cell cross-talk. Our data indicated that DZ2002 affected the function of BMDCs in directing T cell polarization, significantly preventing Th17 differentiation and IL-17 production in the co-culture system.
Furthermore, we demonstrated that in addition to its impacts on DCs, DZ2002 could impede the consequent activation of B cells among PBMCs upon TLR ligand stimulation, affecting both proliferation and Ab production. This observation provided a rational basis for the therapeutic effects of DZ2002 on serum autoantibody levels in NZB/W F1 mice.
Although recent reports have described the effects of an SAHH inhibitor or methylation inhibition on MRL/lpr mice26,43, our study demonstrated that the reversible SAHH inhibitor DZ2002 exerted a therapeutic benefit in lupus-prone female NZB/W F1 mice, another classic animal model of lupus. The therapeutic mechanism of DZ2002 might be attributed to its function in TLR signaling-mediated responses in APCs, which could control both autoantibody accumulation and pathogenic T cell development in SLE.
Author contribution
Study conception and design: Jian-ping ZUO, Wei TANG, and Shi-jun HE. Acquisition of data: Shi-jun HE, Ze-min LIN, Li-fei HOU, Xiao-hui ZHANG, Yan-wei WU, Bing-xin BAI, Xiao-qian YANG, Pei-lan HE, and Feng-hua ZHU. Analysis and interpretation of data: Shi-jun HE, Ze-min LIN, Wei TANG, Jian-ping ZUO. Drafting the article or revising it critically for important intellectual content: Shi-jun HE, Ze-min LIN, Wei TANG, and Jian-ping ZUO.
References
Tsokos GC . Systemic lupus erythematosus. N Engl J Med 2011; 365: 2110–21.
Wentworth J, Davies C . Systemic lupus erythematosus. Nat Rev Drug Discov 2009; 8: 103–4.
Theofilopoulos AN, Dixon FJ . Murine models of systemic lupus erythematosus. Adv Immunol 1985; 37: 269–390.
Perry D, Sang A, Yin Y, Zheng YY, Morel L . Murine models of systemic lupus erythematosus. J Biomed Biotechnol 2011; 2011: 271694.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ . Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006; 25: 417–28.
Hackl D, Loschko J, Sparwasser T, Reindl W, Krug AB . Activation of dendritic cells via TLR7 reduces Foxp3 expression and suppressive function in induced Tregs. Eur J Immunol 2011; 41: 1334–43.
Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T . Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint Bone Spine 2011; 78: 124–30.
Hang L, Slack JH, Amundson C, Izui S, Theofilopoulos AN, Dixon FJ . Induction of murine autoimmune disease by chronic polyclonal B cell activation. J Exp Med 1983; 157: 874–83.
Garrigan K, Moroni-Rawson P, McMurray C, Hermans I, Abernethy N, Watson J, et al. Functional comparison of spleen dendritic cells and dendritic cells cultured in vitro from bone marrow precursors. Blood 1996; 88: 3508–12.
Moser M, Murphy KM . Dendritic cell regulation of TH1-TH2 development. Nat Immunol 2000; 1: 199–205.
Re F, Strominger JL . Heterogeneity of TLR-induced responses in dendritic cells: from innate to adaptive immunity. Immunobiology 2004; 209: 191–8.
Miossec P . Interleukin-17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum 2003; 48: 594–601.
Shih DQ, Targan SR, McGovern D . Recent advances in IBD pathogenesis: genetics and immunobiology. Curr Gastroenterol Rep 2008; 10: 568–75.
Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum 2000; 43: 2455–63.
Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW . Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol 2008; 127: 385–93.
Nalbandian A, Crispin JC, Tsokos GC . Interleukin-17 and systemic lupus erythematosus: current concepts. Clin Exp Immunology 2009; 157: 209–15.
Lombardi V, Van Overtvelt L, Horiot S, Moingeon P . Human dendritic cells stimulated via TLR7 and/or TLR8 induce the sequential production of Il-10, IFN-gamma, and IL-17A by naive CD4+ T cells. J Immunol 2009; 182: 3372–9.
Yu CF, Peng WM, Oldenburg J, Hoch J, Bieber T, Limmer A, et al. Human plasmacytoid dendritic cells support Th17 cell effector function in response to TLR7 ligation. J Immunol 2010; 184: 1159–67.
Wolos JA, Frondorf KA, Esser RE . Immunosuppression mediated by an inhibitor of S-adenosyl-L-homocysteine hydrolase. Prevention and treatment of collagen-induced arthritis. J Immunol 1993; 151: 526–34.
Saso Y, Conner EM, Teegarden BR, Yuan CS . S-Adenosyl-L-homocysteine hydrolase inhibitor mediates immunosuppressive effects in vivo: suppression of delayed type hypersensitivity ear swelling and peptidoglycan polysaccharide-induced arthritis. J Pharmacol Exp Ther 2001; 296: 106–12.
Wolos JA, Frondorf KA, Babcock GF, Stripp SA, Bowlin TL . Immunomodulation by an inhibitor of S-adenosyl-L-homocysteine hydrolase: inhibition of in vitro and in vivo allogeneic responses. Cell Immunol 1993; 149: 402–8.
Wu QL, Fu YF, Zhou WL, Wang JX, Feng YH, Liu J, et al. Inhibition of S-adenosyl-L-homocysteine hydrolase induces immunosuppression. J Pharmacol Exp Ther 2005; 313: 705–11.
Fu YF, Wang JX, Zhao Y, Yang Y, Tang W, Ni J, et al. S-adenosyl-L-homocysteine hydrolase inactivation curtails ovalbumin-induced immune responses [Research Support, Non-US Gov't]. The Journal of pharmacology and experimental therapeutics 2006; 316: 1229–37.
Lawson BR, Manenkova Y, Ahamed J, Chen X, Zou JP, Baccala R, et al. Inhibition of transmethylation down-regulates CD4 T cell activation and curtails development of autoimmunity in a model system. J Immunol 2007; 178: 5366–74.
Fu YF, Zhu YN, Ni J, Zhong XG, Tang W, Re YD, et al. A reversible S-adenosyl-L-homocysteine hydrolase inhibitor ameliorates experimental autoimmune encephalomyelitis by inhibiting T cell activation. The Journal of pharmacology and experimental therapeutics 2006; 319: 799–808.
Tardif V, Manenkova Y, Berger M, Hoebe K, Zuo JP, Yuan C, et al. Critical role of transmethylation in TLR signaling and systemic lupus erythematosus. Clin Immunology 2013; 147: 133–43.
Bahjat FR, Pine PR, Reitsma A, Cassafer G, Baluom M, Grillo S, et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheum 2008; 58: 1433–44.
Tao X, Fan F, Hoffmann V, Longo NS, Lipsky PE . Therapeutic impact of the ethyl acetate extract of Tripterygium wilfordii Hook F on nephritis in NZB/W F1 mice. Arthritis Res Ther 2006; 8: R24.
Hou LF, He SJ, Li X, Yang Y, He PL, Zhou Y, et al. Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum 2011; 63: 2445–55.
Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum 1999; 42: 1644–8.
Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV . Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med 2006; 203: 789–97.
Border WA, Noble NA . Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994; 331: 1286–92. eng.
Suzuki Y, Ito Y, Mizuno M, Kinashi H, Sawai A, Noda Y, et al. Transforming growth factor-beta induces vascular endothelial growth factor-C expression leading to lymphangiogenesis in rat unilateral ureteral obstruction. Kidney Int 2012; 81: 865–79.
Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007; 282: 9358–63.
Zhai YP, Lu Q, Liu YW, Cheng Q, Wei YQ, Zhang F, et al. Over-production of nitric oxide by oxidative stress-induced activation of the TGF-beta1/PI3K/Akt pathway in mesangial cells cultured in high glucose. Acta pharmacol Sin 2013; 34: 507–14.
Jung Y, Byeon SE, Yoo DS, Lee YG, Yu T, Yang Y, et al. 8-(Tosylamino)quinoline inhibits macrophage-mediated inflammation by suppressing NF-kappaB signaling. Acta pharmacol Sin 2012; 33: 1037–46.
Bekeredjian-Ding I, Jego G . Toll-like receptors — sentries in the B-cell response. Immunology 2009; 128: 311–23.
Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ . Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med 2005; 202: 321–31.
Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol 2010; 184: 1840–8.
Afzali B, Lombardi G, Lechler RI, Lord GM . The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol 2007; 148: 32–46.
Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ . Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum 2007; 56: 2936–46.
Wang QQ, Yang HZ, Liu HZ, Mi S, Zhang XW, Yan HM, et al. Interleukin-17A is involved in development of spontaneous pulmonary emphysema caused by Toll-like receptor 4 mutation. Acta Pharmacol Sin 2011; 32: 1045–54.
Yang ML, Gee AJ, Gee RJ, Zurita-Lopez CI, Khare S, Clarke SG, et al. Lupus autoimmunity altered by cellular methylation metabolism. Autoimmunity 2013; 46: 21–31.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (No 81072652, 81273524, 81273525); the National Science & Technology Major Project “New Drug Creation and Manufacturing Program”, China (2012ZX09102-101-006); and the Science & Technology Commission of Shanghai Municipality, China (11431921102).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary figures were available on the APS's website.
Supplementary information
Rights and permissions
About this article
Cite this article
He, Sj., Lin, Zm., Wu, Yw. et al. Therapeutic effects of DZ2002, a reversible SAHH inhibitor, on lupus-prone NZB×NZW F1 mice via interference with TLR-mediated APC response. Acta Pharmacol Sin 35, 219–229 (2014). https://doi.org/10.1038/aps.2013.167
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.167
Keywords
This article is cited by
-
DZ2002 alleviates corneal angiogenesis and inflammation in rodent models of dry eye disease via regulating STAT3-PI3K-Akt-NF-κB pathway
Acta Pharmacologica Sinica (2024)
-
A novel tricyclic BTK inhibitor suppresses B cell responses and osteoclastic bone erosion in rheumatoid arthritis
Acta Pharmacologica Sinica (2021)
-
Reversible SAHH inhibitor protects against glomerulonephritis in lupus-prone mice by downregulating renal α-actinin-4 expression and stabilizing integrin-cytoskeleton linkage
Arthritis Research & Therapy (2019)
-
DZ2002 ameliorates fibrosis, inflammation, and vasculopathy in experimental systemic sclerosis models
Arthritis Research & Therapy (2019)
-
Carbon sequestration in an expanded lake system during the Toarcian oceanic anoxic event
Nature Geoscience (2017)

{kind=link}
{kind=link}