
Abstract
Fragile X syndrome (FXS) is caused mostly by expansion and subsequent methylation of the CGG repeat in the 5′UTR of the FMR1 gene, resulting in silencing of the gene, absence of FMRP and development of the FXS phenotype. The expansion also predisposes the CGG repeat and the flanking regions to further instability that may lead to mosaics between a full mutation and a premutation or, rarely, a normal or deleted allele. Here, we report on a 10-year-old boy with no FXS phenotype, who has a normal CGG tract, although he inherited the maternal expanded allele that causes FXS in his two brothers. Southern blotting demonstrated that the mother carries a premutation allele (∼190 CGG), whereas the propositus shows a normal 5.2 kb fragment after HindIII digestion and a smaller 2.2 kb fragment after double HindIII-EagI digestion, without any apparent mosaicism in peripheral blood leukocytes. PCR and sequence analysis of the FMR1 5′UTR revealed an allele of 43 repeats, with two interspersed AGG triplets in position 10 and 25 and an exceptional CCG triplet in position 17. This latter creates an abnormal EagI site compatible with the smaller 2.2 kb fragment observed with Southern blotting. Haplotype analysis proved that the rearranged allele originated from the maternal expanded allele. To the best of our knowledge, this is the first non-mosaic case of reduction in the CGG tract of the FMR1 gene, resulting in a normal allele.
Similar content being viewed by others

Introduction
The fragile X syndrome (FXS) (OMIM no. 300624) is associated with the presence of a >200 repeat expansion of a CGG trinucleotide sequence in the 5′ noncoding region of the FMR1 gene.1, 2, 3, 4 The subsequent methylation of cytosines in the promoter and in the expanded repeat itself5, 6 causes the loss of FMR1 gene expression.7, 8 This mutational mechanism accounts for the vast majority of FXS cases. Other mutations in the FMR1 gene have been reported: a de novo T → A missense mutation that converts an isoleucine at codon 367 to asparagine, identified in an individual with some of the clinical features of the FXS;9 an inherited 2-base pair substitution that alters the splice acceptor site of exon 210 and several partial or complete deletions, ranging in size from 1 to 13 Mb.10, 11, 12, 13, 14, 15, 16, 17, 18, 19
The polymorphic CGG sequence expansion apparently predisposes the repeat itself, as well as its flanking regions, to further instability that may lead to mosaic conditions. Mosaicism for a full mutation and a premutation is not uncommon in FXS, occurring with a frequency variably estimated at 12–41% of all cases.20, 21, 22 On the other hand, mosaicism for a full mutation with a partially deleted or with a normal allele has been rarely reported in cases of FXS.18 Direct sequencing of the deletion breakpoint and Southern blotting analysis suggest that two independent events occur in the regression from a full mutation to a normal or a deleted allele: the deletion encompassing the CGG tract and the loss of DNA methylation.23 The transcription of an unmethylated and reduced FMR1 allele was not impaired in lymphocytes of a mosaic FXS patient.24
The molecular mechanisms responsible for the CGG repeat expansion and, more rarely, for reductions in the FMR1 gene, are complex and still unclear. One of these is probably slipped strand mispairing (SSM): DNA polymerase replication of GC-rich repetitive sequences is error-prone because of transient detachment of the newly synthesized strand and mispairing during DNA replication, due to the repetitive nature of the CGG stretch.25, 26
Here, we describe a normal boy belonging to an FXS family, in whom a non-mosaic, apparently normal FMR1 allele resulted from reduction of the expanded maternal allele. The patient has two FXS brothers with a methylated full mutation. Their clinically normal mother has a large premutation of ∼190 CGG repeats. To the best of our knowledge, this case represents the first documented instance of a reversion to normal size of an expanded FMR1 allele, not in a mosaic state, possibly arising in the maternal germ line.
Materials and methods
Southern blotting and CGG PCR analysis
Informed consent was obtained for all subjects participating in this study. Blood was drawn from various family members, as shown on the pedigree (Figure 1). Genomic DNA was isolated from 10 ml of peripheral blood leukocytes using standard procedures and 10 μg of DNA was digested with HindIII and/or EagI (New England Biolabs). Digested DNA and a size marker were run on a 0.8% agarose gel with 1 × TAE buffer, blotted on Amersham Hybond N+ nylon membrane and hybridized with radioactive Ox1.9 probe.27 After overnight hybridization and subsequent washing, radioactive filters were exposed to films at −80°C with reinforcing screens before development.

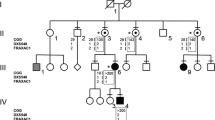
Pedigree of the patient's family with the proband (III-1) indicated by an arrow. Black squares indicate affected males (III-2, III-3) with a methylated full mutation (FM), while II-4 had normal intelligence and carries an unmethylated full mutation (UFM). Haplotype analysis of the family with polymorphic markers DXS548 and FRAXAC1 is indicated below each individual's symbol and shows that the proband has the same haplotype (121–157) of his two affected brothers (III-2 and III-3) and of the maternal uncle (II-4). The number of CGG repeats is also indicated between marker DXS548, which maps 150 kb upstream of the CGG repeat, and marker FRAXAC1, which maps downstream in intron 1.
PCR amplification of the CGG repeats was carried out for all family members with fluorescent primers (see Figure 1). The amplification products were separated on capillary gel electrophoresis using an ABI 3130 sequencer and analyzed with GenMapper v4.0.
Sequence analysis
The DNA of the proband and of a normal control male was amplified, using the following primers for the FMR1 gene promoter, that span the CGG repeat region (GenBank L29074): 1F, 5′-AACTGGGATAACCGGATGCAT-3′ and 2R, 5′-GGCAAAGCCAAAGTGAAGGC-3′; 3F, 5′-CCGTTTCGGTTTCACTTCCG-3′ and 4R, 5′-GGTTACCGCGAAAGATGTTC-3′. PCR amplifications were performed as follows: 30 cycles (1 min at 95°C; 1 min at 55°–60°C, depending on the couple of primers; 1 min at 72°C) with 10% of DMSO, 200 μ M of dNTPs, 2.0 mM of MgCl2, 1 U of Taq polymerase and 1 pmol of each primer in a final volume of 30 μl. A total of 10 μl of each PCR product was visualized with ethidium bromide on a 1.2% agarose gel. To check for the presence of the extra EagI site, part of the amplification product was digested with this enzyme and the digested PCR product was separated on 2% agarose gel.
The 3F-4R amplicon, that in the proband DNA contains the CGG repeats and the second EagI site, was sequenced along with that of a normal control male. Both forward and reverse strands were sequenced using the BigDye Terminator Cycler Sequence kit and protocol (Applied Biosystems). The reactions were denatured for 6 min at 96°C, followed by 27 cycles of 10 s at 96°C, 5 s at 50°C, 4 min at 60°C and extension of 10 s at 60°C. Samples were then purified with 3 M sodium acetate (pH 5.0) and ethanol, denatured and loaded into the ABI 3130 DNA sequencer.
Haplotype analysis
A segregation study was performed on all family members identified in Figure 1. PCR amplification of the polymorphic markers DXS548 and FRAXAC1 was carried out with fluorescent primers.4, 28 Amplifications were performed as follows: 30 cycles (1 min at 95°C; 1 min at 55°; 1 min at 72°C) with 200 μ M of dNTPs, 2.0 mM of MgCl2, 1 U of Taq polymerase and 1 pmol of each primer in a final volume of 20 μl. A total of 10 μl of each PCR product was visualized with ethidium bromide on a 1.2% agarose gel. Alleles were resolved on capillary gel electrophoresis using an ABI 3130 sequencer and analyzed with GenMapper v4.0.
RNA extraction and quantitative RT-PCR
Total RNA was extracted from proband and control male lymphoblastoid cell lines with the single-step acid phenol method, using Trizol (Invitrogen). Two micrograms of total RNA were retro-transcribed to cDNA by MoMLV-RT (Invitrogen) and then used for quantitative fluorescent RT-PCR.29
Results
Figure 1 shows the pedigree of the family with the corresponding haplotype and CGG analysis. The proband (III-1) was a 10-year-old boy referred for genetic testing, because FXS had been previously diagnosed in his two younger brothers (III-2 and III-3). His physical and psychomotor development was normal.
Southern blotting analysis of genomic DNA at the FMR1 locus, together with CGG repeat segregation study, was extended to relevant members of the family. The maternal grandmother (I-1) was found to be carrier of a 31 CGG normal allele and a premutation allele of approximately 70 CGGs. Her premutation was associated with premature ovarian failure (POF) with menopause at 40 years, and was transmitted in four different manners to the offspring. Subject II-4 was apparently not mentally retarded and carried an unmethylated full mutation (UFM) with an amplification of 0.7–1.0 kb (corresponding to approximately 265–365 CGGs). The molecular and epigenetic characterization of this UFM will be reported elsewhere. His twin brother (II-3) inherited the normal maternal allele of 31 CGGs. The proband's maternal aunt (II-2) had a 31 CGG paternal allele and a 0.4 kb premutation allele (∼165 CGGs), and the proband's unaffected mother (II-1) carried a 31 CGG paternal allele and a 0.5 kb band, corresponding to a large premutation of approximately 190 CGGs (Figure 2, lane 3). This premutation allele was further expanded in her FXS sons (III-2 and III-3), who carried a 0.7–2.4 kb (265–830 CGGs) and 1.2–2.3 kb (430–795 CGGs) methylated full mutations, respectively. Upon parental request, the proband was also tested to rule out the possibility of being a premutation carrier. Surprisingly, on the Southern blotting after HindIII/EagI double digestion, we observed a smaller band (2.2 kb) compared with the expected 2.5 kb band (Figure 2, lane 4). We first suspected a deletion that was ruled out by Southern blotting analysis with HindIII digestion alone, showing a normal band of 5.2 kb (Figure 2, lane 2). The absence of extra bands, confirmed on repeated blots with longer exposures, ruled out the possibility of mosaicism with larger alleles in the range of premutation and/or full mutations, either methylated or unmethylated.

Southern blot analysis of the proband's and his mother's DNAs digested with HindIII and EagI (+) or HindIII alone (−), hybridized with radioactive Ox1.9 probe. Lane 1 corresponds to a normal control female with two normal bands of 2.5 and 5.2 kb bands corresponding to the active and inactive X chromosomes, respectively. Lane 3 corresponds to the proband's mother (II-1), carrier of a large premutated allele of 0.5 kb (∼190 CGGs). Lanes 2 and 4 correspond to proband III-1, cut with HindIII alone (5.2 kb) or HindIII and EagI, respectively. Only double digestion (lane 4) reveals the presence of a 2.2 kb fragment, 300 bp shorter than expected.
PCR amplification of the CGG repeat showed the presence of 43 triplets, which did not correspond to the normal maternal allele. These findings suggested that the expanded maternal allele had undergone a partial deletion of the CGG repeat, with the incidental introduction of an extra EagI restriction site (CGGCCG) downstream the canonical site, which maps in the CpG island of the FMR1 promoter. Our hypothesis was confirmed by sequence analysis of the CGG repeat that showed a sequence of 43 triplets with an exceptional CCG repeat at position 17, corresponding to the postulated EagI extra site (Figure 3a). Furthermore, two AGG repeats were observed at positions 10 and 25, respectively. The presence of the extra EagI site in the proband's DNA accounted for the smaller (2.2 kb) HindIII/EagI fragment observed on the Southern blotting hybridized with the Ox1.9 probe. This result was confirmed by digesting with EagI the PCR product spanning the CGG repeat of the proband and of a normal male (Figure 3b). Sequence analysis of the FMR1 promoter region demonstrated the presence of the canonical EagI restriction site. This finding was also confirmed by digesting the PCR product of the FMR1 promoter with EagI, showing the expected normal band both in the proband and in a normal control male (data not shown).

Analysis of CGG repeats sequence with primers 3F and 4R. (a) Sequence analysis of the index case shows a 43 triplet repeat allele with two interspersed AGG triplets on position 10 and 25 (data not shown) and a CCG triplet on position 17. This unusual variant introduces a second EagI restriction site (CGGCCG). (b) The presence of an extra EagI site is confirmed by cutting the PCR product with EagI (+). The uncut PCR products of a control male (∼330 bp) and of the index case (∼370 bp) can be seen in lanes 2 and 4, respectively, but while the fragment of the control male is not cut by EagI (lane 1), two fragments of 210 and 160 bp are visibile when the proband's DNA is digested with EagI (lane 3). In lane 3, a residual uncut band of 370 bp reflects a partial digestion.
Haplotype analysis of polymorphic markers DXS548 and FRAXAC1, flanking the CGG repeat, demonstrated that the rearranged allele of the proband was derived from the maternal expanded allele, the same that had undergone expansion in his two FXS brothers (Figure 1). Thus, our case represents a bona fide example of reverse mutation from an expanded FMR1 allele (in the mother) to a normal-size allele (in the offspring). Results of haplotype analysis exclude the occurrence of unequal crossing-over and suggest the occurrence of a contraction event in cis in the maternal germline. Finally, measurement of FMR1 mRNA by real-time RT-PCR demonstrated a normal level in the proband, relative to a normal control male, in accordance with his normal phenotype (data not shown).
Discussion
We report on a normal boy belonging to an FXS family, probably representing the first described case of reversion to normal of an expanded CGG repeat in the FMR1 gene, with no evidence of mosaicism. This reduction resulted in a 43-repeat allele, containing an exceptional CCG triplet at position 17, which introduced an extra EagI restriction site. This unique situation would have gone unnoticed with PCR analysis or with HindIII digestion only, and was clarified with double HindIII/EagI digestion, followed by Southern blotting analysis. An unequal crossing-over event was excluded through the analysis of polymorphic markers, DXS548 (about 150 kb upstream of the CGG repeat) and FRAXAC1 (in intron 1). The proband and his two affected brothers, all inherited the same haplotype (121–157) present in the maternal expanded allele. We think that the most likely explanation of this apparent inconsistency is a sporadic mutation in the maternal germline. However, we cannot rule out the possibility that the reduced allele was already present in a low-level mosaic state in the mother or that the reduction took place during early embryogenesis.
In this family, there is another peculiarity, namely the presence of an apparently normal male (II-4) harboring an UFM, which will be the object of a separate report. This latter finding might be due to mosaicism in the germline of I-1, who may be a carrier of larger premutations, transmitted to her daughters (II-1 and II-2), as well as UFM such as that inherited by II-4. The contemporary presence of a reduced allele in the proband III-1 and of an UFM in his uncle II-4 is exceptional, although it is difficult to envisage a common cause underlying the two events.
FXS is most often associated with the presence of an expanded CGG repeat sequence within the first exon of the FMR1 gene, which usually results in local DNA hypermethylation and transcrptional silencing of the gene. While this mutational mechanism is largely prevalent, others have been described, including point mutations and deletions of variable size. Deletions, like expansions, could be the consequence of the instability of the FMR1 CGG repeats, once they exceed a certain size, around 55–60 CGG triplet repeats. Some patients have been reported with a mosaicism for a full mutation in association with an allele of normal size or an allele of reduced size, indicating that the instability is not restricted to the repeat itself but may extend to the flanking regions. In our case, there is no apparent mosaicism in lymphocytes and the reduction event may have originated in the maternal germline. The phenotypically normal mother appears to carry a large premutation in peripheral blood leukocytes, with no evidence of a reduced allele. On the other hand, from the analysis of intact ovaries of full mutation fetuses, Malter et al30 demonstrated that only UFM alleles can be detected in oocytes. Similarly, the testis of a 13-week full mutation fetus showed no evidence of premutations, whereas a 17-week full mutation fetus exhibited some germ cells with premutations.30 This study suggested that expanded alleles may contract in the immature testes. Thus, as already mentioned above, we cannot exclude the possibility that the proband inherited an expanded allele from his mother, which underwent a reversion to normal size during the early stages of embryogenesis.
The presence of deletions encompassing the CGG repeat, as well as sequences flanking the CGG tract, has been noted in a small number of FXS males.12, 15, 16, 31 Interestingly, in many of these events, the 5′ breakpoint mapped to a ‘deletion hotspot’ region.31 It was demonstrated that CpG methylation and DNA replication may actually mediate the formation of these deletion events.32 Also, stabilizing AGG interruptions can be lost when replication-mediated CGG deletions occur. On characterizing a small deletion, it was noted that the location of the 5′ breakpoints in their patients corresponded to a very CpG-rich area.31 Therefore, we surmised that a CpG-rich region may be predisposed to rearrangements, resulting in deletions which encompass the promoter sequence preceding the CGG repeat tract. This also suggests that such deletions may occur as a result of intramolecular recombination involving direct repeats or double-strand break repair between the promoter region and the CGG repeat tract. On the other hand, SSM results in an increase or decrease of CGG repeat number depending on misalignment of either the nascent or template strand. The mutation rate is inversely correlated to the size of the repeat unit33 and directly related with the total repeat length.34 An ‘expansion bias’ would be expected, as amplifications increase the frequency of further slippage. However, reductions are also to be expected and, in fact, one such instance was observed in a normal FMR1 allele of 54 CGG repeats.35 An alternative mechanism could be represented by intramolecular recombination among prematurely terminated sequences and Okazaki fragments, resulting from the difficulty of the DNA polymerase to replicate repetitive GC-rich sequences.36 Unequal crossing-over between sister chromatids should also be considered, but this hypothesis has been ruled out in our case.
A relevant question posed by our findings relates to the origin of the CGG deletion, as the mother does not have a deleted allele in her somatic cells. This suggests that the CGG contraction was de novo, likely resulting from an intramolecular event involving the expanded repetitive tract either in the maternal germline or during the early embryogenesis of III-1. This deletion did not include the flanking regions of the CGG repeat and left intact the coding region of the gene, thus allowing normal transcription and production of the FMRP protein. This explains the normal phenotype of the proband. If we had performed only the HindIII digestion, yielding an apparently normal 5.2 kb band on Southern blotting, we would have considered the proband a normal male who inherited the normal FMR1 allele of his mother. However, the methylation status was tested by HindIII/EagI double digestion and revealed a 2.2-kb band, about 300 bp shorter than the expected 2.5 kb band. By PCR, the number of triplet repeats was found to be 43, that is, different from 31, present in the normal maternal allele. By chance, the reduction event introduced an extra EagI restriction site within the CGG tract, as demonstrated by sequence analysis and EagI digestion of the PCR products of the FMR1 promoter and 5′UTR region. Deletion events across the FMR1 locus appear to be infrequent, but they will be occasionally found during routine molecular investigation of patients with developmental delay of unknown causes. The only reliable test that would detect such cases is a Southern blotting assay with double HindIII/EagI digestion. When counseling carriers of CGG tract expansions who have apparently normal offspring, PCR fragment length and haplotype analysis, as well as double HindIII/EagI digestion, may be considered to avoid missing a contraction event, such as the one reported here.
References
Kremer EJ, Pritchard M, Lynch M et al: Mapping of DNA instability at the Fragile X to a trinucleotide repeat sequence p(CGG)n. Science 1991; 252: 1711–1714.
Oberlé I, Rousseau F, Heitz D et al: Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991; 252: 1097–1102.
Yu S, Pritchard M, Kremer E et al: Fragile X genotype characterized by an unstable region of DNA. Science 1991; 252: 1179–1181.
Verkerk AJMH, Pieretti M, Sutcliffe JS et al: Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991; 65: 905–914.
Vincent A, Heitz D, Petit C, Kretz C, Oberlé I, Mandel JL : Abnormal pattern detected in fragile X patients by pulsed field gel electrophoresis. Nature 1991; 329: 624–626.
Hansen RS, Gartler SM, Scott CR, Chen SH, Laird CD : Methylation analysis of CGG sites in the CpG island of the FMR1 gene. Hum Mol Genet 1992; 1: 571–578.
Pieretti M, Zhang F, Fu YH et al: Absence of expression of the FMR1 gene in fragile X syndrome. Cell 1991; 66: 817–822.
Sutcliffe JS, Nelson DL, Zhang F et al: DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1992; 1: 397–400.
de Boulle K, Verkerk AJMH, Reyniers E et al: A point mutation in the FMR1 gene associated with fragile X mental retardation. Nat Genet 1993; 3: 31–35.
Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL : Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet 1995; 10: 483–485.
Gronskov K, Hjalgrim H, Bjerager MO, Brondum-Nielsen K : Deletion of all CGG repeats plus flanking sequences inFMR1 does not abolish gene expression. Am J Hum Genet 1997; 61: 961–967.
Hammond LS, Macias MM, Tarleton JC, Shashidahar Pai G : Fragile X syndrome and deletion in FMR1: new case and review of the literature. Am J Med Genet 1997; 72: 430–434.
Wolff DJ, Gustashaw KM, Zurcher V et al: Deletion in Xq26.3–q27.3 including FMR1 result in a severe phenotype in a male and variable phenotypes in females depending upon the X inactivation pattern. Hum Genet 1997; 100: 256–261.
Orrico A, Galli L, Dotti MT, Plewnia K, Censini S, Federico A : Mosaicisism for full mutation and normal-sized allele of the FMR1 gene: a new case. Am J Med Genet 1998; 78: 341–344.
Grasso M, Faravelli F, Nigro CL et al: Mosaicis for the full mutation and a microdeletion involving the CGG repeat and flanking sequences in the FMR1 gene in eight fragile X patients. Am J Med Genet 1999; 85: 311–316.
Petek E, Kroisel PM, Schuster M, Zierler H, Wagner K : Mosaicism in a fragile X male including a de novo deletion in the FMR1 gene. Am J Med Genet 1999; 84: 229–232.
Garcia Arocena D, de Diego Y, Oostra BA, Willemsen R, Mirta Rodriguez M : A fragile X case with an amplification/deletion mosaic pattern. Hum Genet 2000; 106: 366–369.
Fan H, Booker JK, McCandless SE, Shashi V, Fleming A : Mosaicism for an FMR1 gene deletion in a fragile X female. Am J Med Genet 2005; 136: 214–217.
Han XD, Powell BR, Phalin JL, Chehab FF : Mosaicism for a full mutation, permutation and deletion of the CGG repeats results in 22% FMRP and elevated FMR1 mRNA levels in a high-functioning fragile X male. Am J Med Genet 2006; 140: 1463–1471.
Nolin SL, Glicksman A, Houck Jr GE, Brown WT, Dobkin CS : Mosaicism in fragile X affected males. Am J Med Genet 1994; 51: 509–512.
Rousseau F, Heitz D, Tarleton J et al: A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2253 cases. Am J Hum Genet 1994; 55: 225–237.
Merenstein SA, Sobesky WE, Taylor AK, Riddle JE, Tran HX, Hagerman RJ : Molecular-clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet 1996; 64: 388–394.
Weiss A, Keshet I, Razin A, Cedar H : DNA demethylation in vitro: involvement of RNA. Cell 1996; 86: 709–718.
de Graaff E, De Vries BBA, Willemsen R et al: The fragile X phenotype in a mosaic male with a deletion showing expression of the FMR1 protein in 28% of the cells. Am J Med Genet 1996; 64: 302–308.
Levinson G, Gutman GA : Slipped strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 1987; 4: 203–221.
Chiurazzi P, Kozak L, Neri G : Unstable triplets and their mutational mechanism: size reduction of the CGG repeat vs germline mosaicism in the fragile X syndrome. Am J Med Genet 1994; 51: 517–521.
Nakahori Y, Knight SJL, Holland J et al: Molecular heterogeneity of the fragile X syndrome. Nucleic Acids Res 1991; 19: 4355–4359.
Richards RI, Holman K, Kozman H et al: Fragile X syndrome: genetic localization by linkage mapping of two microsatellite repeats FRAXAC1 and FRAXAC2 which immediately flank the fragile site. J Med Genet 1991; 28: 818–823.
Pietrobono R, Pomponi MG, Tabolacci E, Oostra BA, Chiurazzi P, Neri G : Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res 2002; 30: 3278–3285.
Malter HE, Iber JC, Willemsen R et al: Characterization of the full fragile X syndrome mutation in fetal gametes. Nat Genet 1997; 15: 165–169.
de Graaff E, Rouillard P, Willems PJ, Smits AP, Rousseau F, Oostra BA : Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum Mol Genet 1995; 4: 45–49.
Edamura KN, Pearson CE : DNA methylation and replication: implications for the ‘deletion hotspot’ region of FMR1. Hum Genet 2005; 118: 301–304.
Schloetterer C, Tautz D : Slippage synthesis of imple sequence DNA. Nucleic Acids Res 1992; 20: 211–215.
Weber JL, Wong C : Mutation of human short tandem repeats. Hum Mol Genet 1993; 2: 1123–1128.
Fu YH, Kuhl DPA, Pizzuti A et al: Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67: 1047–1058.
Kuhl DPA, Caskey CT : Trinucleotide repeats and genome variation. Curr Opin Genet Dev 1993; 3: 404–407.
Acknowledgements
This work was supported by TELETHON grant (GGP06224), PRIN 2005 grant (no. 2005060575) and Conquer Fragile X Foundation grant to GN.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tabolacci, E., Pomponi, M., Pietrobono, R. et al. A unique case of reversion to normal size of a maternal premutation FMR1 allele in a normal boy. Eur J Hum Genet 16, 209–214 (2008). https://doi.org/10.1038/sj.ejhg.5201949
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201949
Keywords
This article is cited by
-
Contraction of fully expanded FMR1 alleles to the normal range: predisposing haplotype or rare events?
Journal of Human Genetics (2017)
-
Recent advances in assays for the fragile X-related disorders
Human Genetics (2017)
-
EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders
European Journal of Human Genetics (2015)
-
Clinical utility gene card for: fragile X mental retardation syndrome, fragile X-associated tremor/ataxia syndrome and fragile X-associated primary ovarian insufficiency
European Journal of Human Genetics (2011)
-
Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations
European Journal of Human Genetics (2008)
