
Abstract
Induction of apoptosis in tumour cells, either by direct activation of the death receptor pathway using agonistic antibodies or recombinant ligands, or direct triggering of the Bcl-2-regulated intrinsic apoptosis pathway by small molecule drugs, carries high hopes to overcome the shortcomings of current anticancer therapies. The latter therapy concept builds on a more detailed understanding of how Bcl-2-like molecules maintain mitochondrial integrity and how BH3-only proteins and Bax/Bak-like molecules can undermine it. Means to unleash the apoptotic potential of BH3-only proteins in tumour cells, or bypass the need for BH3-only proteins by blocking possible interactions of Bcl-2-like prosurvival molecules with Bax and/or Bak allowing their direct activation, constitute interesting options for the design of novel anticancer therapies.
Similar content being viewed by others
Introduction
In this review, we aim to summarize our current knowledge of the physiological function of BH3-only proteins. We will discuss current issues of how cell death may be initiated by BH3-only proteins and briefly summarize the evidence indicating that apoptosis acts as a barrier against malignant disease. In the main part of this review, we try to give a comprehensive overview on the role of each individual BH3-only protein in cell death regulation, as delineated mainly from knockout mouse studies. We supplement this information with a summary of the available evidence implicating each individual gene in the pathology of neoplastic disease and the effectiveness of anticancer therapy.
Cell Death Regulation by BH3-only Proteins
A number of developmental or experimental stimuli, including growth factor deprivation, ultraviolet (UV) and γ-irradiation, anoxia or anticancer drugs can lead to cell death via the ‘intrinsic’ Bcl-2-regulated apoptosis pathway.1, 2 Disruption of the outer mitochondrial membrane plays a central role in this death pathway, because it results in the release of apoptogenic factors such as cytochrome c or smac/Diablo from the inner membrane space into the cytosol and subsequent caspase activation. Binding of cytochrome c to the adapter molecule Apaf-1 triggers the formation of the ‘apoptosome’, a multimeric complex containing Apaf-1, cytochrome c, (d)ATP and pro-caspase-9 molecules. Autocatalytic activation of caspase-9 through dimerization then leads to activation of the so-called ‘effector’ caspases (caspases-3, -6 and -7) and death of the cell.1, 2
Members of the Bcl-2 family either promote or prevent apoptosis. The prosurvival Bcl-2 family members include Bcl-2 itself, Bcl-XL, Bcl-w, A1 and Mcl-1. They share four Bcl-2 homology domains (BH1–BH4) among each other, with the exception of Mcl-1 that contains only three BH domains.3 These proteins are crucial for cell survival, as the loss of any of these proteins causes abnormal death of certain cell populations such as lymphocytes or melanocytes in bcl-2−/−mice4 or erythroid progenitors and neurons in bcl-x−/− mice.5 Deficiency in the mcl-1 gene causes death of early-stage embryos, and conditional deletion of this gene in the T- or B-cell lineage leads to rapid loss of mature lymphocytes. MxCre-mediated deletion of mcl-1 also causes rapid loss of committed myelocytic progenitors and haemopoietic stem cells.6, 7 These mouse model systems impressively demonstrate that all these genes are essential for keeping cells alive. The proapoptotic Bcl-2 family members can be subdivided into two groups: Bax-like molecules (Bax, Bak, Bok/Mtd, Bcl-G and Bfk) contain three or at least two BH regions (BH1, BH2 and BH3) and display a high degree of similarity to prosurvival Bcl-XL in their three-dimensional structure, whereas the so-called ‘BH3-only’ proteins (Bad, Bik/Blk/Nbk, Hrk/DP5, Bid, Bim/Bod, Noxa, Bmf and PUMA/Bbc3) only share the BH3 domain with each other and the rest of the Bcl-2 protein family.3
BH3-only proteins are essential for cell death initiation, as based on genetic and biochemical studies, whereas the Bax/Bak-like proteins are critical further downstream in apoptosis signalling at the level of mitochondrial outer membrane permeabilization (MOMP). A large body of studies suggests that different apoptotic stimuli can activate distinct but sometimes overlapping sets of BH3-only proteins.3 Apoptosis-independent functions have been proposed for certain BH3-only proteins such as the control of S-phase progression after DNA damage by Bid.8, 9 The physiological relevance of these observations still needs confirmation, ideally in an animal model system. The proapoptotic activity of BH3-only proteins is tightly controlled by diverse transcriptional and post-translational mechanisms. For example, PUMA and Noxa are transcriptionally induced in response to DNA damage by p53 whereas other BH3-only proteins, such as Bid or Bad, are thought to be mainly regulated at the post-translational level.2
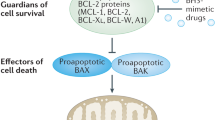
The molecular mode of BH3-only protein action is still under intense investigation. BH3-only proteins bind to a hydrophobic groove on the surface of Bcl-2-like molecules, formed by their BH1, BH2 and BH3 domains and thereby antagonize their prosurvival function.10 This physical interaction strictly depends on a functional BH3-domain within the BH3-only protein. In one model of BH3-only protein function, this interaction is thought to block the ability of Bcl-2-like prosurvival proteins to keep proapoptotic Bax/Bak in check, preventing them from undergoing oligomerization and causing loss of mitochondrial outer membrane integrity and cell death (Figure 1). David Huang and colleagues found that different BH3-only proteins have distinct, only partially overlapping, binding preferences for their prosurvival Bcl-2-like relatives.11 Their study suggests that certain BH3-only proteins can only antagonize a very distinct subset of Bcl-2-like molecules in vivo (e.g. Noxa can only block Mcl-1 and A1), whereas others, such as Bim and PUMA, are able to bind with similar affinities to all known Bcl-2 prosurvival proteins and, therefore, may have broader physiological functions than other BH3-only proteins. In support of their idea, they also demonstrate in a follow-up study that Mcl-1 physically interacts with Bak in the outer mitochondrial membrane until displaced by a BH3-only protein. In response to UV-irradiation, Noxa is strongly induced and interacts with Mcl-1 targeting it for proteasomal degradation. This event frees a substantial amount of Bak able to oligomerize, but overexpression of Noxa is not sufficient to kill the cell. Cell death initiation by Noxa in murine embryonic fibroblasts (MEFs) in response to UV-irradiation requires the activity of additional BH3-only proteins to antagonize other prosurvival Bcl-2 homologues present in the cell. Consistent with this idea, overxpression of Noxa readily induced apoptosis in MEF lacking Bcl-XL that can neutralize Bak in the absence of Mcl-1.12

A possible mode of BH3-only protein action based on their selective affinities for individual Bcl-2 prosurvival homologues (see text for details)
An alternative model, proposed by Don Newmeyer's group, suggests that Bcl-2-like prosurvival molecules act as a sink for BH3-only proteins that are proposed to exist in two flavours: ‘direct activators’ (Bid and Bim) and ‘de-repressors’ (all others). In this model, members of both classes compete for binding to Bcl-2 or prosurvival homologues (Figure 2). Excess of ‘de-repressors’ would prevent sequestration of ‘direct activators’ by prosurvival homologues of Bcl-2 freeing them to bind and activate Bax/Bak-like molecules directly.13 This model would predict that ‘de-repressors’ must have higher binding affinities to prosurvival Bcl-2 family members or need to be present in vast excess over Bim or Bid. The latter may be achieved, for example, by transcriptional upregulation of PUMA or Noxa in response to DNA damage. Alternatively, at comparable ratios, post-translational modifications causing conformational change leading to altered affinities on either side may influence the life–death balance. However, as direct binding of Bim or PUMA to Bax or Bak with physiologically relevant affinity has so far not been reported, a ‘hit and run’ mode of interaction needs to be postulated and further investigations will be required to clarify this issue.

Alternative model describing BH3-only protein function based on the direct interaction of tBid or Bim with Bax (see text for details)
Genetic experiments, however, clearly demonstrate that BH3-only proteins act upstream of Bax/Bak, as enforced overexpression of BH3-only proteins triggers cytochrome c release and apoptosis only in fibroblasts proficient for Bax or Bak but not in those lacking both molecules.14 More recent studies suggest that this redundancy may only be restricted to cell death induced by certain BH3-only proteins, such as Bid or Bim, whereas others may preferentially induce cell death using one or the other Bax-like protein, maybe even in a cell type and/or stimulus-dependent manner. For example, Nbk/Bik and PUMA appear to kill in a strictly Bax-dependent manner.15, 16 In light of the previously introduced models, Nbk/Bik may fail to activate Bak, because it has a very low binding affinity for Mcl-1 (Figure 1). However, PUMA can bind to Mcl-1 and should therefore be able to activate Bak and also be able to kill by engaging Bak. Maybe PUMA kills only via displacement of p53 from Bcl-XL, as recently proposed but whether p53 may really be able to directly activate Bax remains highly controversial.17, 18
Taken together, these results indicate that the decision of whether a cell will live or die along the intrinsic apoptosis pathway after exposure to a stress-inducing signal is determined to a large extent by the set of BH3-only proteins activated in response to a given stimulus and whether the cell type-specific panel of Bcl-2 prosurvival homologues can counterbalance this attack.
Apoptosis as a Barrier Against Neoplastic Disease
The link between apoptosis and cancer emerged almost 20 years ago, when Bcl-2, overexpressed in ∼85% of human follicular lymphoma owing to translocation of its gene into the immunoglobulin heavy chain gene locus, was found to inhibit cell death rather than to promote proliferation like classical oncogenes known at that time.
Bcl-2-like proteins have been implicated in the development of other haemopoietic as well as non-haemopoietic malignancies, such as acute promyelocytic leukaemia, breast and pancreatic β-cell cancers, where Bcl-2 or Bcl-XL overexpression has been shown to facilitate tumorigenesis in vivo.19 Overexpression of Bcl-2 in lymphocytes renders mice susceptible to lymphomagenesis, particularly when combined with other oncogenes such as c-myc or pim-1 that drastically accelerate tumorigenesis in double transgenic mice.19 Pan-haemopoietic overexpression of Bcl-2 under control of the vav-gene promoter causes the classic follicular lymphoma phenotype associated with the t(14,18) translocation.20 Bcl-XL, the closest relative of Bcl-2, has been implicated in mouse myeloid and T-cell leukaemias21 and A1, another prosurvival relative of Bcl-2, has been reported to facilitate BCR/abl-mediated leukaemogenesis.22 Mice expressing a mcl-1 transgene under control of its own promoter develop B-cell neoplasias with high frequency, ranging from follicular lymphoma to diffuse large B-cell lymphoma, albeit rather late in life.23
Similar to overexpression of Bcl-2-like proteins, the absence of proapoptotic Bcl-2 relatives can also enhance tumour progression. Somatic frameshift mutations in the bax gene have been described in some cases of colon cancer of the mutator phenotype.24 Moreover, transformation of choroid plexus epithelial cells by transgenic expression of a truncated version of SV40 large T antigen (inactivating only the retinoblastoma protein Rb, but not p53) shows accelerated kinetics on a bax−/− background.25 In addition, haploid loss of bax leads to accelerated mammary tumour development in C3(1)/SV40 large T antigen transgenic mice.26
In summary, overexpression of Bcl-2-like proteins or loss of Bax or Bak function can contribute to tumorigenesis by enhancing survival of otherwise doomed cells, enabling accumulation of additional mutations driving tumour progression.
The observation that Bax and Bak display features of tumour suppressors raises the possibility that proapoptotic molecules may be tumour suppressors by default. Consistent with this idea, loss of Apaf-1 expression, the adapter molecule required for caspase-9 activation downstream of MOMP, was reported to occur frequently in patients with progressive melanoma, and a significant number of melanoma cell lines were reported to display reduced Apaf-1 expression owing to epigenetic silencing that correlated with drug resistance.27 These findings, however, were challenged as other researchers failed to detect the loss of Apaf-1 expression in primary tumour samples and cell lines at similar frequencies, maybe owing to differences in the suitability of certain Apaf-1 antibodies for immunohistochemistry.28, 29 Furthermore, neither loss of apaf-1 nor caspase-9 accelerated c-myc-driven lymphomagenesis in mice,30 suggesting that loss of apoptosome function is unlikely to be a key event in transformation. Apaf-1- or caspase-9-deficient mouse embryonic fibroblasts displayed no enhanced colony-forming and transformation potential in response to oncogenic transformation by E1A/H-ras and H-ras/c-myc in vitro whereas loss of p53 or Bax did.30 Taken together, this indicates that loss of apoptosome function can be compensated for by other cell death mechanisms in vivo and is not a prerequisite for transformation.
Similarly, loss of death receptors (DRs) such as TRAIL-R or CD95/Fas is not tumorigenic per se, although loss of CD95 causes lymphadenopathy.31, 32 The accumulating cells in CD95-deficient lpr mice, however, are polyclonal in nature but Bcl-2 overexpression can synergize in the development of a disease resembling human acute myeloblastic leukaemias in mice.33 Combined inactivation of all DR signalling in T cells by overexpression of a dominant-negative version of the adapter molecule Fas-associated death domain protein (FADD), required for caspase-8 activation downstream of DRs, causes thymic lymphomas in lck-FADD-DN mice, raising the possibility that concerted DR action is required to prevent the rise of lymphatic tumours.34 As loss of FADD is not compatible with embryogenesis in mice, it is unclear whether DR signalling is required to prevent tumorigenesis outside the T-cell compartment. Interestingly, however, loss of caspase-8 expression, required for apoptosis induction by all DRs, was repeatedly described to occur in solid tumours such as childhood neuroblastomas, rhabdomyosarcomas as well as lung and colorectal carcinomas.35, 36, 37 As caspase-8 and FADD do also contribute to other essential processes such as differentiation and proliferation, their tumor suppressor potential may not entirely depend on the mediation of cell death in response to DR ligation.
This indicates that only inactivation of cell death regulators that integrate a wider range of death signals parallel to and/or upstream of mitochondria can cause clonal survival of damaged cells and may therefore promote tumorigenesis. Consistent with this idea, inactivation of the ARF/mdm2/p53 signalling network that regulates cell cycle arrest and apoptosis induction primarily along the Bcl-2-regulated pathway, upstream of mitochondria in response to a wide range of stressors, is the most frequent detectable molecular change in human tumours.38
BH3-only Proteins and Tumour Suppression by p53
A large set of genes transcriptionally regulated by p53, and even p53 itself acting directly at mitochondria, have been discussed to mediate apoptosis in response to DNA damage.39 Several members of the Bcl-2 family are regulated by p53 at the transcriptional level. Bax was the first target in this family described to be induced by p53 in response to DNA damage,40 but studies using knockout mice clearly demonstrated that loss of Bax did not impair p53-mediated apoptotic responses indicating redundancy with other cell death mediators such as Bak 1. Only recently, the connection between p53 and cell death activation by the Bcl-2 family became more clear after novel members of the BH3-only protein family were reported to be regulated in a p53-dependent manner.39
Bid
Bid, which is considered to amplify apoptosis signalling, particularly by linking the DR pathway to the intrinsic pathway and known for quite a long time, was only recently reported to be transcriptionally induced by p53 in response to γ-irradiation.41 MEFs lacking bid were shown to have abnormally reduced sensitivity to certain chemotherapeutic drugs such as 5-fluorouracil (5-FU).41 However, the degree of protection was rather small. Moreover, full Bid activity requires its cleavage, either by caspase-8 downstream of DR activation or in response to genotoxic stress-induced Caspase-2 activation, converting it into the truncated, active form, called tBid. Bid- and caspase-8-deficient lymphocytes as well as caspase-8 fibroblasts were all found to be normally sensitive to p53-dependent apoptosis,42, 43, 44 as were cells from mice lacking caspase-2, with the exception of oocytes.45 Although the overall amount of Bid was observed to increase in MEFs in response to DNA damage, the amount of active tBid appeared to remain unchanged during p53-induced apoptosis.41 These observations indicate that the apoptotic cascade is already highly active at times when Bid is cleaved and activated, demonstrating that it functions as an amplifier of Bax/Bak-induced disturbance of mitochondrial integrity and not as a key initiator in p53-mediated apoptosis. Therefore, somewhat surprisingly, mice lacking Bid spontaneously develop a clonal malignancy that resembles human chronic myelomonocytic leukaemia on a mixed genetic background.46 Myeloid precursors from Bid-deficient animals were reported to be protected from DR-mediated cell death and show enhanced colony-forming potential in vitro, that may constitute the basis for the development of the observed premalignant myeloid hyperplasia in these mice.46 Whether loss of Bid expression can contribute to tumorigenesis and/or drug resistance in humans is only poorly investigated (Table 1). One extensive immunohistochemical survey of Bid protein expression in 254 prostate, 100 colorectal adenocarcinomas, 95 ovarian cancer specimens, 59 brain tumours as well as 50 B-cell non-Hodgkin's lymphomas revealed that, if anything, Bid expression increases in some of these tumours during disease progression.47 Analysis of the cancer cell line panel of the NCI revealed that expression of Bid did not correlate with the responsiveness of these cells to a large number of commonly used anticancer drugs. The only exception, out of 20 drugs tested, were two ribonucleotide reductase-inhibiting agents (Figure 3). Unfortunately, a correlation with the responsiveness of these cells to DR-induced apoptosis was not assessed. Immunohistochemical analysis of Bid expression in hepatocellular carcinoma (HCC) specimens indicates weaker, albeit clearly detectable expression levels in tumourous tissue when compared with matched non-tumourous tissue.48 No correlation with response to radiotherapy could be found in a study analysing Bid protein expression in a set of 98 biopsies of patients with cervix carcinoma by immunohistochemistry. Somewhat surprisingly, in the same study, high Bid expression was even identified as an adverse prognostic factor for disease-specific and metastasis-free survival in patients below 52 years of age.49

Collection of classical and novel anticancer agents and their (currently) known ability to engage BH3-only proteins. Solid arrows indicate a strong activation/interdependence and dashed arrows weak or proposed ones
Inactivating mutations in the bid gene locus have been reported to occur with low frequency in advanced gastric cancers. Analysis of 67 samples revealed four mutations but only one of these was a frame-shift mutation that also caused premature termination of protein translation. Overexpression of this mutant in 293 T cells confirmed impaired proapoptotic potential of Bid in response to 5-FU and TRAIL-R crosslinking.50
In conclusion, these investigations indicate that loss of Bid expression and function is only a very rare event in human cancer that will generally not impede on the effectiveness of conventional anticancer therapy (Figure 3). However, on rare occasions, inactivation of Bid may interfere with TRAIL-R activation-based combination therapy.
PUMA and Noxa – Two Killers on the Run
The first BH3-only protein reported to be a p53 target was called ‘Noxa’, the Greek word for ‘damage’, and cloned using a differential display technique.51 This short 103 amino acid (aa) long protein showed no sequence homology to other known proteins except for two BH3 domains in mouse Noxa and one in the human counterpart. The mouse Noxa protein is the only Bcl-2 family member known to contain two BH3 domains that may have arisen during incomplete gene duplication. Mutational analysis confirmed that both BH3 domains are functional and contribute to the proapoptotic potential of Noxa. The noxa promoter region contains a functional p53-binding site, as confirmed by promoter luciferase assays.51 Subsequent sequence analysis revealed that human noxa had already been identified some time ago as a protein highly expressed in adult T-cell leukaemia (ATL) cells after PMA treatment. The protein was called APR (for ATL-derived PMA-responsive gene) but no function was assigned to that molecule.52 Human noxa encodes a gene product of 54 amino acids that contains only one BH3 domain. Similar to the murine gene, the human promoter contains a p53 response element and is induced readily following p53 activation,51 but was found to be constitutively expressed in certain tumour cell lines such as Jurkat or HeLa (personal observations).
PUMA (p53 upregulated modulator of apoptosis) was described shortly thereafter by three groups.53, 54, 55 PUMA mRNA and protein were both reported to be induced in normal as well as malignant cells following treatment with actinomycin D, 5-FU, adriamycin or in response to oncogenic stress. Its induction in response to DNA damage depends on p53 as colon carcinoma cells expressing a human papillomavirus (HPV) E6 protein or engineered to lack wt p53 (H1299 and HCT116 cells, respectively) are unable to express PUMA in response to DNA damage.54, 55 Similar to p21waf1, PUMA is induced rapidly and mRNA expression is detectable as shortly as 3 h after and peaks 6 h after doxocycline-regulated induction of p53 and, hence, is induced more rapidly than Noxa or Bax that are induced rather late in this process.54, 55
Consistent with their role as p53 target genes and their ability to be activated also by p53-independent processes (hypoxia, serum deprivation and glucocorticoids), cells derived from PUMA- or Noxa-deficient mice show increased resistance to a range of p53-dependent and, in case of PUMA deficiency, also p53-independent stress stimuli, albeit in a cell and tissue type-dependent manner.56, 57, 58
Thymocytes and myeloid progenitors from PUMA-deficient animals are abnormally resistant to DNA damage caused by the treatment with anticancer drugs or by γ-irradiation. Although upregulation of noxa mRNA was also observed in these cells in response to these stimuli, most cell types from Noxa-deficient mice were normally sensitive to these death stimuli. PUMA deficiency protected thymocytes also against certain p53-independent cell death stimuli, such as cytokine deprivation or treatment with glucocorticoids, the pan-kinase inhibitor staurosporine and most potently, the phorbol ester PMA. Similar results were obtained in an analysis of PUMA-deficient bone marrow-derived pre-B cells, mature peripheral B and T cells as well as bone marrow-derived myeloid progenitors.56, 57, 58
A possible explanation for the apparent lack of protection of lymphocytes in the absence of Noxa may be the fact that it appears to display a very limited binding spectrum to Bcl-2-like prosurvival molecules. Mcl-1 and A1 are the only prosurvival homologues that can be bound and neutralized efficiently by Noxa.11, 12 Lymphocytes, however, depend on a mix of Mcl-1, Bcl-2 and Bcl-XL for survival and therefore one might speculate that PUMA, reported to bind all three molecules with high affinity, can potently antagonize all these molecules, whereas Noxa is probably unable to be equally potent.
Taking all these studies into account, it appears to be certain that PUMA and Noxa are essential for mediating the effects of anticancer therapy based on the induction of DNA damage (Figure 3). Our own in vivo studies indicate that, in the presence of a functional p53 pathway, PUMA is the rate-limiting BH3-only protein mediating DNA damage and glucocorticoid-induced apoptosis of primary lymphocytes, in a cell and tissue type-dependent manner together with Bim that is not a primary p53 target.59 Again, Noxa appears to be dispensable for lymphocyte death in vivo but plays a major role in other tissues such as the gastrointestinal tract as well as UV irradiation-induced apoptosis in MEF.57, 59 Whether PUMA or Noxa can contribute to all these forms of apoptosis in primary p53-deficient tumours, maybe in a p73-dependent manner (as suggested by in vitro studies) remains to be firmly established.60 Induction of noxa mRNA and protein, however, can be induced in a p53-independent manner by application of proteasome inhibitors such as Velcade (PS-341) in melanoma cell lines in vitro but not healthy melanocyte cultures from foreskin.61 C8161 melanoma xenotransplants treated with Velcade also displayed induction of Noxa protein paralleling tumour regression.61 Expression of other BH3-only proteins such as Bim, also regulated in part via the proteasome, appeared unaltered in malignant melanocytes positive for Bim protein expression and treated with proteasome inhibitors, suggesting that depending on the origin of the tumour Velcade may exert its antitumorigenic effect by recruiting/activating different BH3-only proteins61 (Figure 3). It appears that induction of Noxa is required to induce apoptosis in these tumour cells as antisense oligonucleotide treatment reduced both induction of Noxa protein levels and cell death in response to proteasome inhibition.61 This is somewhat surprising as protein levels of Mcl-1, Noxas primary antiapoptotic target, also increased in response to this treatment suggesting that Noxa should be completely neutralized. Usually, binding of Noxa to Mcl-1, for example, in response to UV irradiation targets this Bcl-2 prosurvival homologue for proteasomal degradation and this drop in Mcl-1 expression is required to sensitize cells for the action of additional BH3-only proteins that can neutralize other survival factors such as Bcl-XL12. In case of malignant melanocytes, however, the increase in Noxa protein may be strong enough to outweigh the molarity of Mcl-1 allowing Noxa-mediated neutralization of additional prosurvival proteins and the induction of apoptosis. The only other Bcl-2 homologue known to bind to Noxa, albeit with lower affinity, is A1 but its expression was not analysed in this study.61
Are PUMA or Noxa Required for p53-Mediated Tumour Suppresson?
Mutations in the p53 gene are among the most frequent genetic alterations observed in human tumours,62 and patients diagnosed with the rare familial Li-Fraumeni cancer syndrome harbour germ line mutations in one p53 allele predisposing them to develop a range of cancers owing to loss of the wild-type (wt) p53 allele in the emerging clone of neoplastic cells.63, 64 The Li–Fraumeni syndrome is characterized by a high incidence and early onset of leukaemia and sarcoma, a range of tumours observed also in mouse models genetically engineered to lack one or both alleles of p5365, 66 or having their wt p53 replaced with a mutant form found at high frequency in human cancers.67, 68 These mice develop tumours with 100% penetrance. It is unclear whether this is due to loss of all p53-activated processes or only loss of apoptosis. Loss of the cyclin-dependent kinase inhibitor p21waf1, the major cell cycle inhibitor activated by p53, causes only a low incidence of tumours and these arise mostly late in life (>16 months)69 as does loss of the key initiator of p53-driven scenescence ink4a.70 The concept that only the proapoptotic arm of p53 function needs to be lost for oncogene-driven tumorigenesis to occur71 has been challenged. In mice expression of a cell cycle competent but apoptosis-defective mutant of p53, found in tumour patients, did cause tumorigenesis significantly later, when compared with p53−/− animals, indicating that cell cycle arrest and the maintenance of chromosomal stability do play an important role in p53-mediated tumour suppression.72 Furthermore, although Bcl-2 overexpression can inhibit p53-induced apoptosis in lymphocytes, lymphomas develop in bcl-2 transgenic mice much more slowly and with much lower incidence compared to p53−/− mice.39
Although not tumorigenic per se, there is evidence that loss of PUMA can promote tumorigenesis under certain experimental conditions. Deregulated expression of the c-myc proto-oncogene results in the increased expression of PUMA in MEFs following E1A overexpression. In line with these observations, puma−/− myeloid progenitor cells and MEFs are highly resistant to apoptosis induced by c-myc expression plus growth factor deprivation, similar to the level of protection afforded by loss of p53.58
Studies in bone marrow and splenic B cells derived from transgenic mice expressing c-myc under control of the Ig heavy chain enhancer (Eμ-myc) demonstrated that c-myc plus cytokine deprivation-induced apoptosis is mediated, at least in part, via the ARF/p53/PUMA pathway.73 Eμ-myc transgenic mice develop B-cell lymphomas with 100% incidence within the first year of life.74 This lymphomagenesis is accelerated by loss of p53 (even loss of one allele) or loss of ARF.75 Consistently, loss of a single copy of puma drastically accelerates c-myc-induced B-cell lymphomagenesis (Andreas Strasser, personal communication) and when haemopoietic stem cells (HSCs) from Eμ-myc transgenic mice, infected with a puma-specific shRNA construct were transferred into normal recipients, lymphomas also arose with a drastically reduced latency and disease onset was comparable to the one observed in the absence of p53.76 Interestingly, the Eμ-myc lymphomas that carry a PUMA-specific shRNA maintained wt p53 status and could undergo cell cycle arrest in response to γ-irradiation.76 In contrast, p53 is frequently inactivated or lost in Eμ-myc lymphomas,75 it therefore appears that the pressure to lose p53 is obviated by loss of PUMA. This indicates that when cell cycle control is deregulated (e.g. by c-myc overexpression) p53's proapoptotic function is its most critical action in tumour suppression. However, in the absence of mutations that deregulate cell cycling, other functions of p53 appear also critical for tumour suppression.
Whether this concept also holds true for other tumour types and other oncogenic mutations remains to be investigated in more detail. MEFs infected with E1A and ras-encoding retroviruses injected into immuno-compromised NIH Swiss athymic nude mice formed tumours much more rapidly when PUMA expression was downregulated by specific short hairpin siRNAs in these cells.76 Tumour formation was reported to occur as rapidly as in the absence of p53 but without alterations in cell cycle regulation. PUMA downregulation did not accelerate tumorigenesis in MEFs overexpressing mutant Ras alone. These cells underwent senescence and did not form tumours, which is in sharp contrast to cells overexpressing mutant Ras in a p53−/− background which are highly tumorigenic.76 This indicates that in the context of ras-induced tumorigenesis, p53 suppresses transformation mostly by inducing cell senescence.
Interestingly, loss of Bim but not PUMA facilitates malignant growth of transformed mouse baby kidney cells overexpressing E1A and lacking p53 function,77 although PUMA was previously reported to be upregulated in these tumours, at least under hypoxic conditions.78 Bim seems to be a critical suppressor of H-ras driven tumorigenesis of epithelial cells that needs to be inactivated by H-ras/Raf/MAPK-mediated phosphorylation leading to its proteosomal degradation.77 However, a contribution of PUMA in suppression of E1A/H-ras driven tumorigenesis in the context of functional p53 cannot be excluded based on these experiments.
Until now, no convincing data has been provided that loss of Noxa may facilitate tumour formation. In mice bearing a liver-specific deletion of c-jun, the development of chemically induced HCC is significantly delayed, revealing the oncogenic potential of c-jun in an in vivo model.79 c-Jun, a central component of the transcription factor AP-1, is a major regulator of proliferation and apoptosis in liver cells and loss of c-Jun in HCC leads to an increased number of apoptotic cells that display increased mRNA levels of p53 and noxa. Induction of Noxa in the absence of c-jun seems to be specific, as the expression of other proapoptotic p53 target genes, such as Bax, PUMA or Apaf-1, was unaffected.79 However, it was not formally demonstrated that the observed upregulation of noxa mRNA was the consequence of p53 activation, leaving the possibility that Noxa and p53 may be independently activated by loss of c-jun. Taken together, these observations indicate that c-jun promotes tumour cell survival by inhibiting the proapoptotic action of p53.
Collectively, these findings suggest that inactivation of the proapoptotic function of p53 may not be sufficient for tumour development but that two or more of its activities, such as cell cycle arrest and apoptosis have to be impaired. Interestingly, studies with knock-in mice that express an apoptosis-defective, but cell cycle inhibitory competent mutant of p53 (R172P) indicate an equally prominent role for these two p53 functions in the suppression of thymic lymphoma development.72 Cross-breeding experiments generating puma−/− /p21−/− or noxa−/−/p21−/− double-mutant mice may reveal whether loss of apoptosis induction and cell cycle arrest is sufficient to reconstitute the tumour phenotype observed in p53−/− animals.
Do mutations of PUMA and Noxa promote tumorigenesis in humans?
A number of studies have investigated whether the puma or noxa genes are mutated or lost in a broad range of human cancers, focusing on the idea that tumours with functional p53 may be the most likely candidates for having lost PUMA or Noxa (Table 1).
The chromosomal locus 19q13.3, harbouring the puma gene, is frequently lost in human gliomas, neuroblastomas and certain B-cell lymphomas.80, 81, 82 Deletion of the long chromosomal arm, 19q, is also a frequent event in human head and neck squamous cell carcinomas (HNSCCs) and in lung cancer. Samples from 15 HNSCCs and 15 lung cancer patients were collected and analysed for their puma status.83 No mutation of the puma gene itself has been found in any of the tumours analysed. A similar study also failed to provide a relationship between clinical and pathological features and PUMA expression in sporadic colorectal cancer.83 However, a recent report provides evidence that PUMA expression decreases during the development of malignant cutaneous melanoma. Performing immunohistochemistry on 107 primary melanomas, 51 metastatic tumours and 64 dysplastic nevi, Karst et al.84 demonstrated that PUMA expression decreases during disease progression (mean expression in dysplastic nevi>primary melanomas>metastatic tumours) and correlated with the overall as well as disease-specific 5-year survival rates of the patients. Unfortunately, the p53 mutation status was not assessed in these samples, but previous reports indicated that loss of p53 is a rather rare event in melanoma.85 This may indicate that progressive reduction in PUMA expression levels during melanoma development may be owing to a molecular mechanism that does not involve p53 loss such as hypermethylation of the gene locus.
So far no relationship between Noxa expression and malignant diseases has been observed. Different human cancers, including 78 colon adenocarcinomas, 53 advanced gastric adenocarcinomas, 83 non-small-cell lung cancers, 76 breast carcinomas, 33 urinary bladder transitional cell carcinomas and 90 HCCs, were tested for their noxa status.86 Apart from one transitional cell carcinoma of the urinary bladder, which carried a somatic mutation in noxa, all tumour samples contained wild-type noxa sequences (Table 1). Moreover, when analysed in vitro, the observed mutation had no impact on the proapoptotic activity of Noxa.86
Taken together, these data indicate that loss of p53's proapoptotic activity alone is insufficient to promote tumorigenesis in a broad range of cell types in both humans and mice, fostering the hypothesis that at least one additional or even more p53 functions need to be impaired in order to recapitulate the Li–Fraumeni pathophysiology. It remains possible that impaired p53-mediated apoptosis alone may be sufficient to promote tumours in tissues with high basal cell proliferation, such as certain haematopoietic cell types or cells of the gastrointestinal system. Mutational analysis of puma and noxa in aggressive leukaemia/lymphomas with wt p53 status may help test this hypothesis. In addition to mutation, one may also have to consider epigenetic changes that may impact on the expression of PUMA or Noxa in tumour cells.
The others: Bim/Bod, Bmf, Blk/Bik/Nbk, Bad and Hrk/DP5
The majority of BH3-only proteins are regulated independently of p53 in response to cell stress by means as diverse as transcriptional activation, sub-cellular sequestration to cytosolic scaffold proteins or cytoskeletal components and proteasomal degradation. For some proteins, more than one regulatory mechanism may apply in a stimulus and cell type-dependent manner.
Bim/Bod
The means of Bim regulation include dynein-light chain-mediated cytoskeletal sequestration,87 transcriptional induction in response to growth factor withdrawal88 and proteasomal degradation after ERK-mediated phosphorylation and subsequent ubiquitinylation.89 At the moment, it is unclear how important the individual modes of regulation are under physiological conditions and whether the different modes may be used in a cell type-specific manner. Generation of animal models carrying knock-in mutations impairing individual modes of regulation will be suitable to clarify these issues.
In normal physiology, Bim is crucial for negative selection of B- and T-cell precursors and the regulation of peripheral lymphocyte homeostasis.3 The hyperplasia caused by the extended lifespan of Bim-deficient lymphoid and myeloid cells facilitates tumorigenesis on a tumour-prone background. Loss of just one allele of bim causes c-myc transgenic animals to develop B-cell lymphomas much more rapidly when compared with c-myc transgenic bim+/+ littermates. This accelerated lymphomagenesis does not require loss of the ARF/p53 pathway, frequently observed in c-myc transgenic mice, indicating that Bim acts in a separate cell death cascade.74 Bim appears to be directly regulated by c-myc as indicated by decreased levels of this protein in genuine c-myc lymphomas in mice.74 Mutants of the c-myc gene that can be found in some Burkitt lymphoma patients with poor prognosis appear to have lost this ability.90 Fetal liver stem cells transduced with these mutant versions of c-myc develop B-cell lymphomas much more rapidly when compared with tumours induced by transduction with wt c-myc. The most striking feature of these c-myc mutants is that they are unable to block Bcl-2 function by increasing Bim expression levels in MEFs and haemopoietic stem cells but still maintain the ability to promote proliferation and activation of ARF and p53.90 Loss of Bim or p53 allows wt c-myc to drive B-cell lymphomagenesis with similar kinetics than c-myc mutants and tumours caused by the mutants no longer need to loose wt p53. Consistent with these observations, immunohistochemical analysis revealed that 6/7 Burkitt lymphomas from patients expressing mutant c-myc scored negative for Bim expression whereas all lymphomas derived from patients with wt c-myc retained reactivity with anti-Bim antibodies.90
Taken together, this indicates that c-myc usually activates parallel apoptotic pathways to prevent tumorigenesis and that inactivation of one of these pathways to apoptosis, either the one regulated by p53 or the one activated by Bim, is sufficient to promote tumorigenesis once cell cycle progression is deregulated.
Another elegant study confirms that Bim also acts as a tumour suppressor in H-ras-driven carcinogenesis in epithelial cells. Loss of Bim allows the tumorigenic outgrowth of mouse baby kidney-derived epithelial cells that have been transformed with the viral E1A oncogene and a dominant negative version of p53.77 Under standard conditions, those cells do not give rise to tumours once transplanted into nude mice unless apoptosis is impaired, for example, by loss of Bax/Bak or Bim. Interestingly, loss of PUMA failed to promote tumorigenesis in the same model system excluding that p53-independent regulation of PUMA contributes significantly to tumour progression in this system. In the same study, Bim has also been identified as the key regulator of paclitaxel responsiveness of these tumours as application of this drug leads to its rapid accumulation. Consistently, bim−/− tumours resist paclitaxel treatment in vivo. The data provided strongly suggest that oncogenic H-ras may confer resistance of tumours to paclitaxel therapy by preventing accumulation of Bim. The molecular basis underlying this observation is that H-ras-mediated activation of the MAPK signalling cascade causes phosphorylation of Bim on Ser69 that promotes its proteasomal degradation.91 Application of proteasome inhibitors, one of which currently in early clinical trial for the treatment of multiple myeloma (Velcade, PS 341), restored Bim accumulation and paclitaxel sensitivity in H-ras overexpressing tumour cells not only in vitro but also in vivo suggesting additive beneficial effects of this combination regimen in the treatment of human tumours (Figure 3).
The relevance of these findings made in animal models is underlined by the recent observations that loss of Bim also occurs in human tumours and tumour cell lines (Table 1). Loss of the chromosomal region 2q13, harbouring the bim gene, can be observed in mantle cell lymphomas, a malignancy derived from pre-germinal center CD5+ B cells.92 Although a limited number of patient samples displayed loss of one bim allel (sufficient to confer partial resistance of mouse lymphocytes to factor-deprivation), 3/5 MCL cell lines had lost both allels and the other two cell lines tested were heterozygous for bim.92 Malignant B cells may be particularly prone to lose functional Bim, as we recently observed loss of Bim mRNA and protein expression in 2/5 human multiple myeloma cell lines, too (unpublished observations). Bim protein expression appears to be frequently absent in renal cell carcinomas (RCCs) (see below) and can be lost in melanoma cell lines derived from patients with metastatic lesions,61 suggesting that this BH3-only protein also possesses tumour suppressor potential in humans and its loss may be related to progression of disease and maybe also drug resistance. Recent investigations demonstrate that the antitumorigenic effects of Gleevec, a kinase inhibitor used in the treatment of Philadelphia chromosome positive (Ph+) leukaemias, where the c-abl tyrosine kinase is hyperactivated owing to translocation from chromosome 9 to the break point cluster (bcr) region on chromosome 22, giving rise to the oncogenic BCR/Abl fusion protein, largely depends on Bim. Cell lines established from Ph+ chronic myelogenous leukaemia patients during blast crisis displayed decreased levels of Bim when compared with acute myeloid leukaemia (AML) cell lines. Consistently, Bim expression was found to be lower in Ph+ acute lymphocyte leukaemia (ALL) when compared to Ph− ALL cells suggesting the BCR/Abl may elevate, directly or indirectly, the turn over of Bim owing to phosphorylation-dependent degradation.93 Treatment of the Ph+ K562 erythroleukaemia cell line with Gleevec/Imatinib causes a strong induction of Bim protein expression by a combination of transcriptional activation and inhibition of proteasomal degradation (usually enforced by MAPK-mediated phosphorylation of Bim) along with dephosphorylation of Bad and transcriptional induction of Bmf. Downregulation of Bim by RNA interference confers partial resistance to the drug and cell lines derived from bim−/−bad−/− double-deficient fetal liver cells, transformed with bcr-c-abl, were almost completely resistant to Gleevec treatment. Consistent with a prominent role of Bim in the mediation of the antitumorigenic effect of Gleevec, primary leukaemia cells from non-responsive patients failed to upregulate Bim ex vivo (Andreas Strasser, CSHL Conference on Programmed Cell Death 2005). Interesingly, Bim but not PUMA mRNA was found to be induced in a significant portion of children with acute lymphoblasitc leukaemia undergoing glucocorticoid monotherapy. These observations are consistent with previously published in vitro data implicating Bim as a glucocorticoid receptor target and the execution of dexamethasone-induced apoptosis in leukaemic and normal lymphocytes.59, 94
Taken together, we can anticipate that loss of Bim function may be frequently involved in human tumorigenesis and drug resistance to novel generation compounds such as Velcade, Gleevec or histone deacetylase (HDAC) inhibitors, but also evergreens such as glucocorticoids, which in part execute their antitumorigenic effects by inducing or enhancing the expression of Bim95, 96 (Figure 3).
Bmf
In contrast to Bim, little is known about the biology of the Bmf, a more recent addition to the BH3-only protein family is reported to be regulated in a similar manner as Bim. Both molecules share a highly conserved dynein light chain-binding motif near their N-termini that targets Bmf to the actin cytosekeleton and Bim to microtubules, respectively.97 Bmf can be released from the cytoskeleton in response to UV radiation or loss of adhesion and/or integrin signals preceding a distinct form of cell death, called anoikis, observed only in fibroblasts, epithelial or endothelial cells preventing detached cells from colonizing elsewhere.
In haemopoietic cells, Bmf is widely expressed and has been implicated in cytokine withdrawal-induced cell death of granulocytes where the protein was reported to accumulate during in vitro culture;56 however, granulocyte numbers are normal in bmf−/− mice (AV. unpublished). Alternative splicing of bmf may also contribute to regulate its in vivo function. Two additional splice variants of bmf (termed bmf II and III) were found to be expressed in normal and malignant human B cells, derived from patients with B-cell lymphocytic leukaemia98 and multiple myeloma cell lines (unpublished observations).
Surprisingly, these novel variants of Bmf lack a functional BH3 domain and Bmf III also contains a different carboxy terminus. Overexpression of Bmf II or Bmf III in Hela cells increased their colony-forming potential, whereas Bmf, as previously reported, acted proapoptotic. B-CLL cells undergo rapid apoptosis in vitro and this cell death correlated with enhanced mRNA expression of the full-length form of Bmf, containing the BH3 domain necessary for apoptosis induction, but not the other two isoforms.98 In line with the lifespan-limiting role of Bmf, a recent study describes that treatment of oral and esophageal squamous carcinoma cell lines with novel HDAC-inhibitors causes upregulation of Bmf and concomitant tumour cell death (Figure 3). Reduction of bmf mRNA and protein by RNA-interference abrogated the antitumorigenic effects of HDAC inhibitors enabling clonal survival of tumour cells in vitro.99
The tumour suppressor potential of Bmf remains to be investigated in an in vivo model and so far only circumstance evidence exists that it may be involved in the prevention of tumorigenesis (Table 1), as the human bmf gene is located on chromosome 15q14, a region harbouring a so far unidentified tumour suppressor that is frequently lost in patients with advanced stages of breast, lung and colon carcinomas.100, 101
Blk/Bik/Nbk
Although being widely expressed in immature and mature lymphocytes, myeloid cells and nucleated erythrocytes Blk, the murine orthologue of Bik/Nbk, appears redundant for cell death induced by a broad range of apoptotic. Apoptosis induced by cytokine withdrawal, the gulcocorticoid dexamethasone, phorbol ester, ionomycine, the topoisomerase II inhibitor etoposide or B-cell receptor crosslinking is not impaired in blk−/− lymphocytes.102 Furthermore, loss of blk failed to prevent degenerative phenotypes caused by loss of bcl-2 such as polycystic kidney disease and leukopenia indicating that Blk, in contrast to Bim, does not mediate apoptosis by neutralizing Bcl-2 in these tissues.102 Interestingly, although Blk appears to have overlapping functions with Bim in spermatogenesis (but not haemopoiesis) as combined loss leads to infertility, reduced testicular cellularity and loss of spermatozoa presumably because excess spermatogonia cannot be deleted by apoptosis leading to the displacement of Sertoli cells and aborted testicular development.103
So far no spontaneous tumours have been reported to arise in these animals but expression of both, Blk/Nbk as well as Bim was reported to be frequently lost in patients suffering from clear-cell RCCs by Peter Daniels group (Table 1). Concomitant loss of both BH3-only proteins was reported in 6/7 RCC cell lines. Immunohistochemistry confirmed expression of Bik/Nbk in normal kidney epithelium next to malignant tissue that had lost Bik/Nbk expression in 28 patient biopsies where side-to-side evaluation of healthy and malignant tissue was possible.104 Overall, weak expression or complete loss of Bik/Nbk expression was observed in all 57 clear-cell RCCs studied. Fluorescence in situ hybridization analysis revealed that allelic loss and/or methylation was responsible for the loss of Bik/Nbk expression in 9/10 cell lines investigated and treatment with 5′-aza2dC restored mRNA and in most cases also protein expression.104 Bim mRNA expression also increased in response to 5′-aza2dC treated Caki-2 cells suggesting that Bim levels may also be reduced owing to epigenetic silencing. Adenoviral expression of p53 in RCC cell lines failed to restore Bik/Nbk expression in 6/7 cases, although the mouse orthologue Blk was reported to be induced in mouse embryonic fibroblasts by the E1A oncogene in a p53-mediated manner.105 Direct inducible re-expression of Bik/Nbk by means of adenoviral transfer of the gene in RCC cell lines, but not primary renal epithelial cells, caused massive Bax-dependent apoptosis suggesting that the loss of Bik/Nbk expression may facilitate transformation, tumour progression and maybe drug resistance in RCC.104
Single-strand conformation polymorphism analysis and conventional sequencing analysis of bik exons 2–7 in 71 samples of human B-cell lymphomas revealed occasional missense mutations in follicular lymphoma, marginal zone and diffuse large B-cell lymphomas. However, none of the mutations found in the coding region were located in the BH3 domain and the functional impact of the amino-acid changes on the cell death-promoting potential of Bik/Nbk was not evaluated.106 Comparative genomic hybridization performed on human glioma samples revealed a minimal common deletion area mapping to the bik gene locus on chromosome 22q13, harbouring a putative tumour suppressor gene.107
Taken together, loss of Bik/Nbk can be observed in a range of different human tumour entities. Further functional investigations will be needed to evaluate the relevance of this BH3-only protein in cancer pathology and response to anticancer therapy. Intercrossing of blk−/− mice with various tumour-prone mouse strains may be a valuable first step in this direction.
Bad
The BH3-only protein Bad has long been considered as a key player in apoptosis induction in response to cytokine and/or growth factor deprivation. Bad is negatively regulated by phosphatidylinositol-3 (PI3)-kinase/Akt signalling,108 a survival pathway frequently hyperactivated in many human tumour entities, owing to loss of its negative regulator phosphatase and tensin homolog deleted on chromosome 10 (PTEN) or, even more indirectly, loss of the PTEN transcriptional regulator p53.109 Therefore, a prominent role for this BH3-only protein in apoptosis induction and/or tumour suppression has been anticipated for a long time. Loss of Bad protein expression in mice does not support a crucial role for this BH3-only protein in cytokine deprivation-induced death of primary lymphocytes.110 Interestingly, bad−/− mammary epithelial cell cultures were more resistant to the effects of epidermal growth factor deprivation and serum deprivation rendered Bad-proficient lymphocytes more susceptible to the effects of FasL and tumour necrosis factor and MEF wild type for Bad were more susceptible to etoposide treatment. Spontaneous tumorigenesis was observed in about 40% of animals in a cohort of aged bad−/− mice (latency >15 month). B220+ surface Ig+ diffuse large B-cell lymphomas were the most frequent tumour entity observed (>40%), accompanied by a broad range of different haemopoietic malignancies.110 Consistent with its proposed cell death-sensitizing role and a mild delay in γ-radiation-induced death of primary bad−/− thymocytes, Bad-deficient mice succumbed to radiation-induced thymic lymphomas significantly earlier than their wt littermates.110
Activation of Bad by dephosphorylation of Ser112 by application of the EGFR tyrosine kinase inhibitor Iressa/Gefitinib combined with dephosphorylation of Ser 136 mediated by re-expression of PTEN (or a combination of MAPK and PI-3K inhibitors) was reported to inhibit growth and induce apoptosis in MDA-468 breast cancer cells in vitro, but more importantly also prevented tumour outgrowth in a mouse xenograft model in vivo.111 Unfortunately, the role of other BH3-only proteins was not investigated in this study but we can anticipate that simultaneous inhibition of PI-3K/Akt and MAPK signals may, depending on the tumour cell type, also lead to induction of Bim via Foxo3a-mediated gene transcription and dephosphorylation-dependent protein stabilization.111
Mutations of the bad gene have been reported in 2/47 colon adenocarcinoma samples (Table 1). Interestingly, both mutations were located in the BH3 domain, one of which strongly reducing binding to Bcl-2 and Bcl-XL. When overexpressed in 293 T cells, both mutants were less potent in inducing apoptosis then the wt form of Bad.112
Taken together, these studies indicate that Bim and Bad are critical mediators of the effects of new-generation anticancer agents and both genes can be occasionally impaired owing to mutations or epigenetic silencing in human tumours (Figure 3).
Hrk/DP5
DP5 was first identified as a BH3-only protein rapidly induced in response to NGF deprivation of rat sympathetic neurons and its overexpression potently induced apoptosis in cultured neurons.113 DP5 is widely expressed in the developing nervous system during embryogenesis where cell death plays a crucial role in the deletion of neurons with misguided axons but expression appears to be minimal or absent in most other adult tissues with the exception of the brain.113 Neuronal cell death caused by β-amyloid-triggered calcium increase has been proposed to be mediated by DP5, which is upregulated at the mRNA and protein level in neuronal cultures.114 In line with its proposed role in apoptosis in the central nervous system, DP5 was reported to be upregulated strongly in brain ischaemia/reperfusion models of neuronal cell death115 and motor neurons from DP5−/− mice are protected from axotomy-induced cell death.116 The human homologue, Harakiri (Hrk), shares 72% identity with DP5 and was isolated by means of two-hybrid analysis from a HeLa cDNA library using Bcl-2 as bait.117 In contrast to DP5, Hrk appears to be expressed also outside the neuronal system and RNA was detected in haemopoietic tissues as well as pancreas.117 Subsequently, Hrk/DP5 has been reported to be induced in response to IL3-deprivation, in a calcium-dependent manner, in haemopoietic progenitor cell lines such as FDC-P1 promyeloid cells and in human CD34+ haemopoietic progenitors after growth factor withdrawal.118
Inactivation of the hrk gene owing to aberrant DNA methylation has been reported to occur frequently in human gastric and colorectal cancers (Table 1). More than 30% of the investigated tumour cell lines in this study (e.g. DLD1, HT29, MKN28 and KatoIII) had the transcription start site of hrk methylated, impairing its mRNA expression, but this methylation was not found in haemopoietic, liver or pancreatic tumour cell lines.119 Analysis of primary tumour material revealed the very same methylation in colorectal (24%) and gastric cancer (17%) patients whereas no such methylation was found in HCC or normal tissues derived from colon or stomach. Antimethylating agents restored hrk mRNA expression in gastric cancer cell lines paralleling the induction of apoptosis that was synergistically enhanced by simultaneous application of HDAC inhibitors such as trichostatin A119 (Figure 3). Although such treatment did not change the mRNA levels of other BH3-only proteins such as Bid, Bad and Noxa, this remains only a correlative observation as no assays have been performed that would prove that Hrk is essential for the death of these tumour cells. Neverthless, a reduction or loss of Hrk expression owing to promoter and/or exon 1 methylation has also been reported to occur in a significant number of astrocytomas (>19%), primary (>27%) and secondary glioblastomas (>43%). No methylation was found in cells adjacent to the tumour. Immunohistochemical analysis revealed a trend towards decreasing levels of Hrk expression with increasing tumour grade and complete loss of expression associates with loss of heterozygosity (LOH) at 12q13 in the majority of cases.120 Interestingly, silencing of Hrk appears to occur more frequently in p53 competent tumours, suggesting that loss of Hrk expression may relieve the pressure to lose this key tumour suppressor. However, until confirmed in well-defined mouse models of tumorigenesis, this remains pure speculation.
Conclusions
Collectively, all these studies indicate that BH3-only proteins act as a crucial barrier against malignant diseases not only in animal models but also in humans. They, therefore, constitute critical targets mediating the effects of anticancer therapy and loss of their function, owing to mutation, LOH or epigenetic silencing may well contribute to tumour progression and drastically impair the effectiveness of conventional therapeutics but also of new generation antitumour agents targeting the proteasome or certain tyrosine receptor kinase pathways.
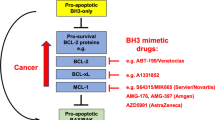
Based on our expanding knowledge of the molecular characteristics and function of BH3-only proteins, promising lead compounds exploiting the characteristics of the α-helical structure of the BH3 domain have been generated by rational drug design. The small molecule drug ABT-737,121 for example, has been modelled to fit into the binding groove of Bcl-XL. Whether drugs like this induce apoptosis by displacing Bax/Bak-like molecules from Bcl-2 prosurvival homologues, allowing Bax/Bak oligomerization, or, alternatively, by causing the displacement of ‘direct activator’ BH3-only proteins, mopped-up by the high levels of Bcl-2 often found in tumour cells, leading to direct Bax activation remains to be firmly established. Regardless of how they induce apoptosis at the molecular level, these drugs truly hold a large promise for more effective anticancer treatment strategies in the near future.
Abbreviations
- Bad:
-
Bcl-2 antagonist of cell death
- Bak:
-
Bcl-2 antagonist/killer
- Bax:
-
Bcl-2-associated protein X
- Bcl-X:
-
Bcl-2-related protein X
- Bcl-2:
-
B cell lymphoma 2
- BH:
-
Bcl-2 homology domain
- Bbc3:
-
Bcl-2 binding component 3
- Bid:
-
Bcl-2 interacting domain death agonist
- Bik:
-
Bcl-2 interacting killer-like
- Bim:
-
Bcl-2 interacting mediator of cell death
- Blk:
-
Bik-like
- Bmf:
-
Bcl-2 modifying factor
- DP5:
-
death protein 5
- Hrk:
-
Harakiri
- Nbk:
-
natural born killer
- PUMA:
-
p53-upregulated modulator of apoptosis
References
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219.
Cory S and Adams JM (2002) The Bcl-2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2: 647–656.
Strasser A (2005) The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5: 189–200.
Veis DJ, Sorenson CM, Shutter JR and Korsmeyer SJ (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75: 229–240.
Motoyama N, Wang FP, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S and Loh DY (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x deficient mice. Science 267: 1506–1510.
Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K and Korsmeyer SJ (2005) Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307: 1101–1104.
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC and Korsmeyer SJ (2003) Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426: 671–676.
Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus RC and Gross A (2005) Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 122: 593–603.
Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A and Korsmeyer SJ (2005) A role for proapoptotic BID in the DNA-damage response. Cell 122: 579–591.
Willis SN and Adams JM (2005) Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell. Biol. 17: 617–625.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 17: 393–403.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM and Huang DC (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19: 1294–1305.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR and Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17: 525–535.
Zong WX, Lindsten T, Ross AJ, MacGregor GR and Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15: 1481–1486.
Yu J, Wang Z, Kinzler KW, Vogelstein B and Zhang L (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 100: 1931–1936.
Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B, Schulze-Osthoff K and Daniel PT (2003) Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 22: 3580–3590.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P and Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 11: 577–590.
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD and Green DR (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309: 1732–1735.
Coultas L and Strasser A (2003) The role of the Bcl-2 protein family in cancer. Semin. Cancer Biol. 13: 115–123.
Egle A, Harris AW, Bath ML, O'Reilly L and Cory S (2004) VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 103: 2276–2283.
Packham G, White EL, Eischen CM, Yang H, Parganas E, Ihle JN and Grillot DAM (1998) Selective regulation of Bcl-X L by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev. 12: 2475–2487.
Nieborowska-Skorska M, Hoser G, Kossev P, Wasik MA and Skorski T (2002) Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase pim-1 in the BCR/ABL-mediated leukemogenesis. Blood 99: 4531–4539.
Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD and Craig RW (2001) MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood 97: 3902–3909.
Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC and Perucho M (1997) Somatic frameshift mutations in the bax gene in colon cancers of the microsatellite mutator phenotype. Science 275: 967–969.
Yin CY, Knudson CM, Korsmeyer SJ and Van Dyke T (1997) Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 385: 637–640.
Shibata MA, Liu ML, Knudson MC, Shibata E, Yoshidome K, Bandey T, Korsmeyer SJ and Green JE (1999) Haploid loss of bax leads to accelerated mammary tumor development in C3(1)/SV40-TAg transgenic mice: reduction in protective apoptotic response at the preneoplastic stage. EMBO J. 18: 2692–2701.
Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C and Lowe SW (2001) Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409: 207–211.
Allen JD, Zhang XD, Scott CL, Boyle GM, Hersey P and Strasser A (2005) Is Apaf-1 expression frequently abrogated in melanoma? Cell. Death Differ. 12: 680–681.
Peltenburg LT, de Bruin EC, Meersma D, Smit NP, Schrier PI and Medema JP (2005) Expression and function of the apoptosis effector Apaf-1 in melanoma. Cell. Death Differ. 12: 678–679.
Scott CL, Schuler M, Marsden VS, Egle A, Pellegrini M, Nesic D, Gerondakis S, Nutt SL, Green DR and Strasser A (2004) Apaf-1 and caspase-9 do not act as tumor suppressors in myc-induced lymphomagenesis or mouse embryo fibroblast transformation. J. Cell. Biol. 164: 89–96.
Diehl GE, Yue HH, Hsieh K, Kuang AA, Ho M, Morici LA, Lenz LL, Cado D, Riley LW and Winoto A (2004) TRAIL-R as a negative regulator of innate immune cell responses. Immunity 21: 877–889.
Nagata S (1999) Fas ligand-induced apoptosis. Ann. Rev. Gen. 33: 29–55.
Traver D, Akashi K, Weissman IL and Lagasse E (1998) Mice defective in two apoptosis pathways in the myeloid lineage develop acute myeloblastic leukemia. Immunity 9: 47–57.
Newton K, Harris AW and Strasser A (2000) FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. EMBO J. 19: 931–941.
Kim HS, Lee JW, Soung YH, Park WS, Kim SY, Lee JH, Park JY, Cho YG, Kim CJ, Jeong SW, Nam SW, Kim SH, Lee JY, Yoo NJ and Lee SH (2003) Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology 125: 708–715.
Shivapurkar N, Toyooka S, Eby MT, Huang CX, Sathyanarayana UG, Cunningham HT, Reddy JL, Brambilla E, Takahashi T, Minna JD, Chaudhary PM and Gazdar AF (2002) Differential inactivation of caspase-8 in lung cancers. Cancer Biol. Ther. 1: 65–69.
Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM and Kidd VJ (2000) Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 6: 529–535.
Hanahan D and Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70.
Michalak E, Villunger A, Erlacher M and Strasser A (2005) Death squads enlisted by the tumour suppressor p53. Biochem. Biophys. Res. Commun. 331: 786–798.
Miyashita T and Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299.
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ and El-Deiry WS (2002) BID regulation by p53 contributes to chemosensitivity. Nat. Cell Biol. 4: 842–849.
Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, Wakeham A, Bouchard D, Yeh WC, McGlade JC, Ohashi PS and Hakem R (2003) Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17: 883–895.
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P and Wallach D (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276.
Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA and Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891.
Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL and Yuan J (1998) Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 12: 1304–1314.
Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, Kutok JL, Alt FW and Korsmeyer SJ (2003) Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. 17: 229–239.
Krajewska M, Zapata JM, Meinhold-Heerlein I, Hedayat H, Monks A, Bettendorf H, Shabaik A, Bubendorf L, Kallioniemi OP, Kim H, Reifenberger G, Reed JC and Krajewski S (2002) Expression of Bcl-2 family member Bid in normal and malignant tissues. Neoplasia 4: 129–140.
Chen GG, Lai PB, Chak EC, Xu H, Lee KM and Lau WY (2001) Immunohistochemical analysis of pro-apoptotic Bid level in chronic hepatitis, hepatocellular carcinoma and liver metastases. Cancer Lett. 172: 75–82.
Green MM, Hutchison GJ, Valentine HR, Fitzmaurice RJ, Davidson SE, Hunter RD, Dive C, West CM and Stratford IJ (2005) Expression of the proapoptotic protein Bid is an adverse prognostic factor for radiotherapy outcome in carcinoma of the cervix. Br. J. Cancer 92: 449–458.
Lee JH, Soung YH, Lee JW, Park WS, Kim SY, Cho YG, Kim CJ, Seo SH, Kim HS, Nam SW, Yoo NJ, Lee SH and Lee JY (2004) Inactivating mutation of the pro-apoptotic gene BID in gastric cancer. J. Pathol. 202: 439–445.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T and Tanaka N (2000) Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058.
Hijikata M, Kato N, Sato T, Kagami Y and Shimotohno K (1990) Molecular cloning and characterization of a cDNA for a novel phorbol-12-myristate-13-acetate-responsive gene that is highly expressed in an adult T-cell leukemia cell line. J. Virol. 64: 4632–4639.
Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L and Chittenden T (2001) Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 98: 11318–11323.
Yu J, Zhang L, Hwang PM, Kinzler KW and Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell. 7: 673–682.
Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7: 683–694.
Villunger A, Scott C, Bouillet P and Strasser A (2003) Essential role for the BH3-only protein Bim but redundant roles for Bax, Bcl-2, and Bcl-w in the control of granulocyte survival. Blood 101: 2393–2400.
Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T and Tanaka N (2003) Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17: 2233–2238.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL and Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328.
Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, Adams JM, Strasser A and Villunger A (2005) BH-only proteins Puma and Bim are rate-limiting for {gamma}-radiation and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood 106: 4131–4138.
Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C and Vousden KH (2004) p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J. Biol. Chem. 279: 8076–8083.
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, Bennett F, Pollock PM, Trent JM, Hendrix MJ, Rizzo P, Miele L and Nickoloff BJ (2005) Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 65: 6282–6293.
Hollstein M, Sidransky D, Vogelstein B and Harris CC (1991) p53 mutations in human cancers. Science 253: 49–53.
Malkin D, Li FP, Strong LC, Fraumeni JFJ, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA and Friend SH (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233–1238.
Srivastava S, Zou ZQ, Pirollo K, Plattner W and Chang EH (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li–Fraumeni syndrome. Nature 348: 747–749.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CAJ, Butel JS and Bradley A (1992) Mice deficient for p53 are developmentally normal but are susceptible to spontaneous tumours. Nature 356: 215–221.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT and Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4: 1–7.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D and Jacks T (2004) Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell 119: 847–860.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK and Lozano G (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li–Fraumeni syndrome. Cell 119: 861–872.
Martin-Caballero J, Flores JM, Garcia-Palencia P, Collado M and Serrano M (2004) Different cooperating effect of p21 or p27 deficiency in combination with INK4a/ARF deletion in mice. Oncogene 23: 8231–8237.
Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW and DePinho RA (2001) Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413: 86–91.
Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM and Lowe SW (2002) Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 1: 289–298.
Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S and Lozano G (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat. Genet. 36: 63–68.
Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA and Cleveland JL (2003) c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol. Cell Biol. 23: 7256–7270.
Egle A, Harris AW, Bouillet P and Cory S (2004) Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 101: 6164–6169.
Eischen CM, Weber JD, Roussel MF, Sherr CJ and Cleveland JL (1999) Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 13: 2658–2669.
Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ and Lowe SW (2004) Suppression of tumorigenesis by the p53 target PUMA. Proc. Natl. Acad. Sci. USA 101: 9333–9338.
Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM and White E (2005) Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7: 227–238.
Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K and White E (2004) Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 18: 2095–2107.
Eferl R and Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3: 859–868.
Shimazaki C, Inaba T and Nakagawa M (2000) B-cell lymphoma-associated hemophagocytic syndrome. Leukaemia Lymphoma 38: 121–130.
Yong WH, Ueki K, Chou D, Reeves SA, von Deimling A, Gusella JF, Mohrenweiser HW, Buckler AJ and Louis DN (1995) Cloning of a highly conserved human protein serine–threonine phosphatase gene from the glioma candidate region on chromosome 19q13.3. Genomics 29: 533–536.
Mora J, Cheung NK, Chen L, Qin J and Gerald W (2001) Loss of heterozygosity at 19q13.3 is associated with locally aggressive neuroblastoma. Clin. Cancer Res. 7: 1358–1361.
Hoque MO, Begum S, Sommer M, Lee T, Trink B, Ratovitski E and Sidransky D (2003) PUMA in head and neck cancer. Cancer Lett. 199: 75–81.
Karst AM, Dai DL, Martinka M and Li G (2005) PUMA expression is significantly reduced in human cutaneous melanomas. Oncogene 24: 1111–1116.
Chin L, Merlino G and DePinho RA (1998) Malignant melanoma: modern black plague and genetic black box. Genes Dev. 12: 3467–3481.
Lee SH, Soung YH, Lee JW, Kim HS, Lee JH, Park JY, Cho YG, Kim CJ, Kim SY, Park WS, Kim SH, Lee JY and Yoo NJ (2003) Mutational analysis of Noxa gene in human cancers. Apmis 111: 599–604.
Puthalakath H, Huang DCS, O'Reilly LA, King SM and Strasser A (1999) The pro-apoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell 3: 287–296.
Dijkers PF, Medemadagger RH, Lammers JJ, Koenderman L and Coffer PJ (2000) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10: 1201–1204.
Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, Fukuda A, Hikita A, Seto H, Okada T, Inaba T, Sanjay A, Baron R, Kawaguchi H, Oda H, Nakamura K, Strasser A and Tanaka S (2003) Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. EMBO J. 22: 6653–6664.
Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP and Lowe SW (2005) Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436: 807–811.
Ley R, Balmanno K, Hadfield K, Weston C and Cook SJ (2003) Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J. Biol. Chem. 278: 18811–18816.
Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S and Seto M (2005) Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 24: 1348–1358.
Kuribara R, Honda H, Matsui H, Shinjyo T, Inukai T, Sugita K, Nakazawa S, Hirai H, Ozawa K and Inaba T (2004) Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitors. Mol. Cell Biol. 24: 6172–6183.
Schmidt S, Rainer J, Riml S, Ploner C, Jesacher S, Achmuller C, Presul E, Skvortsov S, Crazzolara R, Fiegl M, Raivio T, Janne OA, Geley S, Meister B and Kofler R (2005) Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood 2006; 107: 2061–2069.
Zhang Y, Adachi M, Zhao X, Kawamura R and Imai K (2004) Histone deacetylase inhibitors FK228, N-(2-aminophenyl)-4-[N-(pyridin-3-yl-methoxycarbonyl)amino- methyl]benzamide and m-carboxycinnamic acid bis-hydroxamide augment radiation-induced cell death in gastrointestinal adenocarcinoma cells. Int. J. Cancer 110: 301–308.
Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET and Yu Q (2005) Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 102: 16090–16095.
Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DCS and Strasser A (2001) Bmf: a pro-apoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 293: 1829–1832.
Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M and Osorio LM (2004) Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia 18: 41–47.
Zhang Y, Adachi M, Kawamura R and Imai K (2006) Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 13: 129–140.
Wick W, Petersen I, Schmutzler RK, Wolfarth B, Lenartz D, Bierhoff E, Hummerich J, Muller DJ, Stangl AP, Schramm J, Wiestler OD and von Deimling A (1996) Evidence for a novel tumor suppressor gene on chromosome 15 associated with progression to a metastatic stage in breast cancer. Oncogene 12: 973–978.
Schmutte C, Tombline G, Rhiem K, Sadoff MM, Schmutzler R, von Deimling A and Fishel R (1999) Characterization of the human Rad51 genomic locus and examination of tumors with 15q14–15 loss of heterozygosity (LOH). Cancer Res. 59: 4564–4569.
Coultas L, Bouillet P, Stanley EG, Brodnicki TC, Adams JM and Strasser A (2004) Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol. Cell Biol. 24: 1570–1581.
Coultas L, Bouillet P, Loveland KL, Meachem S, Perlman H, Adams JM and Strasser A (2005) Concomitant loss of proapoptotic BH3-only Bcl-2 antagonists Bik and Bim arrests spermatogenesis. EMBO J. 24: 3963–3973.
Sturm IS C, Gillissen B, Siebert R, Janz M, Radetzki S, Jung K, Loening S, Dörken B and Daniel PT (2006) Loss of the tissue-specific proapoptoptic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 13: 619–627.
Mathai JP, Germain M, Marcellus RC and Shore GC (2002) Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene 21: 2534–2544.
Arena V, Martini M, Luongo M, Capelli A and Larocca LM (2003) Mutations of the BIK gene in human peripheral B-cell lymphomas. Genes Chrom. Cancer 38: 91–96.
Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD and Sikic BI (2005) High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res. 65: 4088–4096.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y and Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241.
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S and Mak TW (2001) Regulation of PTEN transcription by p53. Mol. Cell 8: 317–325.
Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME and Korsmeyer SJ (2003) Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 100: 9324–9329.
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J and Rosen N (2005) The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 8: 287–297.
Lee JW, Soung YH, Kim SY, Nam SW, Kim CJ, Cho YG, Lee JH, Kim HS, Park WS, Kim SH, Lee JY, Yoo NJ and Lee SH (2004) Inactivating mutations of proapoptotic Bad gene in human colon cancers. Carcinogenesis 25: 1371–1376.
Imaizumi K, Tsuda M, Imai Y, Wanaka A, Takagi T and Tohyama M (1997) Molecular cloning of a novel polypeptide, DP5, induced during programmed neuronal death. J. Biol. Chem. 272: 18842–18848.
Imaizumi K, Morihara T, Mori Y, Katayama T, Tsuda M, Furuyama T, Wanaka A, Takeda M and Tohyama M (1999) The cell death-promoting gene DP5, which interacts with the Bcl-2 family, is induced during neuronal apoptosis following exposure to amyloid β protein. J. Biol. Chem. 274: 7975–7981.
Guan QH, Pei DS, Xu TL and Zhang GY (2005) Brain ischemia/reperfusion-induced expression of DP5 and its interaction with Bcl-2, thus freeing Bax from Bcl-2/Bax dimmers are mediated by c-Jun N-terminal kinase (JNK) pathway. Neurosci. Lett 2006; 393: 226–230.
Imaizumi K, Benito A, Kiryu-Seo S, Gonzalez V, Inohara N, Lieberman AP, Kiyama H and Nunez G (2004) Critical role for DP5/Harakiri, a Bcl-2 homology domain 3-only Bcl-2 family member, in axotomy-induced neuronal cell death. J. Neurosci. 24: 3721–3725.
Inohara N, Ding L, Chen S and Núñez G (1997) harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-XL . EMBO J. 16: 1686–1694.
Sanz C, Benito A, Inohara N, Ekhterae D, Núñez G and Fernandez-Luna JL (2000) Specific and rapid induction of the proapoptotic protein Hrk after growth factor withdrawal in hematopoietic progenitor cells. Blood 95: 2742–2747.
Obata T, Toyota M, Satoh A, Sasaki Y, Ogi K, Akino K, Suzuki H, Murai M, Kikuchi T, Mita H, Itoh F, Issa JP, Tokino T and Imai K (2003) Identification of HRK as a target of epigenetic inactivation in colorectal and gastric cancer. Clin. Cancer Res. 9: 6410–6418.
Nakamura M, Ishida E, Shimada K, Nakase H, Sakaki T and Konishi N (2005) Frequent HRK inactivation associated with low apoptotic index in secondary glioblastomas. Acta Neuropathol. (Berlin) 2005; 110: 402–410.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW and Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435: 677–681.
Acknowledgements
Work in our laboratory is supported by fellowships and grants from the Austrian Science Fund (FWF) and the Austrian Ministry for Education, Science and Culture (bm:bwk). VL is a DOC-FFORTE fellow of the Austrian Academy of Science (OeAW). We are grateful to our colleagues, especially Andreas Strasser, David Vaux, Peter Daniel and Georg Häcker for many interesting discussions and sharing unpublished results as well as to all members of the SFB021 (Proliferation and Cell Death in Tumours) for their input into our research. We apologize to many scientists in this field whose excellent research was not cited but was only referred to indirectly through reviews.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Borner
Rights and permissions
About this article
Cite this article
Labi, V., Erlacher, M., Kiessling, S. et al. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ 13, 1325–1338 (2006). https://doi.org/10.1038/sj.cdd.4401940
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401940
Keywords
This article is cited by
-
Cullin-5 neddylation-mediated NOXA degradation is enhanced by PRDX1 oligomers in colorectal cancer
Cell Death & Disease (2021)
-
Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand?
Cell Death & Disease (2016)
-
How cell death shapes cancer
Cell Death & Disease (2015)
-
Role of CtBP2 in the Apoptosis of Retinal Ganglion Cells
Cellular and Molecular Neurobiology (2015)
-
TRAIL promotes membrane blebbing, detachment and migration of cells displaying a dysfunctional intrinsic pathway of apoptosis
Apoptosis (2013)
