
Abstract
Mechanisms of prostate cancer (CaP) recurrence during a combined androgen blockade (CAB) are poorly understood. Previously, the role of androgen receptor (AR) gene mutations underlying the CAB therapy relapse has been raised. To investigate the hypothesis that AR gene aberrations are involved in CAB relapse, 11 locally recurrent CaP samples from patients treated with orchiectomy and bicalutamide were analyzed for copy number changes and DNA sequence alterations of the AR gene by fluorescence in situ hybridization and single-strand conformation polymorphism, respectively. Altogether, base changes were detected in four tumors (36%). Three of them were missense mutations (G166S, W741C, M749I) and two were silent polymorphisms. Interestingly, none of the tumors had AR amplification. These data suggest that different AR variants are developed and selected for during various types of hormonal treatments, and also, that CAB achieved by orchiectomy and bicalutamide does not act as a selective force for AR amplification.
Similar content being viewed by others

Introduction
Despite increased use of prostate specific antigen (PSA) screening and improved diagnosis of prostate cancer (CaP), 20% to 40% of CaPs are still diagnosed at a clinically advanced stage (Scardino et al., 1994) when cure by surgery is no longer possible. Recurrence rates after radical surgery or radiotherapy range from 25% to 50% (Dennis and Griffiths, 2000). For these patients, palliative hormonal therapy remains the only effective treatment.
Both surgically and chemically induced androgen withdrawal therapies (AWT) cause a 95% reduction in serum testosterone levels, but leave a substantial amount of adrenal androgen precursors in the circulation (Dennis and Griffiths, 2000; Labrie et al, 1993). The concentration of dihydrotestosterone (DHT) in CaP tissue is decreased only by about 60%, because adrenal androgens are effectively converted into DHT in prostate. To further inhibit action of the remaining androgens at the level of androgen receptor (AR), antiandrogens are generally combined with AWT to reach combined androgen blockade (CAB) (Labrie et al, 1993). Although disputed, CAB therapy seems to provide slight improvement in 5-year survival over AWT (Prostate Cancer Trialists’ Collaborative Group, 2000), and it has become an approved treatment for advanced CaP. Up to 90% of CaPs initially respond to hormonal therapy (Palmberg et al, 1999), but eventually, they acquire a therapy-resistant phenotype, resulting in disease recurrence after a few months or years.
Occurrence of mutations in the AR has been suggested to underlie CaP recurrence during CAB therapy (Taplin et al, 1995, 1999). Interestingly, accumulation of mutations at the codon 877 of the AR has been observed in CaPs relapsed during CAB achieved by AWT and flutamide (Taplin et al, 1999). However, previous reports on the role of AR in CaP progression during CAB therapy have shortcomings because they have not studied the possible involvement of other AR-related mechanisms, such as AR amplification, which has been found in approximately 30% of AWT refractory CaPs, and they have not examined AWT+bicalutamide refractory CaP samples, although bicalutamide acts as a pure androgen antagonist lacking the agonistic effects of flutamide, and in adjunction with AWT, might provide more complete androgen blockade than other antiandrogens in clinical use.
Therefore, it can be hypothesized that the frequency and spectrum of AR mutations in AWT+bicalutamide refractory CaPs may differ from those in AWT+flutamide refractory ones. To test this hypothesis, we established the frequency of AR mutations and amplifications in CaP relapsed during first-line CAB achieved by orchiectomy and bicalutamide.
Results and Discussion
Antiandrogens combined with AWT have proven to be effective in the treatment of advanced CaP (Prostate Cancer Trialists’ Collaborative Group, 2000). Before long, however, resistance to this therapy develops, and an enigmatic problem is the process by which cancer cells loose their hormonal control and escape CAB.
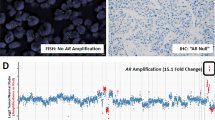
AR protein is a key mediator of growth signaling in the prostate, and to evaluate whether AR aberrations are associated to CaP progression despite ongoing CAB, we screened the coding region of the AR gene, as well as analyzed the AR gene copy number, of 11 CaP tissue samples from patients who failed orchiectomy+bicalutamide treatment. Interestingly, none of the tumors showed AR amplification, whereas amino acid changes and silent base substitutions were detected in 27% and 18% of the tumors, respectively (Table 1).
Previous studies have demonstrated that approximately one-third of the advanced CaPs develop hypersensitivity to adrenal androgens during AWT by amplifying the AR gene (Bubendorf et al, 1999; Koivisto et al, 1997; Linja et al, 2001). In the present study, all tumors had a normal AR gene copy number. To some extent this finding could be biased by the small sample size, but in our opinion, a more likely explanation for the total absence of AR amplification among the orchiectomy+bicalutamide treated tumors is the diminished availability of androgens at the AR. Namely, in androgen deficient milieu caused by AWT, AR gene amplification can be developed and selected for to maintain androgen dependent growth of CaP, but when bicalutamide is combined to block the binding of residual androgens to the AR, an increase in receptor copy number is not feasible any more due to the lack of active ligand at the AR. Then, other molecular mechanisms are evolved to cause therapy resistance (Koivisto et al, 1998; Sadar et al, 1999).
Another important mechanism that may contribute to failure of CAB therapy is a mutation of the AR gene (Koivisto et al, 1998; Sadar et al, 1999). The selection of AR variants during bicalutamide treatment has not been studied previously, but mutations in the AR have been documented in 31% to 50% of AWT+flutamide refractory CaPs (Taplin et al, 1995, 1999). For example, Taplin et al (1999) have identified AR mutations in bone marrow metastases from 5 of 16 (31%) CaP patients treated by AWT+flutamide. These mutations resided in codon 877 of the AR gene and were found to be activated by flutamide, but not by bicalutamide. The frequency of mutations may, however, be an underestimate, because the transactivating domain of the AR was not studied. Other studies (Han et al, 2001; Tilley et al, 1996) have indicated that the transactivating domain may harbor up to 50% of all mutations when the whole AR gene was screened in hormone-refractory CaP.
In the present study, using SSCP and sequencing, we analyzed the coding sequence of the AR gene from 11 CaP samples (Table 1). Five base changes, of which three lead to amino acid changes, were detected in four tumors (36%). Four base substitutions were located in the ligand binding domain and one in the transactivating domain. The mutations were novel, except M749I that has previously been identified in prostate cancer by Takahashi et al (1995). The alterations were somatic, because the nonmalignant tissue of the mutation-positive cases was found to harbor wild-type AR.
Our data does not confirm the accumulation of AR mutations at the codon 877 reported by Taplin et al (1999), which is most likely due to the different antiandrogen used (bicalutamide versus flutamide) in the present study. This observation indicates that different types of AR variants are developed and selected for during bicalutamide and flutamide treatments and supports the data by Han et al (2001), who suggested that changes in the hormonal environment may drive the selection of spontaneous somatic mutations that provide a growth advantage for CaP. Although the consequences of the detected mutations for AR function in vivo is beyond the scope of the present study and remains an issue for further study, based on previous reports on the function of AR variants found in prostate cancer (Buchanan et al, 2001; Culig et al, 1993; McDonald et al, 2000; Sack et al, 2001; Taplin et al, 1995; Thompson et al, 2001; Wang et al, 2000; Zhao et al, 2000), it is tempting to predict that the amino acid changes in the ligand binding domain detected here alter the specificity of hormone binding, such that activation of transcription can take place in the presence of not only androgens, but also of other steroid hormones and antiandrogens used in therapy.
Finally, several investigators have reported the paradoxical response (decrease in PSA, symptoms, and objective signs) on withdrawal of bicalutamide in approximately 30% of the patients with progression on luteinizing hormone-releasing hormone analogue and bicalutamide treatment (Kelly, 1998; Sadar et al, 1999). It has been hypothesized that the androgen receptor probably mutates and recognizes the antiandrogen as a stimulator. In the present study, we were not able to confirm this hypothesis, because none of the tumors with mutated AR showed decreased serum PSA level or alleviation of symptoms as a result of antiandrogen withdrawal after clinical or biochemical tumor recurrence.
In summary, we have for the first time studied the frequency of mutated and amplified AR variants in orchiectomy+bicalutamide refractory CaP and found that AR mutations (4/11, 36%) outnumber AR amplification (0%). This is a novel and interesting observation suggesting that different types of AR alterations in CaP are selected for during various types of hormonal therapies.
Materials and Methods
Patient and Tumor Specimens
Seventy-nine patients, who experienced a local tumor recurrence during hormonal therapy and were treated with transurethral resection between 1994 and 1999 at the Tampere University Hospital, were identified from the database of the Department of Pathology. Twenty-two percent (17/79) of these patients experienced a relapse during first-line CAB therapy achieved by a combination of orchiectomy and bicalutamide (Casodex; dose 150 mg/d, Zeneca Pharmaceuticals, Macclesfield, United Kingdom). After histological review of the samples, 11 samples from eight patients contained enough malignant material and were included in the analyses.
All CaP samples were local recurrences. Local tumor recurrence was evidenced by symptoms of urethral obstruction and an increase in serum PSA levels. TNM (tumor node metastasis)-stage of the tumors at the time of diagnosis and histological differentiation of the recurrent tumors are summarized in Table 1. The clinical data were collected from the patient records. For DNA extraction and fluorescence in situ hybridization (FISH), representative tumor areas containing more than 80% of malignant cells were selected by histopathological examination of hematoxylin and eosin-stained slides. Nonmalignant tissue was obtained from all mutation positive patients. The research protocol was approved by the Ethical Committee of the Tampere University Hospital.
Analysis of Gene Amplification by FISH
FISH was performed to sections of the formalin-fixed tissues, and at least two sections were analyzed in separate hybridizations. Five-μm sections were transferred onto TechMate slides (Dako A/S, Glostrup, Denmark) and fixed overnight at 56°C. Two-color FISH was performed using Spectrum Orange-labeled AR probe (Vysis, Downer’s Grove, Illinois) and FITC-labeled probe (DXZ1/BamX7) specific for the centromere of the X chromosome. The treatment protocol for formalin-fixed paraffin-embedded tumors (Paraffin-Pretreatment Reagent; Vysis) was used with modifications. The slides were first deparaffinized and immersed in 0.2N HCl for 20 minutes and incubated in pretreatment-solution at 80°C for 30 minutes and in a protease solution (0.5 mg/0.9% NaCl) for 10 minutes at 37°C. The slides were then postfixed in 10% formalin for 10 minutes, air-dried and denatured on a heat block at 94°C for 5 minutes under coverslip in buffer (100 mM Tris-HCl, pH 8.9, 1 M KCl, 15 mM MgCl2, 500 μg/ml BSA and 0.5% Tween 20 [v/v]). After dehydration, the slides were air-dried and incubated with proteinase K (10 μg/ml PBS; Roche Molecular Biochemicals, Mannheim, Germany) for 10 minutes at 37°C. After dehydration and drying, slides were hybridized overnight in a moist chamber at 37°C. Hybridization mixture contained 1 μl of the both probes and 8 μl hybridization buffer (Vysis). After overnight hybridization, the slides were washed and counterstained with 0.2 μM DAPI. The signals were scored with a Leica DMRB fluorescence microscope (Leica Microsystems, Wetzlar, Germany) equipped with a double-band pass filter using x100 objective. AR signal copy number was counted from at least 50 cancer cells. The AR signals were evaluated in relation to the DXZ1 signals, and the AR gene was considered to be amplified when the AR gene copy number was at least three-folds higher than that of the DXZ1. Control hybridizations included previously analyzed tumors with normal and elevated AR gene copy number.
PCR Single-Strand Conformation Polymorphism and DNA Sequencing
DNA from paraffin-embedded tumor specimens was isolated using standard phenol-chloroform extraction. PCR was performed in a volume of 10 μl using GeneAmp PCR System 9700 (Applied Biosystems Inc., Foster City, California). Sample DNA (25 ng) was amplified in 35 cycles of PCR (94°C for 30 seconds, 55–60°C for 30 seconds, and 72°C for 45 seconds). Reaction mixture contained ×1 PCR buffer (1.5 mM MgCl2), 20 μM of each dNTPs, 0.6 U of AmpliTaqGold DNA polymerase (Applied Biosystems), 0.4 μM of each primer, and 0.4 μCi [α-33P]-dCTP (>2,500 Ci/mmol). PCR fragments were subsequently heat-denatured and run for 15 to 24 hours at 800 V in ×0.5 and ×0.8 MDE gels (FMC BioProducts, Rockland, Maine) containing 10% glycerol. After electrophoresis, the gels were dried and exposed to Kodak BioMax MR film (Kodak, Rochester, New York) for 1 day. The AR gene from normal human DNA and exon 8 from LNCaP cell line, containing a T877A mutation, were amplified and run as controls along with the samples. When band shift was noticed, PCR and SSCP were repeated and aberrant band was cut from the gel, reamplified, and sequenced. The sequencing was carried out with the fmol™ DNA Cycle Sequencing System (Promega Corp., Madison, Wisconsin) using [α-33P]-dCTP label with the same primers as for PCR. All potential mutations were sequenced on both DNA strands. Sequencing gel electrophoresis was performed using standard conditions.
Primers
Primers reported previously for exon 1 of the AR gene by Tilley et al (1996) were used with the exception that the region 40 thru 249 (coordinates from Lubahn et al., 1989) was amplified with 5′-GGGGTAAGGGAAGTAGGTGGA-3′ and 5′-CAGCAGCAGCAAACTGGC-3′, and region 1114 thru 1309 was amplified with 5′-TACAAGTCCGGAGCACTGGA-3′ and 5′-ATGCAGGCTCGCCAGGTC-3′. The DNA fragments in between 249 and 365 and 1410 and 1553 were not analyzed due to their high GC and repeat sequence content. Primers for exons 2 thru 8 were published previously (Koivisto et al, 1997), except that exons 4, 7, and 8 were amplified with the following pairs of primers 5′-GCATTGTGTGT-TTTTGACCACTGATG-3′ and 5′-GCAAAGGAGTTGGGCTGGTTG-3′; 5′-TCTAATGCTCCTTCGTGGGCA-3′ and 5′-CTCTATCAGGCTGTTCTCCCT-3′; and 5′-AACCCTGTTTTTCTCCCT-3′ and 5′-TTTCCCAGAAAGGATCTTGG-3′, respectively.
References
Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G, and Kallioniemi OP (1999). Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res 59: 803–806.
Buchanan G, Yang M, Harris JM, Nahm HS, Han G, Moore N, Bentel JM, Matusik RJ, Horsfall DJ, Marshall VR, Greenberg NM, and Tilley WD (2001). Mutations at the boundary of the hinge and ligand binding domain of the androgen receptor confer increased transactivation function. Mol Endocrinol 15: 46–56.
Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, Eberle J, Bartsch G, and Klocker H (1993). Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol 7: 1541–1550.
Dennis LJ and Griffiths K (2000). Endocrine treatment in prostate cancer. Semin Surg Oncol 18: 52–74.
Han G, Foster BA, Mistry S, Buchanan G, Harris JM, Tilley WD, and Greenberg NM (2001). Hormone status selects for spontaneous somatic androgen receptor variants that demonstrate specific ligand and cofactor dependent activities in autochthonous prostate cancer. J Biol Chem 276: 11204–11213.
Kelly WK (1998). Endocrine withdrawal syndrome and its relevance to the management of hormone refractory prostate cancer. Eur Urol 34 (Suppl 3): 18–23.
Koivisto P, Kolmer M, Visakorpi T, and Kallioniemi OP (1998). Androgen receptor gene and hormonal therapy failure of prostate cancer. Am J Pathol 152: 1–9.
Koivisto P, Kononen J, Palmberg C, Hyytinen E, Tammela T, Trapman J, Isola J, Visakorpi T, and Kallioniemi O-P (1997). Androgen receptor gene amplification: A possible molecular mechanism for failure of androgen deprivation therapy in prostate cancer. Cancer Res 57: 314–319.
Labrie F, Belanger A, Dupont A, Luu-The V, Simard J, and Labrie C (1993). Science behind total androgen blockade: From gene to combination therapy. Clin Invest Med 16: 475–492.
Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, and Visakorpi T (2001). Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res 61: 3550–3555.
Lubahn DB, Brown TR, Simental JA, Higgs HN, Migeon CJ, Wilson EM, and French FS (1989). Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity. Proc Natl Acad Sci USA 86: 9534–9538.
McDonald S, Brive L, Agus DB, Scher HI, and Ely KR (2000). Ligand responsiveness in human prostate cancer: Structural analysis of mutant androgen receptors from LNCaP and CWR22 tumors. Cancer Res 60: 2317–2322.
Palmberg C, Koivisto P, Visakorpi T, and Tammela TL (1999). PSA decline is an independent prognostic marker in hormonally treated prostate cancer. Eur Urol 36: 191–196.
Prostate Cancer Trialists’ Collaborative Group (2000). Maximum androgen blockade in advanced prostate cancer: An overview of the randomised trials. Prostate Cancer Trialists’ Collaborative Group. Lancet 355: 1491–1498.
Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR Jr, Weinmann R, and Einspahr HM (2001). Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci USA 98: 4904–4909.
Sadar MD, Hussain M, and Bruchovsky N (1999). Prostate cancer: Molecular biology of early progression to androgen independence. Endocr Relat Cancer 6: 487–502.
Scardino PT, Beck JR, and Miles BJ (1994). Conservative management of prostate cancer. N Engl J Med 330: 1831.
Takahashi H, Furusato M, Allsbrook WC Jr, Nishii H, Wakui S, Barrett JC, Boyd J (1995). Prevalence of androgen receptor gene mutations in latent prostatic carcinomas from Japanese men. Cancer Res 55: 1621–1624.
Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, and Balk SP (1999). Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res 59: 2511–2515.
Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, and Keer HN (1995). Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 332: 1393–1398.
Thompson J, Saatcioglu F, Janne OA, and Palvimo JJ (2001). Disrupted amino- and carboxyl-terminal interactions of the androgen receptor are linked to androgen insensitivity. Mol Endocrinol 15: 923–935.
Tilley WD, Buchanan G, Hickey TE, and Bentel JM (1996). Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res 2: 277–285.
Wang C, Young WJ, and Chang C (2000). Isolation and characterization of the androgen receptor mutants with divergent transcriptional activity in response to hydroxyflutamide. Endocrine 12: 69–76.
Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, and Feldman D (2000). Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med 6: 703–706.
Acknowledgements
This study was financially supported by the Medical Research Fund of Tampere University Hospital, the Finnish Cancer Institute (to PAK and E-RH), the Finnish-Norwegian Medical Research Fund (to PAK), and the Pirkanmaa Regional Fund of the Finnish Cultural Foundation (to KH).
We thank Mrs. Sinikka Kekkonen and Mrs. Lila Nikkola for their skillful technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haapala, K., Hyytinen, ER., Roiha, M. et al. Androgen Receptor Alterations in Prostate Cancer Relapsed during a Combined Androgen Blockade by Orchiectomy and Bicalutamide. Lab Invest 81, 1647–1651 (2001). https://doi.org/10.1038/labinvest.3780378
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780378
This article is cited by
-
Androgen Receptor Predicts First and Multiple Recurrences in Non-Muscle Invasive Urothelial Carcinoma of the Bladder
Pathology & Oncology Research (2019)
-
Androgen receptor signaling in prostate cancer
Cancer and Metastasis Reviews (2014)
-
Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches
Oncogene (2013)
-
AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression
Oncogene (2012)
-
Transgelin induces apoptosis of human prostate LNCaP cells through its interaction with p53
Asian Journal of Andrology (2010)
