
Abstract
Cellular prion protein (PrPC), an N-linked glycoprotein, is expressed in a variety of tissues, but its functions remain unclear. PrPC is abundantly expressed in the endocrine pancreas, which regulates blood glucose homeostasis. Therefore, we investigated whether the expression of PrPC was altered in islets of Langerhans in a model of spontaneous type 1 diabetes, the diabetes-prone BioBreeding (BBdp) rat and a model of β-cell adaptation to hyperglycemia, the chronic glucose-infused Sprague Dawley rat. Pancreatic sections from animals aged 7–100 days were stained immunohistochemically and evaluated using light, fluorescence and confocal microscopy. PrPC was ubiquitously expressed in all four major endocrine cell types within islets. Surprisingly, cytosolic inclusions containing PrPC were identified exclusively in a subpopulation of insulin-producing β-cells. The inclusions exhibited different molecular characteristics from the PrP aggregates previously described in vitro in neurons. The frequency of β-cells with PrPC inclusions increased with age and was threefold greater in diabetes-prone rats than in controls at 100 days. Cytosolic PrPC expression in β-cells was suppressed whereas the number and size of PrPC inclusions markedly increased in response to hyperglycemia during the first 2 days of continuous glucose infusion in Sprague Dawley rats. In summary, this is the first report describing in vivo cytosolic PrPC aggregation. These unique PrPC inclusions were β-cell specific, more frequent in diabetes-prone animals, and responded to hyperglycemia in glucose-infused Sprague Dawley rats. These data suggest a potential dysfunction in β-cells of diabetes-prone rats, and point to new avenues for the study of diabetes pathogenesis.
Similar content being viewed by others


Main
Cellular prion protein (PrPC) is expressed mainly in the central nervous system as well as in the pancreatic islets, spleen, testes and other organs.1, 2 The high conservation of PrPC among mammals infers an essential physiological role.3 However, the function of this protein is still unclear. One reason is that PrPC knockout mice display no dramatic change in phenotype.4 To date, several proteins interacting with PrPC have been identified5, 6, 7 and a picture is emerging of a protein involved in multiple cellular functions including signal transduction,8 copper metabolism9 as well as a membrane receptor or ligand.10 Recently, it has been reported that PrPC is also important for the self-renewal of long-term repopulating hematopoietic stem cells.11 Furthermore, emerging evidence suggests that cytosolic PrPC is involved in cell survival, but whether it is a cell protective molecule or cell death trigger is controversial.12
The β-cell is the major cell type within islets of Langerhans with a unique capacity to produce insulin and regulate blood glucose. In type 1 diabetes (T1D), β-cells are selectively destroyed by a progressive inflammatory process called insulitis that involves infiltration of pancreatic islets by mononuclear cells. It is generally thought that the destruction of 80–90% of the β-cells results in hyperglycemia.13 Some studies suggest that islet defects occur in diabetes-prone animals before islet inflammation.14, 15, 16 However, the key events leading to immune-mediated β-cell destruction as well as abnormalities in the target β-cells remain unclear.17
Although it is known that PrPC is abundantly expressed in the endocrine pancreas,1, 18 there are no reports linking PrPC and glucoregulation. In addition, PrPC expression has not been described in animal models of diabetes. Therefore, we investigated whether PrPC expression was altered in endocrine cells in the pancreas of diabetes-prone BB rats or Sprague Dawley rats with hyperglycemia induced by chronic infusion of glucose.
Materials and methods
Animals
BioBreeding control (BBc) and diabetes-prone BioBreeding (BBdp) rats were purchased from the Animal Resources Division of Health Canada, Ottawa. Wistar Furth rats were purchased from Charles River, Laval, QC, Canada. Animals were maintained under specific pathogen-free conditions and were fed with a standard cereal-based rodent diet. Rats were killed by exsanguination while under anesthesia with 3% isoflurane in oxygen. For immunohistochemistry analysis, tissues were fixed in Bouin's solution or universal molecular fixative (UMFIX, Sakura Tissue-Tek, Torrance, CA, USA), embedded and 5 μm serial sections were cut and attached to charged slides. The degree of insulitis was evaluated on H&E stained pancreas sections as described previously.19
Pancreatic sections from glucose-infused Sprague Dawley rats were from a study reported by Topp et al.20 The aim of this study was to evaluate the adaptation of β-cells to hyperglycemia that occurs during chronic glucose infusion. Briefly, male rats were maintained at 22°C and 12 h light–dark cycle, with free access to food and water. Under anesthesia, an indwelling catheter was inserted into the jugular and exteriorized from the neck and connected to a flexible tether/swivel system. Rats were infused (2 ml/h) with either 0.45% saline or 50% dextrose in 0.45% saline for 1, 2, 3, 4, 5 or 6 days. Animals were killed while anesthetized with sodium pentobarbital 35 mg/kg i.p., and the pancreas was rapidly removed and fixed in Bouin's solution overnight. For additional details, refer to Topp et al.20
Antibodies and Immunostaining
Pancreatic sections were heated in 10 mM citric acid buffer (pH 6.0) in a microwave oven for 10 min to retrieve antigen. All incubation steps with antibodies were performed at room temperature. For PrPC detection, a mixture of six monoclonal mouse anti-PrP antibodies, a gift from AW Stuke (German Primate Centre, Goettingen, Germany), was used. The specificity of these antibodies has previously been reported.21, 22 We confirmed further the specificity of the PrP antibodies in our laboratory using Western blots of purified PrPC. All other primary antibodies are shown in Supplementary Table 1. Secondary antibodies for immunohistochemistry were obtained from DAKO Diagnostics (Mississauga, ON, Canada). DAB (Sigma-Aldrich Canada Ltd., Oakville, ON, Canada) at 0.06% and H2O2 at 0.03% were used as substrates for HRP staining and the BCIP/NBT substrate system (DAKO) for AP staining.
Double immunofluorescence staining of PrPC and the following proteins: insulin, glucagon, somatostatin, pancreatic polypeptide, GM-130, γ-tubulin, proteasome, ubiquitin, Hsc70 or vimentin, was performed on paraffin sections fixed with Bouin's solution or UMFIX. Sections were incubated with mouse anti-PrP monoclonal antibody 13F1022 for 2 h followed by incubation with biotin-conjugated rabbit anti-mouse antibody for 30 min. Thereafter, sections were incubated for 2 h with primary antibodies directed against the proteins mentioned above. Avidin-conjugated Cy3 and FITC-conjugated donkey anti-guinea-pig (DAKO), FITC-conjugated donkey anti-rabbit (DAKO), donkey anti-mouse/Alexa488 (Invitrogen Canada Inc., Burlington, ON, Canada) or donkey anti-goat/Alexa488 (Invitrogen) antibodies were applied for 30 min. Nuclei were stained with Hoechst (Sigma), DR (Biostatus Limited, Shepshed, UK) or SYBR Green I (Molecular Probes, Eugene, Oregon, USA) for 1 min. Zeiss LSM 410 and 510 Meta CLSM microscopes, DRAQ5 (633 nm), Cy3 (565 nm), FITC (505 nm) and Hoechst (395 nm) filters and a × 63/1.4 oil Plan-Apochromat objective were used to acquire confocal images. A Zeiss Axioplan2 microscope equipped with × 40/0.75 plan Neofluar and a high-resolution IEEE 1394 FireWire™ digital CCD color camera (MicroPublisher 3.3 RTV; QImaging, Burnaby, BC, Canada) was used for fluorescence imaging. For negative controls, sections were incubated with the secondary antibodies alone or primary antibodies alone. In each case, negative controls showed no staining on pancreatic sections (Figure 1e–f). Coincident fluorescent signals, observed using three different color filters, were considered to be autofluorescence.

PrPC expression in pancreatic tissue of rats. PrPC was expressed in all endocrine cells in the pancreas with no difference in expression between BBc (a) and BBdp (b) animals aged 45 days. However, some cells contained cytosolic PrPC inclusions (inset, arrowhead). Compared with endocrine cells, ganglion cells (black arrow) showed a lower expression of PrPC. Bar=100 μm in each image and 10 μm in the inset. In rats over 70 days of age, the number of islet cells with inclusions increased and the aggregates were more pronounced. The immunofluorescence images show an islet from a BBdp (c) and a Wistar Furth (d) rat, both aged 100 days. Several cells containing cytosolic PrPC inclusions were observed (white arrows). Cell nuclei stained with Hoechst (blue). Bar=25 μm. Negative control sections incubated with primary (e) or secondary (f) antibodies alone showed no staining. Bar=25 μm.
Three-Dimensional Reconstructions and Orthogonal Projections
The image stacks were obtained using a Zeiss 510 Meta CLSM confocal laser scanning microscope in multitrack mode, a × 63/1.4 oil Plan-Apochromat objective, and Zeiss Confocal Microscopy Software, Release 3.2. The three-dimensional (3D) reconstruction and the orthogonal projection were performed with ‘Imaris v.4.1.3’ after data deconvolution with ‘Huygens Essential’.
Image Analysis
All image analyses were performed as described previously.19 Briefly, coded sections were analyzed using a Zeiss Axioplan2 microscope equipped with × 20/0.5 or × 40/0.75 plan Neofluar objectives and a high-resolution IEEE 1394 FireWire™ digital CCD color camera or a × 63/1.4 oil Plan-Apochromat objective and a high-resolution AxioCamHR camera (Zeiss). Northern Eclipse software (Empix, Mississauga, ON, Canada) was used to analyze images. To determine the percentage of islet β-cells containing inclusions, sections were first labeled with a cocktail of non-β-cell antibodies (rabbit anti-glucagon, anti-pancreatic polypeptide and anti-somatostatin (1/1500 each)) using AP-conjugated anti-rabbit pAb and BCIP/NBT substrate. This was followed by labeling for PrPC with a mixture of six anti-PrP mAbs (2 μg/ml) using biotin-conjugated anti-mouse pAb, HRP-conjugated avidin and DAB as a substrate. Six islets from each section were randomly selected at × 50 magnification. Images of selected islets were taken at × 400 magnification and β-cell area was traced and measured. β-Cells containing inclusions from the selected islets were counted. The percentage of cells containing cytosolic aggregates was calculated as follows:

∑ia—sum of β-cell areas; Ac—area of a single β-cell (estimated as 150 μm2); ni—number of β-cells with inclusions.
Statistics
Data are expressed as mean±s.d. or mean±s.e. Student's t-test and ANOVA were used to evaluate the significance of differences between means (GraphPad Prism, Version 4.03). P-values less than 0.05 were considered to be statistically significant.
Results
PrPC Aggregation in Islets of Control and Diabetes-Prone Rats
To investigate whether PrPC expression was altered in the endocrine pancreas in T1D, we first compared the expression of PrPC in islets of diabetes-prone rats with the control, BBc strain aged <50 days. We detected intense PrPC staining in all the endocrine cells of both BBc (Figure 1a) and BBdp (Figure 1b) rats and to a lesser extent in neurons (Figure 1b, black arrow). There was no obvious difference in PrPC staining between the strains in animals younger than 50 days. Surprisingly, we observed cytosolic accumulation of PrPC in aggregates (Figure 1b inset, arrowhead) in islets of both strains. Further evaluation revealed that the number of islet cells with inclusions was increased in animals aged >75 days, and the aggregates were much more pronounced compared with younger animals (Figure 1c).
Cytosolic aggregation of PrPC in islets was also observed in Wistar Furth (Figure 1d) and Sprague Dawley rats. Thus, a certain level of PrPC aggregation in islets was apparent in several rat strains. We then determined the cellular origin and further characterized the nature of these aggregates.
PrPC Aggregation in Islets is Unique to β-Cells
Pancreatic islets contain four major endocrine cell types, α-, β-, δ- and γ-cells, which secrete the hormones glucagon, insulin, somatostatin and pancreatic polypeptide, respectively. To determine which cell type possessed inclusions, we performed double immunofluorescence staining of PrPC and each of the four hormones. PrPC inclusions were detected only in insulin+ cells (Figure 2). There was no colocalization of PrPC aggregates with glucagon, pancreatic polypeptide or somatostatin positive cells (Figure 2). The aggregates were therefore confined to β-cells, the endocrine cell type selectively destroyed in autoimmune T1D. As observed in multiple planes of confocal images stained for insulin and PrP, only a subset of β-cells contained PrP aggregates.

Identification of islet cells associated with PrPC aggregates. The double immunofluorescence analysis shows single islets of BBdp rats aged 100 days stained for PrPC and insulin (β-cell), glucagon (α-cell), pancreatic polypeptide (PP; γ-cell) or somatostatin (δ-cell). PrPC aggregates (white arrows) were localized within insulin+ cells and thus were specifically confined to β-cells. Cell nuclei were stained with Hoechst (blue). Bar=40 μm.
Cytosolic PrPC Aggregates in β-Cells do not Display Characteristics of Aggresomes
Cytosolic protein aggregations are associated with several genetic and degenerative diseases, such as Alzheimer's disease and Huntington's disease.23, 24 Inclusions are often aggregates of misfolded or unfolded proteins that do not bind to each other under physiological conditions. Thus far, cytosolic PrP aggregates have been reported only in studies performed in vitro.25, 26, 27, 28
We performed additional analyses to further characterize the PrPC aggregates in β-cells. All PrP inclusions described in vitro to date were similar to aggresomes.29 We investigated whether PrPC inclusions in β-cells also possessed characteristics of aggresomes, such as the presence of γ-tubulin, vimentin and ubiquitin-proteasome system (UPS).
Each β-cell contained one cytosolic PrPC aggregate. The aggregates were oval and reached a diameter greater than 4 μm in BBdp animals aged >75 days (Figure 3a and Supplementary Figure 1a; Supplementary Movie 1). They were mainly localized at the cell periphery but were also to some extent perinuclear (Supplementary Movie 2). Immune fluorescence double staining for the PrPC and GM130 Golgi marker revealed that the PrPC concentrated in inclusions did not colocalize with the Golgi apparatus (Figure 3b and Supplementary Figure 1b; Supplementary Movie 3). Thus, PrPC was not in the process of being transported through the secretory pathway but was cytoplasmic. Next, we examined whether the aggregated PrPC was associated with the microtubule organization center (MTOC) and UPS. As shown by immunofluorescence labeling, there was no γ-tubulin present in the inclusions (Figure 3c and Supplementary Figure 1c). Thus, the cytosolic PrPC aggregates were not localized to MTOC. In addition, the inclusions did not colocalize with proteasomes and ubiquitin (Figure 3d and e and Supplementary Figure 1d; Supplementary Movie 4). The finding that PrPC inclusions were not associated with the cellular UPS indicates that they are not recognized as misfolded proteins designated for degradation. Further, PrPC aggregates were not surrounded by vimentin (Supplementary Figure 2). Thus, the PrPC inclusions in β-cells did not display characteristics of aggresomes.

PrPC inclusions are cytosolic and do not display characteristics of an aggresome. The 3D reconstruction (a–d) and 2D projections (e and f) were prepared with ‘Imaris’ after data deconvolution with ‘Huygens Essential’. (a) PrPC (red) was mainly localized on the membrane (black arrows) and a cytosolic inclusion (yellow arrows). As shown by double immunofluorescence staining the aggregate did not colocalize with (b) Golgi apparatus (green). Neither did PrPC localize to the MTOC as revealed by (c) γ-tubulin staining (green), nor did it colocalize with (d) proteasomes (green) or (e) ubiquitin (green) (f) Hsc70 (green) was highly expressed in islets, but there was no colocalization with the cytoplasmic PrPC inclusions (yellow arrows). Nuclei were counterstained with (a) SYBR green or (c, e and f) DR. Distance between the ticks in the grid is 5 μm. Bars: (e) 5 μm; (f) 30 μm.
As the aggresome-like PrP structures described in vitro also colocalized with Hsc70,26, 27 we addressed whether PrPC qinclusions in β-cells contained this cytosolic chaperone. Although Hsc70 in islet cells was highly expressed, it did not colocalize with PrPC inclusions (Figure 3f). These observations demonstrate that the cytosolic PrPC aggregates in β-cells exhibit distinct molecular characteristics from the aggresome-like PrP structures described in vitro and thus represent a new and unique subcellular PrPC entity.
Cytosolic PrPC Aggregation in β-Cells Increases with Age and is more Frequent in Diabetes-Prone Rats
We next investigated whether there was an association between the development of autoimmune T1D and the aggregation of PrPC in β-cells using the BBdp rat, an animal model of spontaneous T1D. The pancreatic histopathology in the BBdp rats closely resembles that seen in human T1D.30 In BBdp rats from the Ottawa colony, insulitis begins around 50 days of age and is well established by approximately 70 days of age. The average age at onset of T1D is around 90 days. To assess whether the appearance of the inclusions differed between BBc and BBdp rats, we stained for PrPC in sections from animals at 30, 45, 70 and 100 days of age (n=5–9 per group) and counted cells containing PrPC inclusions (six islets/animal). The number of islet cells containing inclusions increased with age, showing a positive correlation only in BBdp rats (P<0.02) (Figure 4a). The difference between the rat strains was most striking at 100 days. To assess the exact number of β-cells with inclusions at this age, we double-stained sections from BBc and BBdp animals (n=9) for PrPC and islet non-β-cell hormones and counted β-cells containing inclusions. The frequency of β-cells with PrPC inclusions was approximately three times greater in BBdp (27.1±8.2%) compared with BBc (9.8±3.4%) rats (mean±s.d.; P<0.0001) (Figure 4b and Supplementary Figure 3). These results strongly suggest that increased formation of cytosolic PrPC aggregates is favored in β-cells of diabetes-prone rats. Interestingly, ganglion cells in the same sections showed remarkably weaker PrPC expression compared with islet cells at all ages and neither ganglion cells nor brain tissue contained PrPC inclusions (Figure 4c and d).

Age-dependent appearance of inclusions and expression of PrPC. (a) The number of islet cells with inclusions increased with age as shown in animals aged 30, 45, 70 and 100 days (n=5–9 per group; mean±s.e.). A positive correlation was found in the BBdp strain (P<0.02) but not in BBc rats (GraphPad Prism V 4.03). (b) Pancreatic sections from 100 d rats were double labeled with monoclonal anti-PrP antibodies and a cocktail of non-β-cell antibodies (anti-glucagon, anti-pancreatic polypeptide and anti-somatostatin). (c) PrPC staining intensity in ganglion cells (black arrow) was notably weaker compared with endocrine cells (red arrow) which also contained aggregates (yellow arrows). Ganglion cells did not contain PrPC aggregates. (d) PrPC staining on cerebellum from a BBdp rat aged 100 days. (e–f) PrPC expression in islet cells of a BBdp rat aged 11 (e) and 100 days (f). In the younger animal, PrPC was distributed ubiquitously in the islet and there were no aggregates. In contrast, a pronounced cytosolic PrPC aggregation (white arrows) and strong membrane staining (yellow arrows) was seen in β-cells of the 100-day-old animal. Bar=10 μm (c) and 20 μm (e and f).
We also observed a different expression pattern of PrPC at different ages in BBdp and BBc rats. In younger animals (<30 days), PrPC was ubiquitously localized in the cytoplasm and plasma membrane (Figure 4e), whereas in older animals (>75 days), PrPC was mainly localized to the plasma membrane and inclusions. This pattern was particularly evident in β-cells from BBdp animals older than 75 days, which showed strong membrane staining and pronounced cytosolic aggregates of PrPC reaching a diameter up to 5 μm (Figure 4f).
Cytosolic PrPC Aggregation in β-Cells Responds to Hyperglycemia
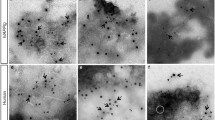
β-Cells play a key role in glucose homeostasis. To investigate whether PrPC expression in β-cells was linked to glucoregulation, we investigated the effect of hyperglycemia on PrPC expression in β-cells by analyzing pancreatic tissue from rats infused chronically with glucose, a model of β-cell adaptation to hyperglycemia.20 We examined the expression pattern as well as cytosolic aggregation of PrPC in β-cells of male Sprague Dawley rats infused with either saline or 50% glucose (2 ml/h) for 0, 1, 2, 3, 4, 5 and 6 days (n=45 per group).
These analyses revealed that during the first 48 h of glucose challenge, there was nearly complete suppression of cytosolic PrPC expression in β-cells, whereas its expression on the plasma membrane did not change. In contrast, untreated animals displayed a normal expression of cytosolic PrPC in islet β-cells (Figure 5a and b). Saline-infusion slightly enhanced the expression of cytosolic PrPC in islets. However, it did not result in suppression of cytosolic PrPC or increased formation of aggregates as observed in glucose-infused rats. After 1 day of glucose challenge when blood glucose was ∼25 mmol/l, cytosolic PrPC in β-cells was almost undetectable (Figure 5c). The same pattern of cytosolic PrPC expression was observed after 2 days of glucose infusion (blood glucose ∼25 mmol/l) and pronounced cytosolic PrPC aggregation was identified in β-cells (Figure 5d). At day three when blood glucose returned to preinfusion levels (∼10 mmol/l) β-cells displayed a low expression of cytosolic PrPC. At day 4 and 5, cytosolic PrPC expression increased, reaching the level observed in islets of saline-infused or untreated rats by day 6 (Figure 5e–h). The size and number of inclusions at day 6 were similar to day 0. Of note, non-β-cells did not show any changes in PrPC expression in response to glucose challenge. In summary, under hyperglycemic conditions, the cytosolic expression of PrPC in β-cells was suppressed while the aggregation was remarkably increased. These results clearly demonstrate that cytosolic expression and aggregation of PrPC in rat β-cells is a physiological process that responds to changes in blood glucose levels.

Time-dependent expression and aggregation of PrPC in glucose-infused Sprague Dawley rats. Untreated (a) as well as saline-infused (b) animals showed cytosolic PrPC expression in pancreatic islets. In contrast, glucose-infused rats showed remarkable changes of cytosolic PrPC expression in β-cells. At day 1 (c) and 2 (d) of glucose challenge, β-cells in animals with high blood glucose exhibited low cytosolic PrPC expression, yet displayed a high frequency of β-cells with cytosolic PrPC inclusions. After 72 h (e), when blood glucose in the animals had returned to preinfusion levels, β-cells showed upregulation of cytosolic PrPC expression and pronounced PrPC aggregation. Cytosolic PrPC expression was further increased at day 4 (f) and 5 (g) reaching the normal level by day 6 (h). Bar=20 μm.
Discussion
PrPC is expressed on the cell membrane, anchored to a glycosylphosphatidylinositol moiety in a wide variety of cells and tissues.1, 2, 11, 31 Its physiological role is still described as a black box, enigmatic and obscure.6, 32 To our knowledge, there are only two reports describing the expression of PrPC in islet cells.18, 33 However, the expression of PrPC in islets of Langerhans has not been linked with glucoregulation nor has it been reported in animal models of diabetes.
In this study, we demonstrate that PrPC is strongly expressed in islets and forms β-cell-specific cytosolic aggregates. Infusion with glucose specifically altered cytosolic PrPC expression in β-cells. There is only one report linking expression of PrPC and glucose concentration.34 Hypoglycemic conditions resulted in an upregulation of PrPC expression in N18 mouse neuroblastoma cells in the first 24 h followed by a decline. In the present study, the expression of cytosolic PrPC in β-cells of Sprague Dawley rats decreased after the first 24 h of glucose challenge, and gradually returned to normal levels after 4 days. Topp et al20 reported that blood glucose in the infused animals was highest (∼25 mmol/l) on day 1 and 2 and decreased by day 3. Insulin increased ∼12-fold during the first 24 h and remained ∼8-fold higher than control values by day 5. These data clearly show that the suppression of cytosolic PrPC expression and increased aggregation in rat β-cells was influenced by blood glucose concentration. The lack of colocalization of Hsc70, ubiquitin and proteasomes with the aggregates is consistent with the proper folding of PrPC within the inclusions. Thus, the inclusions may function as storage depots to sequester excess cytosolic PrPC in β-cells during periods of high blood glucose.
As we detected moderate cytosolic PrPC aggregation in several rat strains, we propose that the aggregation of PrPC in rat β-cells is a normal physiological process possibly associated with blood glucose regulation. The significantly higher proportion of β-cells with PrPC inclusions and pronounced PrPC aggregation in BBdp rats aged >50 days, when islet inflammation becomes visible, could be associated with dysregulated blood glucose. Interestingly, we did not observe a correlation between insulitis score and the number of inclusions in β-cells of BBdp rats. This indicates a previously unrecognized dysfunction in β-cells of BBdp rats that could precede autoimmune β-cell destruction, a point that requires additional evaluation. In accordance with our findings, results from human35 and animal36 studies also suggest that aberrant β-cell activity might precede diabetes.
In conclusion, this study demonstrates altered metabolism of PrPC in β-cells in two animal models associated with glucose dysregulation. We show for the first time the in vivo accumulation of PrPC in cytosolic inclusions, which were restricted to β-cells in pancreatic islets. These cellular PrPC aggregates have different molecular characteristics from the cytosolic aggresome-like inclusions of PrP described previously in vitro26, 27, 28 and thus, reveal a unique subcellular PrPC structure. The appearance of the inclusions was age dependent, and more frequent in BBdp rats compared with the control strain. Moreover, hyperglycemic conditions resulted in pronounced accumulation of PrPC in inclusions. These findings suggest a previously unrecognized physiological role for PrPC in β-cells that is closely associated with glucose homeostasis. Our data raise the possibility of a β-cell dysfunction in BBdp rats that precedes the development of islet inflammation. These intriguing associations could shed further light on the physiological processes leading to both diabetes and prion diseases.
References
Brown KL, Ritchie DL, McBride PA, et al. Detection of PrP in extraneural tissues. Microsc Res Tech 2000;50:40–45.
Oesch B, Westaway D, Walchli M, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985;40:735–746.
Wopfner F, Weidenhofer G, Schneider R, et al. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 1999;289:1163–1178.
Bueler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993;73:1339–1347.
Strom A, Diecke S, Hunsmann G, et al. Identification of prion protein binding proteins by combined use of far-Western immunoblotting, two dimensional gel electrophoresis and mass spectrometry. Proteomics 2006;6:26–34.
Lasmezas CI . Putative functions of PrP(C). Br Med Bull 2003;66:61–70.
Bragason BT, Palsdottir A . Interaction of PrP with NRAGE, a protein involved in neuronal apoptosis. Mol Cell Neurosci 2005;29:232–244.
Mouillet-Richard S, Ermonval M, Chebassier C, et al. Signal transduction through prion protein. Science 2000;289:1925–1928.
Kramer ML, Kratzin HD, Schmidt B, et al. Prion protein binds copper within the physiological concentration range. J Biol Chem 2001;276:16711–16719.
Gauczynski S, Hundt C, Leucht C, et al. Interaction of prion proteins with cell surface receptors, molecular chaperones, and other molecules. Adv Protein Chem 2001;57:229–272.
Zhang CC, Steele AD, Lindquist S, et al. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci USA 2006;103:2184–2189.
Roucou X, LeBlanc AC . Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med 2005;83:3–11.
Rood PP, Bottino R, Balamurugan AN, et al. Facilitating physiologic self-regeneration: a step beyond islet cell replacement. Pharm Res 2006;23:227–242.
Homo-Delarche F . Is pancreas development abnormal in the non-obese diabetic mouse, a spontaneous model of type I diabetes? Braz J Med Biol Res 2001;34:437–447.
Rosmalen JG, Leenen PJ, Pelegri C, et al. Islet abnormalities in the pathogenesis of autoimmune diabetes. Trends Endocrinol Metab 2002;13:209–214.
Wang GS, Karamchandani J, Pulido O, et al. Altered islet homeostasis before classic insulitis in BB rats. Diabetes Metab 2002;28:3S90–3S97; discussion 93S108–93S112.
Homo-Delarche F, Drexhage HA . Immune cells, pancreas development, regeneration and type 1 diabetes. Trends Immunol 2004;25:222–229.
Amselgruber WM, Buttner M, Schlegel T, et al. The normal cellular prion protein (PrP(c)) is strongly expressed in bovine endocrine pancreas. Histochem Cell Biol 2006;125:441–448.
Wang GS, Gruber H, Smyth P, et al. Hydrolysed casein diet protects BB rats from developing diabetes by promoting islet neogenesis. J Autoimmun 2000;15:407–416.
Topp BG, McArthur MD, Finegood DT . Metabolic adaptations to chronic glucose infusion in rats. Diabetologia 2004;47:1602–1610.
Muller H, Strom A, Hunsmann G, et al. Separation of native prion protein (PrP) glycoforms by copper-binding using immobilized metal affinity chromatography (IMAC). Biochem J 2005;388:371–378.
Krasemann S, Groschup MH, Harmeyer S, et al. Generation of monoclonal antibodies against human prion proteins in PrP0/0 mice. Mol Med 1996;2:725–734.
Kopito RR . Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000;10:524–530.
Ross CA, Poirier MA . Protein aggregation and neurodegenerative disease. Nat Med 2004;10:S10–S17.
Yedidia Y, Horonchik L, Tzaban S, et al. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 2001;20:5383–5391.
Ma J, Lindquist S . Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA 2001;98:14955–14960.
Kristiansen M, Messenger MJ, Klohn PC, et al. Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J Biol Chem 2005;280:38851–38861.
Cohen E, Taraboulos A . Scrapie-like prion protein accumulates in aggresomes of cyclosporin A-treated cells. EMBO J 2003;22:404–417.
Johnston JA, Ward CL, Kopito RR . Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998;143:1883–1898.
Mordes JP, Bortell R, Blankenhorn EP, et al. Rat models of type 1 diabetes: genetics, environment, and autoimmunity. Ilar J 2004;45:278–291.
Ballerini C, Gourdain P, Bachy V, et al. Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J Immunol 2006;176:7254–7262.
Derrington EA, Darlix JL . The enigmatic multifunctionality of the prion protein. Drug News Perspect 2002;15:206–219.
Atouf F, Scharfmann R, Lasmezas C, et al. Tight hormonal control of PrP gene expression in endocrine pancreatic cells. Biochem Biophys Res Commun 1994;201:1220–1226.
Shyu WC, Chen CP, Saeki K, et al. Hypoglycemia enhances the expression of prion protein and heat-shock protein 70 in a mouse neuroblastoma cell line. J Neurosci Res 2005;80:887–894.
Kahn SE, Halban PA . Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes 1997;46:1725–1732.
Alarcon C, Leahy JL, Schuppin GT, et al. Increased secretory demand rather than a defect in the proinsulin conversion mechanism causes hyperproinsulinemia in a glucose-infusion rat model of non-insulin-dependent diabetes mellitus. J Clin Invest 1995;95:1032–1039.
Acknowledgements
We thank Andreas W Stuke and Walter Bodemer from the German Primate Centre (Goettingen, Germany) for a generous gift of anti-PrP antibodies and for discussions; John Woulfe and Doug Gray of the Ottawa Health Research Institute, Ottawa, Canada for interest, support and useful discussions. We thank Dr Laura Atkinson at Simon Fraser University, Burnaby, Canada for sending the samples from the Finegood Lab. We also thank Christopher Patrick for reading the manuscript. This work was funded by the Canadian Institutes of Health Research, the Canadian Diabetes Association, and the Juvenile Diabetes Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website (http://www.laboratoryinvestigation.org)
Rights and permissions
About this article
Cite this article
Strom, A., Wang, GS., Reimer, R. et al. Pronounced cytosolic aggregation of cellular prion protein in pancreatic β-cells in response to hyperglycemia. Lab Invest 87, 139–149 (2007). https://doi.org/10.1038/labinvest.3700500
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700500
Keywords
This article is cited by
-
Characteristic distribution and molecular properties of normal cellular prion protein in human endocrine and exocrine tissues
Scientific Reports (2022)
-
Mixed pathologies in pancreatic β cells from subjects with neurodegenerative diseases and their interaction with prion protein
Acta Neuropathologica Communications (2021)
-
Ipomoeassin-F disrupts multiple aspects of secretory protein biogenesis
Scientific Reports (2021)
-
Altered cellular localisation and expression, together with unconventional protein trafficking, of prion protein, PrPC, in type 1 diabetes
Diabetologia (2021)
-
Prion protein modulates glucose homeostasis by altering intracellular iron
Scientific Reports (2018)
