
Abstract
Frequent allelic losses on the long arm of chromosome 22 (22q) in gliomas indicate the presence of tumor suppressor gene (TSG) at this location. However, the target gene(s) residing in this chromosome are still unknown and their putative roles in the development of astrocytic tumors, especially in secondary glioblastoma, have not yet been defined. To compile a precise physical map for the region of common deletions in astrocytic tumors, we performed a high-density loss of heterozygosity (LOH) analysis using 31 polymorphic microsatellite markers spanning 22q in a series of grade II diffuse astrocytomas, anaplastic astrocytomas, primary glioblastomas, and secondary glioblastomas that had evolved from lower grade astrocytomas. LOH was found at one or more loci in 33% (12/36) of grade II diffuse astrocytomas, in 40% (4/10) of anaplastic astrocytomas, in 41% (26/64) of primary glioblastomas, and in 82% (23/28) of secondary glioblastomas. Characterization of the 22q deletions in primary glioblastomas identified two sites of minimally deleted regions at 22q12.3–13.2 and 22q13.31. Interestingly, 22 of 23 secondary glioblastomas affected shared a deletion in the same small (957 kb) region of 22q12.3, a region in which the human tissue inhibitor of metalloproteinases-3 (TIMP-3) is located. Investigation of the promoter methylation and expression of this gene indicated that frequent hypermethylation correlated with loss of TIMP-3 expression in secondary glioblastoma. This epigenetic change was significantly correlated to poor survival in eight patients with grade II diffuse astrocytoma. Our results suggest that a 957 kb locus, located at 22q12.3, may contain the putative TSG, TIMP-3, that appears to be relevant to progression to secondary glioblastoma and subsequently to the prognosis of grade II diffuse astrocytoma. In addition, the possibility of other putative TSGs on 22q12.3–13.2 and 22q13.31 that may also be involved in the development of primary glioblastomas cannot be ruled out.
Similar content being viewed by others


Main
Glioblastoma (WHO grade IV) is the most frequent and most malignant tumor of the human nervous system. Despite progress in surgery and adjuvant therapy, glioblastoma patients still have a very poor prognosis due to incomplete resection and resistance to radio- and chemotherapy.1 The majority of glioblastomas develop after a short clinical history and without clinical or histopathological evidence of a less malignant precursor lesion (primary or de novo glioblastoma). Another type of glioblastoma, the so-called secondary glioblastoma, progressively develops more slowly from low-grade diffuse (WHO grade II) or anaplastic astrocytoma (WHO grade III).1 Recent genetic analyses have shown that these glioblastoma subtypes develop through different genetic pathways;2, 3, 4 primary glioblastomas occur in older patients characterized by EGFR amplification/overexpression, PTEN mutation, homozygous p16INK4a deletion, and loss of heterozygosity (LOH) throughout chromosome 10,3, 5, 6 while secondary glioblastomas develop in younger patients and typically show frequent p53 mutations, LOH preferentially on chromosomes 10q and 19q,3, 5, 6, 7 and promoter methylation of the RB1 gene.8
LOH on 22q seems to be of interest, since this allelic loss is a common event in gliomas, occurring with a frequency varying between 11 and 39%. Interestingly, 22q LOH appears to be uncommon in low-grade astrocytomas, but is more frequently detected in anaplastic astrocytomas and glioblastomas. In addition to its link with glioma, 22q LOH correlates with the metastatic ability of pancreatic endocrine tumors.9 Further, allelic loss on 22q was rarely observed in ductal carcinomas, in situ, of the breast, but was observed in up to 66% of invasive ductal and 75% of lobular carcinomas,10, 11 and occurs more frequently in the advanced stage of various other cancers, suggesting an association between 22q LOH and tumor progression.
Recent deletion mapping in gliomas has demonstrated several common regions of overlap residing between the markers D22S533 and D22S689 spanning 22q11.23–12.1, between D22S280 and D22S282 spanning 22q12.3–13.2, and between D22S928 and D22S1169 spanning 22q13.31–13.32.12, 13, 14 So far, several genes mapped to 22q have been evaluated for their potential involvement. The neurofibromatosis type 2 (NF2) gene on 22q12.2 is responsible for NF2 syndrome, in which patients are predisposed to meningiomas, schwannomas, and ependymomas.15 While NF2 mutations have been found in sporadic schwannoma,16 meningioma,17 and spinal cord ependymomas,18 intracranial gliomas usually do not exhibit NF2 mutations.19 The hSNF5/INI1 gene, which has been shown to carry mutations predominately in rhabdoid tumors, maps to 22q11.2, far centromeric of the NF2 gene20 and shows up with a lower frequency in some central nervous system tumors such as medulloblastomas.21 Weber et al,22 however, failed to detect hSNF5/INI1 mutations in astrocytic tumors. 22q may therefore harbor yet unknown tumor suppressor genes (TSGs) relevant for astrocytic tumor development, especially for the malignant transformation of grade II diffuse astrocytoma.
The recent completion of the chromosome 22q sequencing project has permitted the prediction of unknown genes using computer-based approaches, demonstrating the value of a combined approach utilizing data from the human genome project, previous studies, and laboratory-based screening techniques. Using such combined methods, the objective of the present study was to analyze primary and secondary glioblastomas for LOH on 22q, which has been frequently observed in nonselected series of glioblastomas. We narrowed down the location of the putative TSG candidates in secondary glioblastomas, which led us to examine the epigenetic expression status of the tissue inhibitor of metalloproteinases-3 (TIMP-3) gene to investigate its role in astrocytic tumor progression. This study provides some evidence that TIMP-3 is likely to be at least one specific target for LOH on 22q12.3 and that this cytogenetic band is frequently involved in the progression from low-grade astrocytomas to secondary glioblastomas, but does not play a major role in the evolution of primary glioblastomas.
Materials and methods
Tumor Samples and DNA Extraction
In the period 1984–2003, a total of 325 adult supratentorial astrocytic tumors, comprising 67 grade II diffuse astrocytomas, 77 anaplastic astrcytomas, 163 primary glioblastomas, and 18 secondary glioblastomas were surgically removed in the Department of Neurosurgery, Nara Medical University. Tumor samples were fixed in 10% buffered formalin and embedded in paraffin. Pathological diagnosis was performed according to the WHO classification criteria.1 The tumors obtained by biopsy and located in basal ganglia, hypothalamus or thalamus were excluded. In addition, some cases were excluded because of limited availability of tumor or normal material. Suitable tumor series for the present study consisted of 28 grade II diffuse astrocytomas, eight anaplastic astrcytomas, 64 primary glioblastomas, and 18 secondary glioblastomas. To analyze more secondary glioblastomas, we identified patients from the database of our institution. A total of 10 secondary glioblastomas progressed from eight grade II diffuse astrocytomas or two anaplastic astrocytomas were retrieved from other affiliated hospitals. Specimens of 36 grade II diffuse astrocytomas, 10 anaplastic astrocytomas, 64 primary glioblastomas, and 28 secondary glioblastomas were finally examined with a full review of all available clinical data. In all, 64 patients with primary glioblastoma had a preoperative clinical history of less than 3 months (mean, 1.2 months) and histologic diagnosis of a glioblastoma at the first biopsy, without any evidence of a less malignant precursor lesion. A total of 28 patients with secondary glioblastoma had at least two biopsies taken at an interval of over 6 months to avoid a sampling error, with clinical and histologic evidence of progression from grade II diffuse astrocytoma (24 cases) or from anaplastic astrocytoma (four cases). DNA was extracted from paraffin sections, as described previously, to be used for microsatellite analysis.3, 6 Control genomic DNA was extracted from separate tissue blocks not involved with the tumor.
To support the clinical features of ‘primary’ and ‘secondary’ glioblastomas, we have examined the p53 gene mutations, which are considered as major events occurring in secondary glioblastomas, but not in primary glioblastomas.1, 3, 23 Prescreening for mutations by PCR-SSCP analysis was carried out in exons 5–8 of the p53 gene. Sequencing primers used were described previously.23 Samples that showed mobility shifts in SSCP analysis were further analyzed by DNA sequencing. After PCR amplification with the same set of primers, PCR products were sequenced on the Genetic Analyzer 310 (ABI, Foster City, CA, USA) using ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kits (ABI).
LOH Assay
In all, 31 highly polymorphic markers were selected from the Genome Database (http://gdbwww.gdb.org/) and the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/genemap/), their selection based on the published chromosome 22q sequence24 and on the frequency of heterozygosity, as well as by coverage and flanking of the region of interest. For each marker, the sense primer was labeled by a fluorescent dye, and paired normal and tumor DNA samples from each patient were amplified for 30 cycles with an annealing temperature of 56–58°C. Aliquots of the PCR reactions were then mixed with a size standard and formamide, denatured, and subjected to capillary electrophoresis on the Genetic Analyzer 310 (ABI, Foster City, CA, USA) and collected data analyzed with GENESCAN software (ABI). Analyses of each marker were repeated independently at least twice and showed a variation of no more than 3% in allelic ratios. Only samples heterozygous for a given locus were regarded to be informative; loci homozygosity and/or microsatellite instability rendered any particular sample noninformative. Samples were considered to show LOH when a peak allele signal from tumor DNA was reduced by 50% compared with the normal tissue counterpart.25
Chromosome 22q Sequence and Gene Analysis
Where possible, published polymorphic microsatellite markers were physically identified utilizing the July 2003 University of California Santa Cruz (UCSC) GoldenPath draft assembly (genome.cse.ucsc.edu). The names of the polymorphic microsatellites and their linear order, based on the consensus genetic and physical maps of 22q, are shown in Figures 1 and 2. The search for genes and gene candidates located within minimal common regions of deletion (MCRD) was assisted by the use of GeneMap’99 (http://www.ncbi.nlm.nih.gov/genemap99/) and the UCSC Genome Browser on Human, July 2003 Freeze (genome.cse.ucsc.edu).

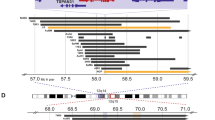
Analysis of allelic loss in chromosome 22q in 64 primary and 28 secondary glioblastomas. Only those cases showing partial allelic loss are shown. A G-banded ideogram of chromosome 22 is shown on the left, and the corresponding polymorphic loci are shown to the right of the ideogram. Case numbers are listed at the top. Rectangular areas indicate the cases showing LOH within the MCRD/S, MCRD/P1, and MCRD/P2, respectively. The results of immunohistochemistry (IHC) for TIMP-3 were recorded as −, positive staining in less than 10% of the tumor cells; +, positive staining in more than 10% of the tumor cells. MCRD, minimal common region of deletion; LOH/inf (%)*, the number of cases with LOH/ the number of informative cases (LOH frequency).

Analysis of 22q LOH in grade II diffuse astrocytomas (A II), anaplastic astrocytomas (AA III), and corresponding all secondary glioblastomas (SGB). Ideogram of chromosome 22 and the location and order of microsatellite markers are on the left, and case numbers are listed at the top. A thick vertical bar at the left refers to the MCRD/S, MCRD/P1, and MCRD/P2, respectively.
Methylation-Specific PCR
DNA methylation patterns in the CpG islands of TIMP-3 were determined by methylation-specific PCR (MSP).26 Sodium bisulfite modification was performed using a CpGenome™ DNA Modification Kit (Intergen, Oxford, UK) according to the manufacturer's protocol with minor modifications.8, 27, 28 The primer sequences, with methylated and unmethylated PCR and MSP conditions, have been previously reported.29 Amplified products were electrophoresed on 2% agarose gels and visualized by ethidium bromide staining.
Immunohistochemical Analysis of TIMP-3 Expression
Expression of TIMP-3 was assessed immunohistochemically, using a monoclonal anti-human TIMP-3 antibody (Oncogene, Cambridge, MA, USA). After deparaffinization, antigen retrieval was carried out using proteinase-K (5 μg/ml). They were then incubated with anti-TIMP-3 at a 1:40 dilution overnight at 4°C. Binding reactions were visualized using a Histofine SAB-PO kit and diaminobenzidine (Nichirei, Tokyo, Japan), and sections were counterstained with hematoxylin.
Statistical Methods
Fisher's exact test was used to examine possible associations between LOH at 22q12.3, TIMP-3 methylation, and TIMP-3 expression. Patient survival probability was calculated by the Kaplan–Meier method, and the significance of the difference between pairs of Kaplan–Meier curves was calculated using the log-rank procedure. Statistical significance was established as P<0.05.
Results
Patient Profiles
The median survival for patients in this series with grade II diffuse astrocytoma, anaplastic astrocytoma, primary glioblastoma, and secondary glioblastoma was 106, 47, 20, and 25 months, respectively. The length of follow-up ranged from 1 to 12.5 years. Recurrence or malignant transformation occurred in 89% of grade II diffuse astrocytoma patients with a median time to recurrence of 88 months, while 90% of anaplastic astrocytoma patients developed regrowth or transformation within a median of 21 months. The p53 mutations were detected in 23 (64%) grade II diffuse astrocytomas, five (50%) anaplastic astrocytomas, eight (13%) primary glioblastomas, and 21 (75%) secondary glioblastomas (data not shown).
Analysis of LOH
AS can be seen in Table 1, 12 of 36 (33%) grade II diffuse astrocytomas, four of 10 (40%) anaplastic astrocytomas, 26 of 64 (41%) primary glioblastomas, and 23 of 28 (82%) secondary glioblastomas exhibited LOH in at least one locus on 22q. Three astrocytomas, one anaplastic astrocytoma, five primary glioblastomas, and four secondary glioblastomas showed loss at every informative locus studied, suggesting a whole chromosome loss in each case. The respective frequencies (LOH cases/informative cases) are listed in Figure 1. The LOH patterns in the 21 primary glioblastomas with partial losses identified two MCRD at 22q12.3–13.2 (MCRD/P1) and at 22q13.31 (MCRD/P2) (Figure 1). The MCRD/P1 derived from five tumors (cases 14, 19, 39, 47, and 54) spanned four markers: D22S277, D22S283, D22S272, and D22S282; the MCRD/P2 derived from 11 tumors (cases 5, 14, 15, 19, 20, 30, 39, 44, 47, 52, and 55) spanned three loci; D22S274, D22S928, and D22S1170. A total of 21 primary glioblastomas exhibited LOH at both MCRD sites, whereas three tumors (cases 22, 41, and 63) had deletions only at MCRD/P1 and two others (cases 30 and 61) had deletions only at MCRD/P2 (Figure 1). The MCRD/P1 deletion spans a 7.5 Mb distance, and the MCRD/P2 deletion maps to a 3 Mb distance on the genetic map.
The MCRD in both grade II diffuse astrocytoma and secondary glioblastoma (MCRD/S) was defined with proximal and distal boundaries marked by D22S1176 and D22S1172, respectively (Figures 1 and 2). This corresponds to the cytogenetic location 22q12.3, and, according to the physical map of human chromosome 22, it comprises 957 kb between markers D22S273 and D22S280.24 The highest frequency of LOH (19 of 23, or 83% of informative secondary glioblastoma cases) was observed at D22S280. Representative cases are depicted in Figure 3. Constitual heterozygosity at D22S1176, 983 kb proximal to D22S280, was retained in cases 29, 50, 70, 73, and 78. Eight cases—28, 29, 48, 50, 59, 62, 72, and 73—retained heterozygosity at D22S1172, which is approximately 465 kb distal to D22S280. These results indicate that a locus at D22S280 in the chromosomal sub-band 22q12.3 may harbor a TSG within a physical map distance of 957 kb as defined by markers D22S273 and D22S280. Interestingly, all of these markers were significantly more frequently deleted in secondary glioblastomas than in other three types of astrocytic tumors. In contrast, 21 primary glioblastomas exhibited LOH at both MCRD/P1 and MCRD/P2, whereas five secondary glioblastomas revealed LOH at both MCRD sites, although there was no significant difference. In addition, seven primary glioblastomas exhibited extensive 22q LOH spanning three MCRD sites; this extensive LOH pattern was found in five secondary glioblastomas. This difference was also not significant.

GeneScan electrophorogram of several 22q markers for normal and tumor genotypes in three cases. The numbers in the right corner of each panel refer to the ratio of larger to shorter allele. Arrows indicate LOH. Markers are indicated at the bottom of the panels. (a) Primary glioblastoma case 47 shows LOH for D22S282 and D22S928, while retaining heterozygosity for D22S280. (b) For secondary glioblastoma case 59, D22S280 exhibits allelic loss, whereas D22S282 and D22S928 exhibit retention of both alleles. (c) Grade II diffuse astrocytoma case 48 retains heterozygosity for D22S1176 and D22S280, while the corresponding secondary glioblastoma shows allelic loss for these markers. Both of these tumors have retained heterozygous alleles for D22S1172.
TIMP-3 Promoter Hypermethylation
TIMP-3 promoter methylation was detected in eight of 36 (22%) grade II diffuse astrocytomas, two of 10 (20%) anaplastic astrocytomas, 18 of 64 (28%) primary glioblastomas, and in 20 of 28 (71%) secondary glioblastomas (Table 1, Figures 1 and 2). This epigenetic alteration was significantly more frequent in secondary glioblastomas than in other types of tumors analyzed (Table 1). In secondary glioblastomas, 18 out of 22 tumors (82%) showing MCRD/S also had methylation of the gene, whereas two out of six tumors (33%) without LOH were methylated in the promoter region (Figure 2). In contrast, only three out of 11 (27%) grade II diffuse astrocytomas, one of three anaplastic astrocyomas (33%), and three of nine (33%) primary glioblastomas demonstrating allelic loss in this region concurrently exhibited gene methylation.
All six secondary glioblastomas that had progressively developed from either a grade II diffuse astrocytoma or an anaplastic astrocytoma with TIMP-3 hypermethylation preserved the methylated sequence of this gene. In contrast, hypermethylation was indicated in 14 secondary glioblastomas that had originated from 13 grade II diffuse astrocytomas and one anaplastic astrocyoma showing nonmethylated promoter sequence. Methylated and unmethylated control DNAs showed the expected fragment sizes of 116 bp and 122 bp, respectively (Figure 4).

Results of methylation-specific PCR of CpG islands of the TIMP-3 promoter in glioblastomas. In patient 27, the precursor grade II diffuse astrocytoma (27 II) revealed unmethylated TIMP-3, whereas TIMP-3 in the corresponding secondary glioblastoma (27 IV) is methylated. In patient 49, promoter hypermethylation was found in both the low-grade precursor (49 II) and the progressively malignant samples (49 IV). In primary glioblastoma case 20 (20IV), only unmethylated TIMP-3 was present. N.C., normal control DNA from normal blood; P.C., positive control for methylated DNA; U, PCR product amplified by unmethylated-specific primers; M, PCR product amplified by methylated-specific primers.
TIMP-3 Immunohistochemistry
Cytoplasmic immunoreactivity to TIMP-3 was observed in more than half (the mean; ∼60%) of neurons and in glial cells in peritumoral brain tissues (24 cases), whereas TIMP-3 expression was detected at differential level of staining (20–50%, the mean; ∼30%) in four other specimens without tumor cells. The majority of grade II diffuse astrocytomas (31 of 36, 86%) showed TIMP-3 expression in more than 10% of tumor cells, whereas 18/28 (64%) secondary glioblastomas showed loss of the expression. Thus, the downregulation of the expression was detected during progression from grade II diffuse astrocytoma to secondary glioblastoma. For other tumors, eight of 10 (80%) anaplastic astrocytomas, and 53 of 64 (83%) primary glioblastomas showed TIMP-3 expression in more than 10% of tumor cells (Table 1, Figure 5).

(a) TIMP-3 immunohistochemistry showing cytoplasmic immunoreactivity in the majority of tumor cells from grade II diffuse astrocytoma (case 27). (hematoxylin counterstain, × 250). (b) In the same case, the progressed secondary glioblastoma exhibits grade II diffuse tumor cell clusters with loss of TIMP-3 expression. (hematoxylin counterstain, × 250). (c) The primary glioblastoma (case 20) shows extensive tumor cell areas with marked immunoreactivity for TIMP-3 (hematoxylin counterstain, × 250).
Methylation Status of TIMP-3 and Correlation with TIMP-3 Expression and LOH at MCRD/S
We detected a close correlation between the loss of TIMP-3 expression, as detected by immunohistochemistry, and concurrent promoter methylation with LOH at MCRD/S in secondary glioblastomas (P=0.0006). A total of 18 secondary glioblastomas with both methylation and LOH showed loss of TIMP-3 expression, whereas all four tumors with TIMP-3 expression showed an unmethylated gene status and retention at MCRD/S (Figure 2). No significant correlation between TIMP-3 expression and methylation status with or without LOH at this region was seen in other types of astrocytic tumors.
Correlation of TIMP-3 and LOH with Survival Data
No clear association between TIMP-3 methylation/LOH at MCRD/S and survival was detected in primary and secondary glioblastomas. We found that those patients with grade II diffuse astrocytomas revealing TIMP-3 methylation had a significantly shorter median survival when compared to those patients with tumors showing an unmethylated promoter sequence (91.5±9.9 months vs 113.6±4.7 months, respectively; P=0.0106). Similarly, those patients with grade II diffuse astrocytoma revealing TIMP-3 methylation indicated significantly shorter relapse-free survival compared to those patients with tumors showing unmethylated sequence (73.1±8.6 months vs 94.4±4.8 months, respectively; P=0.0189). Multivariate analysis showed that, after adjustment for age and extent of resection, TIMP-3 methylation was still associated with patient survival time (hazard ratio, 3.16; 95% CI, 1.12–8.97; P=0.030) and relapse-free survival (hazard ratio, 2.79; 95% CI, 1.02–7.69; P=0.047). However, the number of cases available for the grade II diffuse astrocytomas was limited and needs additional verification. There was also a tendency toward earlier death in patients with grade II diffuse astrocytomas showing 22q LOH as compared to those maintaining the alleles, although we could not find a statistically significant correlation (P=0.054, log-rank test), nor did loss or maintenance of expression correlate with survival in all types of astrocytic tumors. Figure 6 show the Kaplan–Meier survival curves for all 36 patients with grade II diffuse astrocytoma.

(a) The Kaplan–Meier curves of the probability of having no recurrence for 36 patients with grade II diffuse astrocytoma in relation to the methylation status. A statistically significant difference in recurrence rate was observed between the methylation-positive and methylation-negative groups (P=0.0189, log-rank test). M (+), methylated TIMP-3 group; M (−), unmethylated TIMP-3 group. (b) The Kaplan–Meier curves for overall survival (P=0.0106, log-rank test) in relation to the methylation status of all 36 patients with grade II diffuse astrocytoma. M (+), methylated TIMP-3 group; M (−), unmethylated TIMP-3 group.
Discussion
Utilizing a large number of microsatellite markers, we could show that 23 of 28 (82%) secondary glioblastomas reveal LOH on 22q. This is in contrast to three previous reports that observed 22q LOH in only ∼38% of non-selected glioblastomas. The D22S280 marker at cytogenic band 22q12.3 consistently exhibited the highest frequency of LOH (83%). The other frequently affected regions in our primary glioblastoma series were identified at 22q12.3–13.2 and 22q13.31, which overlaps previous data on gliomas.12, 13, 14 The distinct pattern of allelic loss identified in this chromosomal region appears to be an attractive candidate marker for further evaluation with regard to the discrimination between primary and secondary glioblastomas. Based on our data, one might hypothesize that loss of a purtative TSG on MCRD/S is more likely associated with progression to secondary glioblastoma rather than with primary glioblastoma.
The novel MCRD/S was not precisely recognized by three preceding studies. LOH on 22q is not a genome-wide event, however, and its extremely confined localization to MCRD/S may explain the discrepancy in frequency between our study and the previous ones. Using only three flanking markers -(D22S280 and D22S283 at 22q12.3 and D22S282 at 22q13.2—Ino et al12 mapped a common region of allelic loss between the D22S280 and D22S282 loci, but their series exhibited LOH predominantly in the more telomeric regions spanning 10 Mb of chromosome 22. Compared with the microsatellite markers used previously, markers used in this study were located at different sites at both ends of the D22S280 locus, and LOH was found to be restricted to a more centromeric marker. Both of the other related studies performed in nonselected glioblastoma failed to find evidence for the MCRD/S.13, 14 This could be explained if recognition of the MCRD/S requires inclusion of sufficient numbers of secondary glioblastoma cases and suitable microsatellite markers as was the case in our analyses.
Secondary glioblastomas could be tentatively categorized into two groups based upon the comparative 22q LOH profiles of the corresponding progenitor grade II diffuse astrocytomas. The first subgroup of the tumors already harbored allelic loss in the low-grade phenotype and did not acquire additional allelic loss during the progression to glioblastoma. These data indicate that progression of astrocytoma toward a more aggressive malignant phenotype is associated with an accumulation of genetic changes other than those on 22q. In the second subset, that is, those with no detectable LOH at first presentation, recurrent tumors showed additional allelic loss at 22q12.3 correlated with pathways of linear progression. Taking into account all these considerations, it is possible that alterations in the putative TSG(s) on the locus may represent a late event for tumor progression in the majority of grade II diffuse astrocytomas or possibly an early event in those less prevalent neoplasms with malignant potential.
The human chromosome 22 sequence has been completed and released into the public domain.24 Polymorphic markers are now physically mapped at nucleotide-level resolution; the availability of this information is thus useful for the eventual identification of those genes previously mapped to this chromosome in sporadic neoplasms by allelic deletion assays. Using the UCSC genome browser based on the July 2003 assembly, candidate genes were reviewed after defining a 1-Mb minimal region of deletion. Interestingly, the marker D22S280, which exhibited the highest frequency of LOH in our study, is in close relationship to two known genes, synapsin 3 (SYN3) and TIMP-3. SYN3 belongs to a family of neuron-specific synaptic vesicle-associated phosphoproteins that have been implicated in synaptogenesis and in the modulation of neurotransmitter release, but no association with tumor development has yet been identified.30 The second gene, TIMP-3, is embedded within an intron of the SYN3 gene. TIMP-3 is a member of the family of multifunctional secreted proteins that blocks the activity of metalloproteinases, therefore regulating matrix composition, cell growth, invasion and migration.31 It has also been shown to inhibit vascular endothelial growth factor (VEGF)-mediated angiogenesis and to induce apoptosis.32, 33 TIMP-3 thus appears to be an attractive candidate for genetic analysis of a potential role in the tumorigenesis of secondary glioblastoma.
Methylation-associated gene silencing and loss of expression has been demonstrated in various different cancers, including cancers of the kidney and lung.29, 34 Our methylation analysis also suggested the potential relation between loss of the expression and aberrant hypermethylation in a subset of astrocytic tumors. The promoter region of TIMP-3 was methylated in the majority (82%) of secondary glioblastomas with 22q12.3 loss and we demonstrated that promoter methylation is significantly associated with reduced TIMP-3 expression. A minority of the primary glioblastoma and grade II diffuse astrocytoma samples with allelic loss in this specific region of 22q, however, revealed TIMP-3 promoter methylation, but no correlation with immunohistochemical expression. We also saw a higher frequency of hypermethylation in secondary glioblastoma than in primary glioblastoma. This suggest that methylation of the gene may not be of major importance in primary glioblastoma—at least, in tumors without LOH—and silencing of this gene may contribute more to progression to secondary glioblastoma.
The situation in secondary glioblastoma may be different. Hypermethylation was found in a number of secondary glioblastomas, but not in the corresponding lower grade precursor tumor. LOH of 22q is a relatively early event in grade II diffuse astrocytomas, whereas hypermethylation is a later event. In this case, hypermethylation and inactivation of this gene in grade II diffuse astrocytomas that have already lost a 22q allele might contribute more effectively to the development of a more malignant phenotype. In this respect, detection of TIMP-3 alteration and/or allelic loss at 22q12.3 might be a useful screening tool for identifying and understanding astrocytoma progression. As a result of the small number of patients with secondary glioblastoma analyzed and the relatively short median survival of 25 months in this study, we were able to show only a trend toward TIMP-3 methylation and diminished survival; we could not provide statistically significant correlations. When the methylation of this gene was detected in grade II diffuse astrocytomas, however, a statistically significant correlation between methylation status, disease-free survival, and overall survival became apparent. This suggests that the frequency of TIMP-3 methylation reflects disease progression and that accumulating methylation can serve as a sort of molecular clock in astrocytoma progression.
We should point out, however, that the modest number of cases with the gene alteration and/or allelic loss evaluated here does not permit definitive conclusions regarding the role of methylation in the gene and LOH on the region in primary glioblastoma. Our results indicate that TIMP-3 is epigenetically altered only in a small subset of primary glioblastomas in spite of the fact that LOH on 22q is observed in a large number of these tumors. A considerable fraction of chromosomal deletion in primary glioblastoma is due to a high degree of genetic instability and may not relate to specific gene silencing.35 Therefore, one or more still undefined genes on 22q could be the major TSG target(s) of inactivation by allelic loss.
Although most primary glioblastomas showed a comparatively wide common deletion from D22S277 to D22S1170, three tumors demonstrated two different deleted regions with retention of an intact segment between them. This suggests that two putative TSG loci could be located in a large interval on the distal portion of 22q. The MCRD/P1 region spans a 7.5 Mb physical distance at 22q12.3–13.2 and overlaps with a previously detected deletion area.12, 14 The other MCRD/P2 region was confined to a 3 Mb physical distance at 22q13.31 that partially overlaps with an area of deletion previously detected by Oskam et al13 The cytogenetic band at 22q13.1–3 appears to be a focal ‘hot spot’ for LOH in many types of human cancer, including breast, colon, ovarian, and head and neck cancers.11, 36, 37, 38, 39 Lida et al10 were, in fact, able to map a 2 cM region of allelic loss in breast cancer to the same cytogenetic band. Similarly, Allione et al11 reported six regions along chromosome 22q, ranging from 3 to 6 cM in size, two of which overlap those intervals identified in this study, and, in oral squamous cell carcinoma, allelic deletion seems to be restricted to D22S274,39 a marker included within the region of deletion identified in our study. Thus, the commonality and number of deletions at 22q13.1–3 suggest the presence of either a cluster of TSGs, each of which is involved in one tumor type, or a few TSGs involved in the etiology of many tumor types. The UCSC genome browser further indicates that the MCRD/P1 and MCRD/P2 contain 108 and 11 genes/gene candidates, respectively. Several of these genes are likely to be involved in cellular proliferation and/or differentiation and may constitute excellent eligibility for a predicted primary glioblastoma gene, including MKL1, EP300, and DJ1042K10.2, within MCRD/P1. MKL1 participates in a fused gene product generated by a translocation event in acute megakaryoblastic leukemia.40 The EP300 transcriptional coactivator protein is specifically inhibited by the adenovirus oncoprotein E1A and somatic mutations linked to EP300 alterations have been reported in a small proportion of colorectal, breast and pancreatic cancers.41 The genes DJ1042K10, with a RUN domain that was found in Rap and Rab GTPases. Hartmann et al,14 however, did not find evidence of somatic mutations in EP300, and DJ1042K10 and MKL1 in a series of astrocytic tumors, although epigenetic silencing of those genes was not excluded.
References
Kleihues P, Cavenee WK . Pathology and Genetics of Tumours of the Nervous System. International Agency for Research on Cancer (IARC Press): Lyon, 2000.
Lang FF, Miller DC, Koslow M, et al. Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg 1994;81:427–436.
Watanabe K, Tachibana O, Sata K, et al. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 1996;6:217–223; discussion 223–214.
von Deimling A, von Ammon K, Schoenfeld D, et al. Subsets of glioblastoma multiforme defined by molecular genetic analysis. Brain Pathol 1993;3: 19–26.
Tohma Y, Gratas C, Biernat W, et al. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol 1998;57:684–689.
Fujisawa H, Reis RM, Nakamura M, et al. Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Lab Invest 2000;80:65–72.
Nakamura M, Yang F, Fujisawa H, et al. Loss of heterozygosity on chromosome 19 in secondary glioblastomas. J Neuropathol Exp Neurol 2000;59:539–543.
Nakamura M, Yonekawa Y, Kleihues P, et al. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest 2001;81:77–82.
Wild A, Langer P, Celik I, et al. Chromosome 22q in pancreatic endocrine tumors: identification of a homozygous deletion and potential prognostic associations of allelic deletions. Eur J Endocrinol 2002;147:507–513.
Iida A, Kurose K, Isobe R, et al. Mapping of a new target region of allelic loss to a 2-cM interval at 22q13.1 in primary breast cancer. Genes Chromosomes Cancer 1998;21:108–112.
Allione F, Eisinger F, Parc P, et al. Loss of heterozygosity at loci from chromosome arm 22Q in human sporadic breast carcinomas. Int J Cancer 1998;75:181–186.
Ino Y, Silver JS, Blazejewski L, et al. Common regions of deletion on chromosome 22q12.3–q13.1 and 22q13.2 in human astrocytomas appear related to malignancy grade. J Neuropathol Exp Neurol 1999;58:881–885.
Oskam NT, Bijleveld EH, Hulsebos TJ . A region of common deletion in 22q13.3 in human glioma associated with astrocytoma progression. Int J Cancer 2000;85:336–339.
Hartmann C, Numann A, Mueller W, et al. Fine mapping of chromosome 22q tumor suppressor gene candidate regions in astrocytoma. Int J Cancer 2004; 108:839–844.
Martuza RL, Eldridge R . Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med 1988;318: 684–688.
Jacoby LB, MacCollin M, Louis DN, et al. Exon scanning for mutation of the NF2 gene in schwannomas. Hum Mol Genet 1994;3:413–419.
Ueki K, Wen-Bin C, Narita Y, et al. Tight association of loss of merlin expression with loss of heterozygosity at chromosome 22q in sporadic meningiomas. Cancer Res 1999;59:5995–5998.
Ebert C, von Haken M, Meyer-Puttlitz B, et al. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol 1999;155:627–632.
Rubio MP, Correa KM, Ramesh V, et al. Analysis of the neurofibromatosis 2 gene in human ependymomas and astrocytomas. Cancer Res 1994;54:45–47.
Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394:203–206.
Sevenet N, Lellouch-Tubiana A, Schofield D, et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet 1999;8:2359–2368.
Weber M, Stockhammer F, Schmitz U, et al. A mutational analysis of INI1 in sporadic human brain tumors. Acta Neuropathol (Berl) 2001;101:479–482.
Nakamura M, Watanabe T, Yonekawa Y, et al. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C → A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis 2001;22:1715–1719.
Dunham I, Shimizu N, Roe BA, et al. The DNA sequence of human chromosome 22. Nature 1999; 402:489–495.
Nakamura M, Kishi M, Sakaki T, et al. Novel tumor suppressor Loci on 6q22–23 in primary central nervous system lymphomas. Cancer Res 2003;63:737–741.
Herman JG, Graff JR, Myohanen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996;93:9821–9826.
Nakamura M, Sakaki T, Hashimoto H, et al. Frequent alterations of the p14ARF and p16INK4a genes in primary central nervous system lymphomas. Cancer Res 2001;61:6335–6339.
Nakamura M, Watanabe T, Klangby U, et al. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol 2001;11:159–168.
Bachman KE, Herman JG, Corn PG, et al. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res 1999;59: 798–802.
Kao HT, Porton B, Czernik AJ, et al. A third member of the synapsin gene family. Proc Natl Acad Sci USA 1998;95:4667–4672.
Birkedal-Hansen H, Moore WG, Bodden MK, et al. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med 1993;4:197–250.
Ahonen M, Baker AH, Kahari VM . Adenovirus-mediated gene delivery of tissue inhibitor of metalloproteinases-3 inhibits invasion and induces apoptosis in melanoma cells. Cancer Res 1998;58: 2310–2315.
Qi JH, Ebrahem Q, Moore N, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med 2003;9:407–415.
Michael M, Babic B, Khokha R, et al. Expression and prognostic significance of metalloproteinases and their tissue inhibitors in patients with small-cell lung cancer. J Clin Oncol 1999;17:1802–1808.
von Deimling A, Fimmers R, Schmidt MC, et al. Comprehensive allelotype and genetic anaysis of 466 human nervous system tumors. J Neuropathol Exp Neurol 2000;59:544–558.
Castells A, Gusella JF, Ramesh V, et al. A region of deletion on chromosome 22q13 is common to human breast and colorectal cancers. Cancer Res 2000;60:2836–2839.
Reis PP, Rogatto SR, Kowalski LP, et al. Quantitative real-time PCR identifies a critical region of deletion on 22q13 related to prognosis in oral cancer. Oncogene 2002;21:6480–6487.
Lin H, Pizer ES, Morin PJ . A frequent deletion polymorphism on chromosome 22q13 identified by representational difference analysis of ovarian cancer. Genomics 2000;69:391–394.
Miyakawa A, Wang XL, Nakanishi H, et al. Allelic loss on chromosome 22 in oral cancer: possibility of the existence of a tumor suppressor gene on 22q13. Int J Oncol 1998;13:705–709.
Ma Z, Morris SW, Valentine V, et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 2001; 28:220–221.
Gayther SA, Batley SJ, Linger L, et al. Mutations truncating the EP300 acetylase in human cancers. Nat Genet 2000;24:300–303.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (#15590311).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakamura, M., Ishida, E., Shimada, K. et al. Frequent LOH on 22q12.3 and TIMP-3 inactivation occur in the progression to secondary glioblastomas. Lab Invest 85, 165–175 (2005). https://doi.org/10.1038/labinvest.3700223
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700223
Keywords
This article is cited by
-
The novel histone deacetylase inhibitor pracinostat suppresses the malignant phenotype in human glioma
Molecular Biology Reports (2022)
-
Tissue inhibitor of matrix metalloproteinase-3 has both anti-metastatic and anti-tumourigenic properties
Clinical & Experimental Metastasis (2020)
-
Timp3 deficiency affects the progression of DEN-related hepatocellular carcinoma during diet-induced obesity in mice
Acta Diabetologica (2019)
-
IL-32 gamma reduces lung tumor development through upregulation of TIMP-3 overexpression and hypomethylation
Cell Death & Disease (2018)
-
TIMPs: versatile extracellular regulators in cancer
Nature Reviews Cancer (2017)
