
Abstract
Inhibition of glutamate carboxypeptidase II (GCP II; NAALADase) produces a variety of effects on glutamatergic neurotransmission. The aim of this study was to investigate effects of GCP II inhibition with the selective inhibitor, 2-PMPA, on: (a) development of tolerance to the antinociceptive effects, (b) withdrawal, and (c) conditioned reward produced by morphine in C57/Bl mice. The degree of tolerance was assessed using the tail-flick test before and after 6 days of twice daily (b.i.d.) administration of 2-PMPA and 10 mg/kg of morphine. Opioid withdrawal was measured 3 days after twice daily morphine (30 or 10 mg/kg) administration, followed by naloxone challenge. Conditioned morphine reward was investigated using conditioned place preference with a single morphine dose (10 mg/kg). High doses of 2-PMPA inhibited the development of morphine tolerance (resembling the effect of 7.5 mg/kg of the NMDA receptor antagonist, memantine) while not affecting the severity of withdrawal. A high dose of 2-PMPA (100 mg/kg) also significantly potentiated morphine withdrawal, but inhibited both acquisition and expression of morphine-induced conditioned place preference. Memantine inhibited the intensity of morphine withdrawal as well as acquisition and expression of morphine-induced conditioned place preference. In addition, 2-PMPA did not affect learning or memory retrieval in a simple two-trial test, nor did it produce withdrawal symptoms in morphine-dependent, placebo-challenged mice. Results suggest involvement of GCP II (NAALADase) in phenomena related to opioid addiction.
Similar content being viewed by others


INTRODUCTION
Over the last decade, research has provided compelling evidence that glutamate receptors are crucially involved in the phenomena related to opioid tolerance, dependence, and reward (Bisaga and Popik, 2000). Glutamate, a major excitatory neurotransmitter in the brain, stimulates both ionotropic and metabotropic glutamate receptors (Monaghan et al, 1989; Conn and Pin, 1997). Antagonists of the ionotropic N-methyl-D-aspartate (NMDA) receptor complex, including memantine, the low affinity, and highly voltage-dependent clinically available NMDA receptor antagonist, inhibit the development of morphine tolerance (Trujillo and Akil, 1991; Marek et al, 1991; Popik et al, 2000a) and reverse pre-existing tolerance so that opiate-tolerant animals treated with NMDA receptor antagonists become sensitive to doses of morphine that previously did not evoke antinociception (Tiseo and Inturrisi, 1993; Popik et al, 2000a). Antagonists of this receptor complex also attenuate the development (Trujillo and Akil, 1991), expression (Cappendijk et al, 1993), and maintenance (Popik and Skolnick, 1996) of the continuing morphine dependence. The inhibitory effect on the expression of morphine dependence has also been demonstrated for memantine (Popik and Skolnick, 1996) (Bisaga and Popik, 2000).
The current work demonstrates similar effects of compounds that inhibit the function of metabotropic receptors. Thus, an agonist of group II metabotropic receptors for glutamate (mGluRII), (+)-2-aminobicyclo [3,1,0] hexane-2,6-dicarboxylic acid (LY354740), has been shown to inhibit the development of morphine tolerance (Popik et al, 2000b) as well as the expression of morphine dependence (Klodzinska et al, 1999; Vandergriff and Rasmussen, 1999).
In addition to the effects on opioid tolerance and dependence, NMDA receptor antagonists inhibit conditioned reward produced by morphine, a phenomenon thought to be related to drug addiction (Koob and LeMoal, 2001). Conditioned reward can be studied using the conditioned place preference test wherein compounds may affect its acquisition, expression, or both. While the effects on acquisition provide heuristic insight into the role of a given neurotransmitter pathway in the development of conditioned reward, the inhibitory effects on its expression can be regarded as having potential therapeutic impact in the treatment of drug addiction. NMDA receptor antagonists, including memantine, have been shown to inhibit the acquisition of conditioned place preference produced by opiates (Bespalov et al, 1994; Tzschentke and Schmidt, 1995; Popik et al, 1998; Kotlinska and Biala, 1999; Popik and Danysz, 1997) as well as its expression (Bespalov et al, 1994; Tzschentke and Schmidt, 1997; Popik and Danysz, 1997; Popik et al, 1998).
Another, perhaps, more ‘physiological’ way of attenuating glutamate neurotransmission could potentially be achieved by inhibiting the metabolism of N-acetyl-aspartyl-glutamate (NAAG) (Slusher et al, 1999), an endogenous dipeptide, present in the brain in millimolar (0.5–2.7 mM) concentrations (Pouwels and Frahm, 1997) that has been immunohistochemically localized to neurons, particularly those known to be glutamatergic (Williamson and Neale, 1988; Tsai et al, 1990,1993). NAAG has been hypothesized to be involved in neuronal communication as a neurotransmitter, neuromodulator, and precursor of glutamate (Blakely and Coyle, 1988). NAAG is released from neurons after depolarization by a calcium-dependent process upon synaptic stimulation (Tsai et al, 1990; Neale et al, 2000), suggesting its neurotransmitter-like properties.
Importantly, NAAG is hydrolyzed by the neuropeptide glutamate carboxypeptidase (GCP II; EC 3.4.17.21) [N-acetylated-α-linked-acidic dipeptidase (NAALADase)] to liberate N-acetyl-aspartate (NAA) and glutamate (Stauch et al, 1989), and the activity of this enzyme can be inhibited by the recently developed specific inhibitors (Jackson and Slusher, 2001). NAAG itself has been shown to act as a low-potency agonist at NMDA receptors (Koenig et al, 1994; Sekiguchi et al, 1992; Westbrook et al, 1986), and thus, according to these data, an inhibition of its metabolism might result in stimulation of NMDA receptors. However, in many other systems it has been shown to antagonize the effects of NMDA receptor activation (Burlina et al, 1994; Grunze et al, 1996; Puttfarcken et al, 1993), and therefore, according to these findings, an inhibition of its metabolism would actually inhibit NMDA receptors. Thus, as noted by Yamamoto et al (2001a) NAAG acts as an NMDA receptor antagonist at low concentrations but as a low-potency NMDA receptor agonist at high concentrations, and can be regarded as a mixed agonist/antagonist at the NMDA receptor depending on its concentration (Bruno et al, 1998; Thomas et al, 2000).
In addition, an increase in NAAG concentration may decrease glutamatergic tone mediated by presynaptic mGluRII receptors (mGluR3), because another line of evidence indicates that NAAG is an agonist at mGluRII receptors (Wroblewska et al, 1993; Wroblewska et al, 1997) with EC50∼26 μM (Tortella et al, 2000). Lastly, the inhibition of metabolism of NAAG to glutamate could directly produce a reduction of extracellular concentration of glutamate, and via this action may then attenuate the stimulation of both ionotropic and metabotropic receptors for glutamate.
The pharmacological effects of inhibition of GCP II activity have not been investigated until recently, when specific and potent inhibitors of this enzyme were developed. Among them is 2-phosphonomethyl pentanedioic acid (2-PMPA) (Jackson et al, 1996) that potently inhibits GCP II activity with an inhibition constant (Ki) of 0.3 nM. 2-PMPA is selective for GCP II with no apparent affinity for over 100 different receptors, ion channels, transporters, and enzymes including several glutamatergic sites such as NMDA, AMPA, metabotropic glutamate receptors, and glutamate transporters (Slusher et al, 1999).
As a result of its potency and apparent specificity for GCP II, we used 2-PMPA as a prototype compound to explore the role of GCP II inhibition in the development of morphine tolerance, the expression of morphine dependence, and the acquisition and expression of conditioned reward produced by morphine.
METHODS
Subjects
Male C57/BL mice (IMP, Lodz, Poland), 22–24 g of body weight, were group-housed in the standard laboratory cages and kept in a temperature-controlled colony room (21±2°C) with a 12-h light/dark cycle (light on: 07:00, off: 19:00). Commercial food and tap water were available ad libitum. Each experimental group consisted of 7–28 mice per treatment. All mice were used only once.
Apparatus for Experiments 1 and 2
A standardized tail-flick analgesia meter (Columbus, Ohio, USA, model 33), adjusted to a sensitivity of ‘10’ with radiant heat source and connected to an automatic timer was used to assess antinociceptive responses. The intensity of the heat stimulus was adjusted so that the baseline tail-flick latency was ∼3 s. A maximum latency of 10 s (ie cutoff) was used to minimize damage to the tail. The tail withdrawal latency was measured from the start of heat stimulus until the mouse exhibited a flick of the tail. Each response assessment consisted of two separate measurements taken at different portions of the tail (spaced by 1.5–2 cm) and separated by 15 s. The mean of these responses was used for subsequent comparisons.
Morphine antinociceptive potency was investigated with the use of cumulative dose–response curves that allowed for minimization of the animal number used (Paronis and Holtzman, 1991). After adaptation and baseline trials, each mouse was injected s.c. with a low dose of morphine (1 mg/kg). After 30 min, the mouse was retested and injected with the next dose of morphine that was increased by quarter of a log unit. Thus, because the initial dose of morphine was 1.0 mg/kg, the next dose was 1.78 mg/kg, for a cumulative dose of 2.8 mg/kg. This procedure continued until either the mouse did not move its tail within the cutoff time or until there was a plateauing of the dose–response curve, so that the latency did not increase from one dose to the next. Each analgesic responder was not subjected to further tail-flick assessments but was injected with the subsequent dose of morphine, so that every animal received the same total dose of morphine during a given test.
Apparatus for Experiment 4
The place-preference apparatus consisted of three rectangular arms (30×15×20 cm) spaced at 120° from each other, which were all accessible from a triangular (central) platform (Stinus et al, 1990). The apparatus was made of Metaplex and the three arms differed in distinctive visual, tactile, and olfactory cues. Thus, the white arm had a black floor with small holes in it and was marked with peppermint odor, the one black arm with white rough floor was marked with anise odor and, the other black arm with plain black floor had no odor. These distinct cues served as conditioned stimuli (CSs). The use of tactile, texture floor cues allowed mice to be in direct contact with a CS to experience its conditioned effect during preference testing (Vezina and Stewart, 1987 (cf. Cunningham et al, 1992)). The guillotine doors, colored according to the respective wall colors, were inserted during conditioning sessions and removed during the pre-test and post-test. The ceiling of the three arms was made of transparent Plexiglass. During testing, mouse location was monitored through a closed circuit TV camera positioned directly above the apparatus. The testing room had dim indirect lighting, comprising two 15 W bulbs positioned about 1 m above the apparatus. A loudspeaker, also positioned above the apparatus, delivered white noise. The apparatus was kept free of urine and faeces; the floors were repeatedly washed and dried.
Apparatus for Experiment 5
The elevated plus maze (Lister, 1987), made of black painted plywood and consisting of two open arms (5×30 cm) and two enclosed arms (5×30×15 cm), was used to study the effect of 2-PMPA on learning and memory retrieval in mice. The arms extended from a central platform (5×5 cm). The apparatus was elevated to a height of 50 cm above the floor. The open arms were illuminated with two bright lamps. Testing was carried out in an experimental room supplied with a white noise by an experimenter blind to the treatment conditions.
Experimental Design
Effects on morphine tolerance (Experiment 1) and acute effects in the tail-flick test (Experiment 2)
Experiment 1 was carried out to investigate the effect of 2-PMPA on the development of morphine tolerance. On day 1 (test 1), the first measurement of morphine antinociceptive potency was performed, followed by 6 days of b.i.d morphine injections (10 mg/kg, s.c., 7:30 and 17:30) (Elliott et al, 1994; Popik et al, 2000b). Pretreatment with 2-PMPA (30, 50, or 100 mg/kg, i.p.) or memantine (7.5 mg/kg, s.c., a ‘positive control’) was given at 30 min prior to each morphine dose on days 2–7. On day 8 (test 2), the second measurement of morphine antinociceptive potency was carried out. The degree of morphine tolerance was assessed by comparing the morphine antinociceptive potencies (cumulative dose–response curves) obtained in tests 1 and 2.
Experiment 2 was designed to determine whether 2-PMPA might itself produce antinociceptive effects and/or affect the antinociceptive effects of morphine. Morphine (1.5 or 3 mg/kg, s.c.) was administered 30 min after injection of 100 mg/kg of 2-PMPA or placebo, administered i.p. The 3 mg/kg dose of morphine corresponds to the antinociceptive ED50 dose in these test conditions (data not shown).
Effects on morphine withdrawal (Experiment 3)
One week after their arrival to the laboratory, mice started receiving twice daily (b.i.d) s.c. injections of morphine, 30 mg/kg, (7:30 and 17:30) for 3 days as described previously (Popik and Skolnick, 1996). An additional dose of morphine was administered on the morning (07:00 h) of the test day (day 4). One and a half hours later, mice received various doses of 2-PMPA, memantine (7.5 mg/kg, used here as a ‘positive control’), or saline (placebo). Then, 30 min later (ie 2 h after the last dose of morphine), mice were challenged with 4 mg/kg of naloxone (or placebo) (all injections via i.p. route). Immediately afterwards, subjects were placed singularly into glass beakers (21 cm of diameter, 41 cm of height). The typical response of morphine-dependent mouse to naloxone is intense jumping, and thus the number of jumps to the height of 7 cm or more was counted for 10 min by an experimenter blind to the treatment conditions.
Since in Experiments 1 and 4 morphine was used at a dose of 10 mg/kg, the possibility that 100 mg/kg of 2-PMPA might differently affect morphine withdrawal produced by 10 mg/kg of morphine was investigated.
Effects on morphine-induced conditioned place preference (CPP) (Experiment 4)
The experiment was carried out according to an unbiased procedure and consisted of five phases (days): (1) adaptation, (2) pre-test, (3) conditioning with morphine, (4) conditioning with placebo, and (5) post-test. During the adaptation period, mice were carried into the testing room, weighed and handled by the experimenter by moving animals from one standard home cage to another in the proximity of the apparatus. This adaptation phase was intended to reduce the novelty and stress associated with handling, injections, and exposure to the apparatus. During the pre-test, mice were placed individually on the central triangular platform of the apparatus with free access to all three arms for 20 min. The time spent in each arm and the number of arm entries were recorded. For all mice, the two arms registering the most similar preferences were identified and one was then paired with morphine and the other with placebo. After assigning the arms, there were no significant differences between time spent in the ‘to-be’ morphine-paired and the placebo-paired arms during the preconditioning phase. This is an important step in the experimental procedure that avoids any preference bias before conditioning (Manzanedo et al, 2001).
During conditionings, mice randomly assigned to treatment groups were pretreated with placebo, 2-PMPA (100 mg/kg) or memantine (7.5 mg/kg, a ‘positive control’), and injected with morphine (10 mg/kg) or placebo (or, to investigate its putative aversive effects, with 100 mg/kg of 2-PMPA), 20 min before being confined to the respective compartment. The post-test was carried out similarly to the pre-test, with the mice being placed individually on the central triangular platform of the apparatus with free access to all arms for 20 min following treatment with placebo, 2-PMPA (100 mg/kg), or memantine (7.5 mg/kg). The time spent in each arm and the number of arm entries were recorded. The third arm was visited only during the pre-test and post-test (Mucha and Iversen, 1984).
Effects of 2-PMPA on learning and retrieval of memory (Experiment 5)
Although the elevated plus maze has been used mainly in research on anxiety, it also allows investigation of the cognitive effects of drugs. The procedure used was similar to that described by Itoh et al (1991). On the first test, mice were individually placed at the end of one open arm facing away from the central platform. The latency of each mouse to find and enter with its four paws one of the enclosed arms was measured (transfer latency 1 [TL1]). Mice were allowed to explore freely the apparatus for the following 10 s. After 24 h, the second test was carried out. As in the first test, mice were individually placed at the end of one open arm facing away from the central platform and the latency of each mouse to enter one of the enclosed arms was measured again (TL 2). After each mouse, the apparatus was cleaned and dried.
To investigate the effects on learning, mice were pretreated with placebo, 50 and 100 mg/kg of 2-PMPA, or 0.1 mg/kg of MK-801 (a positive control), 20 min before the first test. To evaluate the effects on memory retrieval, mice were treated with 2-PMPA (50 and 100 mg/kg) 20 min before the second test. This closely mimicked the drug treatment of the place preference procedure (see above, Experiment 4). To keep the number and sequence of injections equal among various groups, control mice were treated with placebo instead of active compounds.
Drugs
Morphine HCl (Polfa, Krakow), naloxone HCl, MK-801 [(+)-5-methyl-10,11-dihydroxy-5H-dibenzo(a,d)cyclohepten-5,10-imine maleate] (Sigma-Aldrich St. Louis, MO, USA), and memantine HCl (generous gift of Professor Wojciech Danysz, MERZ & CO, Germany) were dissolved in sterile physiological saline that served as placebo. 2-phosphonomethyl pentanedioic acid (2-PMPA) was synthesized by Guilford Pharmaceuticals as described previously (Jackson et al, 1996) and was dissolved in sterile distilled water and the pH was adjusted to 6±0.25 with 1N NaOH. 2-PMPA was stored at −20°C. All drugs were made fresh the day before experiment and stored at 4°C in the refrigerator. The dose of morphine is expressed as the base, the doses of all other compounds as their respective salts. Doses of morphine, naloxone and memantine were used based on our previous observations (Popik and Skolnick, 1996; Popik et al, 2000b). All compounds were administered in the volume of 10 ml/kg.
Data Presentation and Statistics
For the morphine tolerance study (Experiment 1), latencies (in s) of the tail-flick responses were converted to maximum possible effects (MPEs) (Paronis and Holtzman, 1991), according to the formula: 100×[(postinjectory latency−baseline latency)/(cut-off latency−baseline latency)]. MPE values were used to construct morphine cumulative dose–response curves by nonlinear regression; these curves were used to calculate antinociceptive ED50 values using GraphPad Prism ver. 3.00 (GraphPad Software, CA, USA) software. The ED50 values obtained on tests 1 and 2 were compared among groups, as were the fold shifts (determined by dividing individual test 2 ED50 values by the test 1 ED50 values) with one-way ANOVAs and post hoc Newman–Keul's test.
Effects of 2-PMPA with or without morphine on tail-flick responses (Experiment 2) were compared with the use of area under curve (AUC) assessments on MPE values, which were calculated using trapezoid rule (ΔX*(Y1+Y2)/2) on a series of measurements from 0 to 120 min. These data were subjected to one-way ANOVA and post hoc Newman–Keul's test.
Data from the morphine dependence study (Experiment 3) are expressed as mean±SEM number of jumps per 10 min. Statistical analyses involved one way between subjects ANOVA followed by Newman–Keul's test and Student's t-test.
To assess the effects of 2-PMPA and memantine on acquisition and expression of morphine-induced conditioned place preference (Experiment 4), statistical analysis involved one-way ANOVA followed by Dunnett's test. The magnitude of CPP was assessed as the % preference (pre-test=100%, post-test=X%, (Popik and Danysz, 1997)).
The effects of 2-PMPA and MK-801 on acquisition and retrieval of memory (Experiment 5) were assessed with one-way ANOVAs and post hoc Dunnett's test. The degree of shortening entrance latency is presented for each group by calculating the % difference (TL1=100%, TL2=X%).
Ethics
All experiments were carried out according to the National Institutes of Health Guide for Care and Use of Laboratory Animals (publication No. 85-23, revised 1985) and were approved by the Institute of Pharmacology Animal Care and Use Bioethics Commission.
RESULTS
Effects of 2-PMPA on Development of Morphine Tolerance (Experiment 1)
There were no differences in antinociceptive morphine ED50 values on test 1 among groups (Table 1). Treatment with 10 mg/kg b.i.d. of morphine produced a 6.44-fold increase in the ED50 values as determined on test 2. In contrast, pretreatment with memantine, 50 or 100 (but not 30) mg/kg of 2-PMPA given prior to each dose of morphine attenuated the development of morphine tolerance. The effects of 2-PMPA were related to the dose. This was evidenced by a significant decrease in both test 2 ED50 values (statistically significant for the dose 100 mg/kg) and antinociceptive morphine fold shifts of 2-PMPA for the doses of 100 and 50 mg/kg, as compared with the control group that received placebo+morphine ( Table 1). Similarly, memantine (7.5 mg/kg) produced an inhibition of morphine tolerance.
Effects of 2-PMPA on the Tail-Flick Response and Antinociceptive Effects of Morphine (Experiment 2)
Analysis of AUC revealed that treatment with placebo +1.5 and 3 mg/kg of morphine produced significantly longer tail-flick responses compared to placebo+placebo treatment. In contrast, 100 mg/kg of 2-PMPA+placebo treatment did not affect tail-flick responses as compared to placebo+placebo treatment. Moreover, this dose of 2-PMPA did not affect antinociceptive effects of 1.5 or 3 mg/kg of morphine (Figure 1).

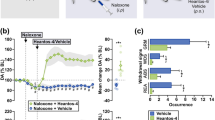
Effects of 2-PMPA on tail-flick responses and on morphine antinociception. Presented are the time courses of tail-flick responses of mice treated with combination of 2-PMPA and morphine. The N is given in brackets. Inset: Presented are mean±SEM AUC values calculated on the same data. One way ANOVA F(5,48)=19.28, P<0.0001, and post hoc Newman–Keul's test revealed that the treatment with placebo+morphine 1.5 mg/kg and with 100 mg/kg 2-PMPA+morphine 1.5 mg/kg differed significantly (**P<0.01) from placebo+placebo treatment. Similarly, treatment with placebo+morphine 3 mg/kg and that with 100 mg/kg 2-PMPA+morphine 3 mg/kg differed significantly (***P<0.001) from placebo+placebo treatment. Effects of 100 mg/kg of 2-PMPA+placebo treatment did not differ from placebo+placebo treatment. Effects of placebo+respective doses of morphine did not differ from the effects of 2-PMPA+respective doses of morphine.
Effects of 2-PMPA and Memantine on Morphine Withdrawal Syndrome (Experiment 3)
Morphine-dependent, naloxone-challenged mice demonstrated robust morphine withdrawal signs (jumps) that were inhibited by 7.5 mg/kg of memantine. 2-PMPA pretreatment at doses 5–50 mg/kg did not affect their severity, however, pretreatment with 100 mg/kg of 2-PMPA potentiated the severity of morphine withdrawal (Table 2). Mice pretreated with 30 mg/kg of morphine and challenged with 2-PMPA (50 or 100 mg/kg) but no naloxone did not exhibit jumps.
Mice treated with 10 mg/kg of morphine, pretreated with placebo, and challenged with naloxone demonstrated severity of withdrawal similar to that produced by 30 mg/kg of morphine. Pretreatment with 100 mg/kg of 2-PMPA did not affect the number of jumps produced by 10 mg/kg morphine treatment ( Table 2, bottom).
Effects on Morphine-Induced Conditioned Place Preference (Experiment 4)
One-way ANOVA performed on pretest values demonstrated differences among groups (P<0.05) and Dunnett's test detected that the pretest scores of mice treated with 2-PMPA before the pre-test differed (were higher than that) from morphine-only treated mice (Table 3). This suggests that for mice treated with 2-PMPA before the pre-test, it might be more difficult to demonstrate a decrease in morphine-induced CPP.
With regard to the effect of conditioning, control mice that received two injections of placebo during conditioning did not change their preference to the respective arm, because the mean±SEM difference between pre-test and post-test time (δ) was 4.6±16.6 s and the resulting preference was 104.2%. Morphine produced robust preference towards the morphine-associated arm, since the δ value for this group was 135.9±17.2 s and the resulting preference was 157.8%. 2-PMPA inhibited place preference produced by morphine, because for the groups treated with this compound during acquisition and expression phases, the δ values were 64±13.1 and 51±19.2 s, respectively (for the resulting preferences (%), see Table 3). Similar inhibition was observed for the groups treated with memantine during acquisition and expression phases, since the respective δ values were 35.7±22.7 and 68.4±28.4 s. 2-PMPA given instead of morphine produced neither preference nor aversion toward the arm associated with its administration (δ=36.2±23 s).
Effects of 2-PMPA on Learning and Memory (Experiment 5)
One-way ANOVAs performed on difference (%) (but not TL1 values), demonstrated a significant difference among groups (Table 4). Although all treatments resulted in shorter TL2 than TL1, only the decrease of TL2 of mice treated with MK-801 was significantly less than that of Placebo.
DISCUSSION
Effects of GCP II Inhibition on Morphine Tolerance
Considering the impact of inhibition of NAAG metabolism on glutamatergic systems, the attenuation of development of morphine tolerance by 2-PMPA was not unexpected. As early as 1991, Marek et al (1991) and Trujillo and Akil (1991) demonstrated the inhibitory effects of kynurenic acid and MK-801 on the development of tolerance to the antinociceptive effects of morphine. Numerous subsequent studies revealed that coadministration of either competitive or noncompetitive NMDA receptor antagonists attenuate and/or reverse the development of tolerance to the antinociceptive effects of morphine. In the mouse, such effects have been shown for a number of NMDA receptor antagonists, including memantine (Lutfy et al, 1993; Elliott et al, 1994; Bilsky et al, 1996; Belozertseva and Bespalov, 1998; Gonzalez et al, 1997; Popik et al, 2000a). Similarly, the stimulation of presynaptic mGluRII receptors with (+)-2-aminobicyclo [3,1,0] hexane-2,6-dicarboxylic acid (LY354740) was shown to inhibit the development of morphine tolerance (Popik et al, 2000b). In the light of these findings, the inhibitory effect of 2-PMPA on morphine tolerance may be explained by its effects at NMDA, mGluRII, or both receptors.
Moreover, the results of Experiment 2 indicate that 2-PMPA, at the dose effectively inhibiting development of morphine tolerance, produced no antinociceptive effect in the tail-flick test, which is in agreement with a recent report of Yamamoto et al that showed lack of an antinociceptive effect of 2-PMPA in another acute pain (hot plate) test (Yamamoto et al, 2001b).
Effects of GCP II Inhibition on Morphine Dependence/Withdrawal
In the light of studies demonstrating that inhibition of GCP II activity inhibits glutamatergic neurotransmission, the results of Experiment 3 are unexpected and somewhat counterintuitive. This is because the expression of opioid withdrawal has been frequently shown to be inhibited by NMDA receptor antagonists and compounds that inhibit mGluR-mediated neurotransmission. Thus, attenuation of the intensity of morphine withdrawal has been reported after acute pretreatment with high- and low-affinity NMDA receptor uncompetitive antagonists, competitive antagonists, glycine, and polyamine site antagonists, as well as NMDAR1 subunit antisense oligonucleotides (Belozertseva et al, 2000; Bristow et al, 1997; Zhu and Ho, 1998; Layer et al, 1996; Popik and Skolnick, 1996; Farzin, 1999; Popik and Danysz, 1997; Brent and Chahl, 1993; Gonzalez et al, 1997). These studies have assessed various classical somatic and autonomic signs of withdrawal, as well as biochemical markers (Bristow et al, 1997; Rasmussen, 1995). Ability of NMDA receptor antagonists to block the expression of opioid withdrawal is consistent with the frequently reported increase in glutamate release in opioid-withdrawn animals (Sepulveda et al, 1998) and facilitation of the withdrawal signs by glutamate receptor agonist administration (Tokuyama et al, 1996). Similarly, stimulation of mGluRII receptors with (+)-2-aminobicyclo [3,1,0] hexane-2,6-dicarboxylic acid (LY354740) was shown to inhibit the expression of morphine dependence (Klodzinska et al, 1999; Vandergriff and Rasmussen, 1999).
The results of Experiment 3, indicating no effect of 2-PMPA on morphine withdrawal or even its intensification with the use of a relatively high dose, suggest that the inhibition of activity of GCP II produces a unique effect on the consequences of repeated exposure to morphine (inhibition of tolerance but not dependence). As mentioned earlier, Yamamoto et al (2001a) suggested that NAAG acts as an NMDA receptor antagonist at low concentrations, but as a low potency NMDA receptor agonist at high concentrations. If this hypothesis were correct, it could be speculated that administration of a high dose (100 mg/kg) of 2-PMPA leads to high concentrations of NAAG that acts as an agonist, rather than an antagonist at NMDA receptors, and thus increases the severity of morphine withdrawal. In support, it should be noted that during opioid withdrawal there is a massive release of glutamate (Rasmussen, 1995; Jhamandas et al, 1996; Sepulveda et al, 1998). The effects of massive glutamate release apparently may not be prevented by NAAG-induced mGluRII stimulation and/or a decrease in glutamate release. Such stimulation of glutamate receptors could increase the severity of morphine withdrawal as was reported for glutamate receptor agonists (Tokuyama et al, 1996). Interestingly, above a certain threshold (5–10 mg/kg), the dose of morphine used to produce dependence appears to have little effect on the intensity of opioid withdrawal (Popik et al, 1998), which Experiment 3 seems to confirm. However, in mice in which dependence was produced with 10 mg/kg of morphine, 100 mg/kg of 2-PMPA did not intensify opioid withdrawal, which suggests that this effect is indeed related to the intensity of neurochemical events associated with the withdrawal state. Martin et al (1997a),(1997b) reported that glutamate release is attenuated by agonists selective for μ-opioid receptors and mGluRII receptors located presynaptically, and that this inhibitory effect is further potentiated in morphine-tolerant animals (Martin et al, 1999). It remains to be assessed if the differential effects of a GCP II inhibitor on morphine tolerance and dependence may be explained solely by the concentration of glutamate. Interestingly, a similar set of effects was recently reported for another indirect inhibitor of glutamate availability, the glutamate transporter activator, (R)-(-)5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) (Nakagawa et al, 2001). As in our report, these authors found inhibitory effects of MS-153 on the development of morphine tolerance, but no effect on morphine antinociceptive actions or expression of morphine dependence.
Alternatively, the results of Experiments 1 and 3 may suggest that morphine tolerance and dependence can be separated, at least at the level of glutamatergic neurotransmission. This has been earlier shown in behavioral (Rahman et al, 1994; Paronis and Woods, 1997a,1997b; Gold et al, 1994) and neurochemical (Lang and Schulz, 1989; Gudehithlu and Bhargava, 1996; Aley and Levine, 1997) studies and experiments with the use of genetically modified mice (Kest et al, 2001; Bohn et al, 2000).
Effects of GCP II Inhibition on Morphine-Induced Conditioned Reward and Learning and Memory
In our study, only one conditioning session with morphine in the compartment distinguished by visual, tactile, and olfactory cues was sufficient to induce significant preference to this compartment, as has been reported by other investigators (Mucha et al, 1982; Bardo and Neisewander, 1986). It has been demonstrated that use of multiple stimuli as cues in the CPP procedure produces stronger conditioning, as opposed to the procedure based on single modality stimulus (Mucha et al, 1982; Barr et al, 1985). This method of establishing CPP using only one drug pairing corresponds better to the literature on humans indicating that the initial drug experience is an important factor contributing to later drug use (Haertzen et al, 1983). Moreover, it eliminates other possible confounding factors like tolerance to the rewarding effect of morphine (Shippenberg et al, 1988).
2-PMPA, at the one, selected dose that significantly affected morphine tolerance and withdrawal inhibited both acquisition and expression of morphine-induced conditioned place preference. This effect resembles the effect of memantine (and other NMDA receptor antagonists, see the Introduction section). Slusher et al, have recently demonstrated that 2-PMPA as well as another GCP II inhibitor, GPI 5693, inhibited both the acquisition and the expression of cocaine-induced CPP in rats (Slusher et al, 2001).
There are various possible interpretations to the effects of compounds on place conditioning. For example, it has been suggested that if a compound by itself produces conditioned aversion, it may attenuate CPP not because of a specific effect on conditioned reward but because subjects may associate an aversive state with the rewarded arm of the apparatus (Carr et al, 1989). In this regard, data from Experiment 4 demonstrate that an effective dose of 2-PMPA did not produce any aversive or rewarding effect by itself when given 20 min before the conditioning, which agrees with observations in rats (Slusher et al, 2001) indicating no effect of 2-PMPA on place conditioning. The dose of memantine used in the present experiment (7.5 mg/kg), that inhibited conditioned morphine reward was also shown not to produce conditioned place preference or aversion (Popik and Danysz, 1997). Moreover, since it is known that the effects of NMDA receptor antagonists on morphine-induced CPP are not because of the state-dependent learning (Tzschentke, 1998), it may be speculated that the effects of 2-PMPA were also probably not because of the state-dependent learning phenomenon.
Another potentially confounding factor could be a direct effect on locomotor activity. Our CPP procedure involved recording the number of visits to arms (a rough measurement of locomotor activity). We found (data not shown) that 2-PMPA did not change the locomotion of mice, which agrees with similar observations in rats (Shippenberg et al, 2000). The unaffected locomotion following 2-PMPA administration is an important issue in interpreting its effects on expression of morphine-induced CPP as it assures that 2-PMPA-treated mice did not simply enter one of the compartments and remain there because their locomotor function was impaired.
Cognitive effects of a compound that modulates conditioned reward should also be carefully considered. For instance, if a compound produced learning or memory impairment, its inhibitory effects on conditioned drug reward would be considered unspecific. This issue has frequently been raised for the effects of NMDA receptor antagonists (Bisaga and Popik, 2000). To assess the effects of 2-PMPA on acquisition and/or retrieval of memory, we used the elevated plus maze model of spatial learning in mice (Itoh et al, 1991). This paradigm is sensitive to the amnesic effects of post-training administration of scopolamine or electroconvulsive shock (Itoh et al, 1990), pretraining effects of scopolamine, MK-801 and diazepam (Itoh et al, 1991), and pretest effects of MK-801 and scopolamine (Hlinak and Krejci, 2000). Results of Experiment 5 demonstrating amnesic effects of MK-801 confirm earlier observations (Itoh et al, 1991); however, we found that in contrast to MK-801, 2-PMPA (50 and 100 mg/kg) affected neither learning nor retrieval of memory. The lack of effects on learning and memory agrees with earlier observations (Slusher et al, 1999) and suggests that the effect of 2-PMPA on acquisition and expression of conditioned morphine reward were not because of its effects on remembering or retrieval of information.
In conclusion, the present study demonstrates that inhibition of activity of GCP II by 2-PMPA inhibits the development of tolerance to the antinociceptive effects of morphine without affecting the nociceptive response per se or interfering with morphine antinociception. Moreover, inhibition of activity of GCP II by 2-PMPA leaves the expression of morphine dependence unaffected (or potentiates it when used at the highest dose). In addition, 2-PMPA inhibited both the acquisition as well as expression of morphine-induced conditioned place preference. In the light of the recent data demonstrating that inhibition of GCP II results in reduction of alcohol intake in rats (Mckinzie et al, 2000) and attenuation of cocaine sensitization (Shippenberg et al, 2000), our findings indicate the therapeutic potential of GCP II inhibition in the management of chronic pain and opioid addiction.
References
Aley KO, Levine JD (1997). Dissociation of tolerance and dependence for opioid peripheral antinociception in rats. J Neurosci 17: 3907–3912.
Bardo MT, Neisewander JL (1986). Single-trial conditioned place preference using intravenous morphine. Pharmacol Biochem Behav 25: 1101–1105.
Barr GA, Paredes W, Bridger WH (1985). Place conditioning with morphine and phencyclidine: dose dependent effects. Life Sci 36: 363–368.
Belozertseva IV, Bespalov AY (1998). Effects of NMDA receptor channel blockers, dizocilpine and memantine, on the development of opiate analgesic tolerance induced by repeated morphine exposures or social defeats in mice. Naunyn-Schmiedeberg's Arch Pharmacol 358: 270–274.
Belozertseva IV, Danysz W, Bespalov AY (2000). Short-acting NMDA receptor antagonist MRZ 2/576 produces prolonged suppression of morphine withdrawal in mice. Naunyn-Schmiedeberg's Arch Pharmacol 361: 279–282.
Bespalov A, Dumpis M, Piotrovsky L, Zvartau E (1994). Excitatory amino acid receptor antagonist kynurenic acid attenuates rewarding potential of morphine. Eur J Pharmacol 264: 233–239.
Bilsky EJ, Inturrisi CE, Sadee W, Hruby VJ, Porreca F (1996). Competitive and non-competitive NMDA antagonists block the development of antinociceptive tolerance to morphine, but not to selective mu or delta opioid agonists in mice. Pain 68: 229–237.
Bisaga A, Popik P (2000). In search of a new pharmacological treatment of drug addiction: N-methyl-D-aspartate (NMDA) antagonists. Drug Alcohol Depend 59: 1–15.
Blakely RD, Coyle JT (1988). The neurobiology of N-acetylaspartylglutamate. Int Rev Neurobiol 30: 39–100.
Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG (2000). mu-Opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408: 720–723.
Brent PJ, Chahl LA (1993). Enhancement of the opiate withdrawal response by antipsychotic drugs in guinea-pigs is not mediated by sigma binding sites. Eur Neuropsychopharmacol 3: 23–32.
Bristow LJ, Hogg JE, Hutson PH (1997). Competitive and glycine/NMDA receptor antagonists attenuate withdrawal-induced behaviours and increased hippocampal acetylcholine efflux in morphine-dependent rats. Neuropharmacology 36: 241–250.
Bruno V, Wroblewska B, Wroblewski JT, Fiore L, Nicoletti F (1998). Neuroprotective activity of N-acetylaspartylglutamate in cultured cortical cells. Neuroscience 85: 751–757.
Burlina AP, Skaper SD, Mazza MR, Ferrari V, Leon A, Burlina AB (1994). N-acetylaspartylglutamate selectively inhibits neuronal responses to N-methyl-D-aspartic acid in vitro. J Neurochem 63: 1174–1177.
Cappendijk SL, de Vries R, Dzoljic MR (1993). Excitatory amino acid receptor antagonists and naloxone-precipitated withdrawal syndrome in morphine-dependent mice. Eur Neuropsychopharmacol 3: 111–116.
Carr GF, Fibiger HC, Phillips AG (1989). Conditioned place preference as a measure of drug reward. In: Liebman J Cooper SJ (eds). The Neuropharmacological Basis of Reward. Clarendon Press: Oxford. pp 264–319.
Conn PJ, Pin JP (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205–237.
Cunningham CL, Niehus DR, Malott DH, Prather LK (1992). Genetic differences in the rewarding and activating effects of morphine and ethanol. Psychopharmacology 107: 385–393.
Elliott K, Minami N, Kolesnikov YA, Pasternak GW, Inturrisi CE (1994). The NMDA receptor antagonists, LY274614 and MK-801, and the nitric oxide synthase inhibitor, Ng-nitro-L-arginine, attenuate analgesic tolerance to the mu-opioid morphine but not to kappa opioids. Pain 56: 69–75.
Farzin D (1999). Modification of naloxone-induced withdrawal signs by dextromethorphan in morphine-dependent mice. Eur J Pharmacol 377: 35–42.
Gold LH, Stinus L, Inturrisi CE, Koob GF (1994). Prolonged tolerance, dependence and abstinence following subcutaneous morphine pellet implantation in the rat. Eur J Pharmacol 253: 45–51.
Gonzalez P, Cabello P, Germany A, Norris B, Contreras E (1997). Decrease of tolerance to, and physical dependence on morphine by, glutamate receptor antagonists. Eur J Pharmacol 332: 257–262.
Grunze HCR, Rainnie DG, Hasselmo ME, Barkai E, Heam EF, McCarley RW et al (1996). NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 16: 2034–2043.
Gudehithlu KP, Bhargava HN (1996). Differential binding of [3H]MK-801 to brain regions and spinal cord of mice treated chronically with morphine. Gen Pharmacol 27: 91–94.
Haertzen CA, Kocher TR, Miyasato K (1983). Reinforcements from the first drug experience can predict later drug habits and/or addiction: results with coffee, cigarettes, alcohol, barbiturates, minor and major tranquilizers, stimulants, marijuana, hallucinogens, heroin, opiates and cocaine. Drug Alcohol Depend 11: 147–165.
Hlinak Z, Krejci I (2000). Oxiracetam prevents the MK-801 induced amnesia for the elevated plus-maze in mice. Behav Brain Res 117: 147–151.
Itoh J, Nabeshima T, Kameyama T (1990). Utility of an elevated plus-maze for the evaluation of memory in mice: effects of nootropics, scopolamine and electroconvulsive shock. Psychopharmacology 101: 27–33.
Itoh J, Nabeshima T, Kameyama T (1991). Utility of an elevated plus-maze for dissociation of amnesic and behavioral effects of drugs in mice. Eur J Pharmacol 194: 71–76.
Jackson PF, Cole DC, Slusher BS, Stetz SL, Ross LE, Donzanti BA et al (1996). Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated alpha-linked acidic dipeptidase. J Med Chem 39: 619–622.
Jackson PF, Slusher BS (2001). Design of NAALADase inhibitors: a novel neuroprotective strategy. Curr Med Chem 8: 949–957.
Jhamandas KH, Marsala M, Ibuki T, Yaksh TL (1996). Spinal amino acid release and precipitated withdrawal in rats chronically infused with spinal morphine. J Neurosci 16: 2758–2766.
Kest B, Hopkins E, Palmese CA, Chen ZP, Mogil JS, Pintar JE (2001). Morphine tolerance and dependence in nociceptin/orphanin FQ transgenic knock-out mice. Neuroscience 104: 217–222.
Klodzinska A, Chojnacka Wojcik E, Palucha A, Branski P, Popik P, Pilc A (1999). Potential anti-anxiety and anti-addictive effects of LY 354740, a selective group II glutamate metabotropic receptors agonist in animal models. Neuropharmacology 38: 1831–1839.
Koenig ML, Rothbard PM, DeCoster MA, Meyerhoff JL (1994). N-acetyl-aspartyl-glutamate (NAAG) elicits rapid increase in intraneuronal Ca2+ in vitro. NeuroReport 5: 1063–1068.
Koob GF, LeMoal M (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: 97–129.
Kotlinska J, Biala G (1999). Effects of the NMDA/glycine receptor antagonist, L-701,324, on morphine- and cocaine-induced place preference. Pol J Pharmacol 51: 323–330.
Lang J, Schulz R (1989). Chronic opiate receptor activation in vivo alters the level of G protein subunits in guinea pig myenteric plexus. Neuroscience 32: 503.
Layer RT, Skolnick P, Bertha CM, Kuehne ME, Popik P (1996). Structurally modified ibogaine analogs exhibit differing affinities for NMDA receptors. Eur J Pharmacol 309: 159–165.
Lister RG (1987). The use of a plus maze to measure anxiety in the mouse. Psychopharmacology 92: 180–185.
Lutfy K, Hurlbut DE, Weber E (1993). Blockade of morphine-induced analgesia and tolerance in mice by MK-801. Brain Res 616: 83–88.
Manzanedo C, Aguilar MA, Rodriguez-Arias M, Minarro J (2001). Effects of dopamine antagonists with different receptor blockade profiles on morphine-induced place preference in male mice. Behav Brain Res 121: 189–197.
Marek P, Ben-Eliyahu S, Gold M, Liebeskind JC (1991). Excitatory amino acid antagonists (kynurenic acid and MK-801) attenuate the development of morphine tolerance in the rat. Brain Res 547: 77–81.
Martin G, Nie Z, Siggins GR (1997a). Metabotropic glutamate receptors regulate N-methyl-D-aspartate-mediated synaptic transmission in nucleus accumbens. J Neurophysiol 78: 3028–3038.
Martin G, Nie Z, Siggins GR (1997b). mu-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci 17: 11–22.
Martin G, Przewlocki R, Siggins GR (1999). Chronic morphine treatment selectively augments metabotropic glutamate receptor-induced inhibition of N-methyl-D-aspartate receptor-mediated neurotransmission in nucleus accumbens. J Pharmacol Exp Ther 288: 30–35.
Mckinzie DL, Li T-K, Mcbride WJ, Slusher BS (2000). NAALADase inhibition reduces alcohol consumption in the alcohol-preferring (P) line of rats. Addict Biol 5: 411–416.
Monaghan DT, Bridges RJ, Cotman CW (1989). The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Ann Rev Pharmacol Toxicol 29: 365–402.
Mucha RF, Iversen SD (1984). Reinforcing properties of morphine and naloxone revealed by conditioned place preferences: a procedural examination. Psychopharmacology 82: 241–247.
Mucha RF, Van der Kooy D, O’Shaughnessy M, Bucenieks P (1982). Drug reinforcement studied by the use of place conditioning in the rat. Brain Res 243: 91–105.
Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M (2001). Inhibition of morphine tolerance and dependence by MS-153, a glutamate transporter activator. Eur J Pharmacol 419: 39–45.
Neale JH, Bzdega T, Wroblewska B (2000). N-Acetylaspartyl-glutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. J Neurochem 75: 443–452.
Paronis CA, Holtzman SG (1991). Increased analgesic potency of mu agonists after continuous naloxone infusion in rats. J Pharmacol Exp Ther 259: 582–589.
Paronis CA, Woods JH (1997a). Ventilation in morphine-maintained rhesus monkeys. I: effects of naltrexone and abstinence-associated withdrawal. J Pharmacol Exp Ther 282: 348–354.
Paronis CA, Woods JH (1997b). Ventilation in morphine-maintained rhesus monkeys. II: tolerance to the antinociceptive but not the ventilatory effects of morphine. J Pharmacol Exp Ther 282: 355–362.
Popik P, Danysz W (1997). Inhibition of reinforcing effects of morphine and motivational aspects of naloxone-precipitated opioid withdrawal by NMDA receptor antagonist, memantine. J Pharmacol Exp Ther 280: 854–865.
Popik P, Kozela E, Danysz W (2000a). Clinically available NMDA receptor antagonists memantine and dextromethorphan reverse existing tolerance to the antinociceptive effects of morphine in mice. Naunyn-Schmiedeberg's Arch Pharmacol 361: 425–432.
Popik P, Kozela E, Pilc A (2000b). Selective agonist of group II glutamate metabotropic receptors, LY354740, inhibits tolerance to analgesic effects of morphine in mice. Brit J Pharmacol 130: 1425–1431.
Popik P, Mamczarz J, Fraczek M, Widla M, Hesselink M, Danysz W (1998). Inhibition of reinforcing effects of morphine and naloxone-precipitated opioid withdrawal by novel glycine site and uncompetitive NMDA receptor antagonists. Neuropharmacology 37: 1033–1042.
Popik P, Skolnick P (1996). The NMDA antagonist memantine blocks the expression and maintenance of morphine dependence. Pharmacol Biochem Behav 53: 791–798.
Pouwels PJ, Frahm J (1997). Differential distribution of NAA and NAAG in human brain as determined by quantitative localized proton MRS. NMR Biomed 10: 73–78.
Puttfarcken PS, Handen JS, Montgomery DT, Coyle JT, Werling LL (1993). N-acetyl-aspartylglutamate modulation of N-methyl-D-aspartate-stimulated [3H]norepinephrine release from rat hippocampal slices. J Pharmacol Exp Ther 266: 796–803.
Rahman AF, Takahashi M, Kaneto H (1994). Morphine dependence with or without tolerance in formalin-treated mice: further evidence for the dissociation. Jpn J Pharmacol 66: 277–280.
Rasmussen K (1995). The role of the locus coeruleus and N-methyl-D-aspartic acid (NMDA) and AMPA receptors in opiate withdrawal. Neuropsychopharmacology 13: 295–300.
Sekiguchi M, Wada K, Wenthold RJ (1992). N-acetylaspartylglutamate acts as an agonist upon homomeric NMDA receptor (NMDAR1) expressed in Xenopus oocytes. FEBS Lett 311: 285–289.
Sepulveda MJ, Hernandez L, Rada P, Tucci S, Contreras E (1998). Effect of precipitated withdrawal on extracellular glutamate and aspartate in the nucleus accumbens of chronically morphine-treated rats: an in vivo microdialysis study. Pharmacol Biochem Behav 60: 255–262.
Shippenberg TS, Emmett Oglesby MW, Ayesta FJ, Herz A (1988). Tolerance and selective cross-tolerance to the motivational effects of opioids. Psychopharmacology 96: 110–115.
Shippenberg TS, Rea W, Slusher BS (2000). Modulation of behavioral sensitization to cocaine by NAALADase inhibition. Synapse 38: 161–166.
Slusher BS, Thomas A, Paul M, Schad CA, Ashby CR (2001). Expression and acquisition of the conditioned place preference response to cocaine in rats is blocked by selective inhibitors of the enzyme N-acetylated-alpha-linked-acidic dipeptidase (NAALADase). Synapse 41: 22–28.
Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A et al (1999). Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat Med 5: 1396–1402.
Stauch BL, Robinson MB, Forloni G, Tsai G, Coyle JT (1989). The effects of N-acetylated alpha-linked acidic dipeptidase (NAALADase) inhibitors on [3H]NAAG catabolism in vivo. Neurosci Lett 100: 295–300.
Stinus L, Le Moal M, Koob GF (1990). Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience 37: 767–773.
Thomas AG, Vornov JJ, Olkowski JL, Merion AT, Slusher BS (2000). N-Acetylated alpha-linked acidic dipeptidase converts N-acetylaspartylglutamate from a neuroprotectant to a neurotoxin. J Pharmacol Exp Ther 295: 16–22.
Tiseo PJ, Inturrisi CE (1993). Attenuation and reversal of morphine tolerance by the competitive N-methyl-D-aspartate receptor antagonist, LY274614. J Pharmacol Exp Ther 264: 1090–1096.
Tokuyama S, Wakabayashi H, Ho IK (1996). Direct evidence for a role of glutamate in the expression of the opioid withdrawal syndrome. Eur J Pharmacol 295: 123–129.
Tortella FC, Lin Y, Ved H, Slusher BS, Dave JR (2000). Neuroprotection produced by the NAALADase inhibitor 2-PMPA in rat cerebellar neurons. Eur J Pharmacol 402: 31–37.
Trujillo KA, Akil H (1991). Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251: 85–87.
Tsai G, Slusher BS, Sim L, Coyle JT (1993). Immunocytochemical distribution of N-acetylaspartylglutamate in the rat forebrain and glutamatergic pathways. J Chem Neuroanat 6: 277–292.
Tsai G, Stauch BL, Vornov JJ, Deshpande JK, Coyle JT (1990). Selective release of N-acetylaspartylglutamate from rat optic nerve terminals in vivo. Brain Res 518: 313–316.
Tzschentke TM (1998). Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56: 613–672.
Tzschentke TM, Schmidt WJ (1995). N-methyl-D-aspartic acid-receptor antagonists block morphine-induced conditioned place preference in rats. Neurosci Lett 193: 37–40.
Tzschentke TM, Schmidt WJ (1997). Interactions of MK-801 and GYKI 52466 with morphine and amphetamine in place preference conditioning and behavioural sensitization. Behav Brain Res 84: 99–107.
Vandergriff J, Rasmussen K (1999). The selective mGlu2/3 receptor agonist LY354740 attenuates morphine- withdrawal-induced activation of locus coeruleus neurons and behavioral signs of morphine withdrawal. Neuropharmacology 38: 217–222.
Westbrook GL, Mayer ML, Namboodiri MA, Neale JH (1986). High concentrations of N-acetylaspartylglutamate (NAAG) selectively activate NMDA receptors on mouse spinal cord neurons in cell culture. J Neurosci 6: 3385–3392.
Williamson LC, Neale JH (1988). Ultrastructural localization of N-acetylaspartylglutamate in synaptic vesicles of retinal neurons. Brain Res 456: 375–381.
Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH (1997). N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J Neurochem 69: 174–181.
Wroblewska B, Wroblewski JT, Saab OH, Neale JH (1993). N-acetylaspartylglutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J Neurochem 61: 943–948.
Yamamoto T, Nozaki-Taguchi N, Sakashita Y (2001a). Spinal N-acetyl-alpha-linked acidic dipeptidase (NAALADase) inhibition attenuates mechanical allodynia induced by paw carrageenan injection in the rat. Brain Res 909: 138–144.
Yamamoto T, Nozaki-Taguchi N, Sakashita Y, Inagaki T (2001b). Inhibition of spinal N-acetylated-alpha-linked acidic dipeptidase produces an antinociceptive effect in the rat formalin test. Neuroscience 102: 473–479.
Zhu H, Ho IK (1998). NMDA-r1 antisense oligonucleotide attenuates withdrawal signs from morphine. Eur J Pharmacol 352: 151–156.
Acknowledgements
This study was supported by Guilford Pharmaceuticals and by KBN grant 4 P05A 42 17. The authors are grateful to Dr W Danysz, MERZ and Co, Frankfurt/M, Germany for the gift of memantine.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Popik, P., Kozela, E., Wróbel, M. et al. Morphine Tolerance and Reward but not Expression of Morphine Dependence are Inhibited by the Selective Glutamate Carboxypeptidase II (GCP II, NAALADase) Inhibitor, 2-PMPA. Neuropsychopharmacol 28, 457–467 (2003). https://doi.org/10.1038/sj.npp.1300048
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300048
Keywords
This article is cited by
-
N-acetylaspartylglutamate Inhibits Heroin Self-Administration and Heroin-Seeking Behaviors Induced by Cue or Priming in Rats
Neuroscience Bulletin (2017)
-
Reinstatement of Morphine-Conditioned Reward is Blocked by Memantine
Neuropsychopharmacology (2006)
-
NAAG peptidase inhibitors and their potential for diagnosis and therapy
Nature Reviews Drug Discovery (2005)
-
2-MPPA, a selective glutamate carboxypeptidase II inhibitor, attenuates morphine tolerance but not dependence in C57/Bl mice
Psychopharmacology (2005)
