
Abstract
Sensorimotor gating, measured by prepulse inhibition (PPI) of the startle reflex, is reduced in schizophrenia patients and in rats treated with dopamine agonists. Strain and substrain differences in the sensitivity to the PPI-disruptive effects of dopamine agonists may provide insight into the genetic basis for human population differences in sensorimotor gating. We have reported greater sensitivity to the PPI-disruptive effects of the D1/D2 agonist apomorphine in Harlan Sprague–Dawley (SDH) vs Wistar (WH) rats. In the present study, we assessed the inheritance pattern of this phenotypic difference. Sensitivity to the PPI-disruptive effects of apomorphine was compared across parental SDH and WH strains, offspring (F1) of an SDH×WH cross, and subsequent offspring (N2) of an SDH×F1 cross. Apomorphine sensitivity followed a gradient of SDH>N2>F1>WH. Parental SDH and WH strains exhibited comparable sensitivity to the PPI-disruptive effects of phencyclidine. The nature of this gradient of APO sensitivity suggests relatively simple additive effects of multiple genes on the phenotype of PPI sensitivity.
Similar content being viewed by others


INTRODUCTION
The motor response to an intense startling stimulus is normally inhibited by a weak stimulus, or prepulse, that precedes the startling stimulus by 30–500 ms. The degree to which startle amplitude is inhibited by the prepulse is a measure of sensorimotor gating. Prepulse inhibition (PPI) is deficient in specific neuropsychiatric disorders, and may be a useful ‘endophenotype’ (ie a physiological marker that, compared to clinical symptomatology, is more closely linked to the disorder genes) for understanding the genetic basis for these disorders. For example, PPI is significantly reduced in schizophrenia probands and first-degree relatives, compared to unaffected controls without a familial history of schizophrenia (Braff et al, 1978; Cadenhead et al, 2000); in schizophrenia patients, this deficit may be ‘reversed’ by dopamine (DA)-receptor blockade (Kumari et al, 1999; Weike et al, 2000). At a conceptual level, deficient PPI may reflect a loss of important mechanisms that normally ‘protect’ the orderly processing of information (represented experimentally by the prepulse) from disruption by subsequent stimuli within a brief temporal window (represented experimentally by the startling stimulus 100 ms later). Such a loss of ‘information protective’ mechanisms may make an individual vulnerable to abnormalities in information processes, which might contribute to the clinical manifestations of schizophrenia. At a neural level, PPI is regulated by forebrain limbic cortical and ventral striatal circuitry implicated in the pathophysiology of schizophrenia (cf Koch and Schnitzler, 1997; Swerdlow and Geyer, 1998). Thus, a dissection of the genetics of PPI and its neural substrates might be a valuable step toward understanding the biology of the more complex phenotype of schizophrenia.
One of the unique advantages of PPI as a potential intermediate or endophenotype is that it can be studied across species, using identical stimulus parameters to elicit comparable response characteristics. We recently reported differences in PPI and its sensitivity to disruption by DA agonists among two outbred rat populations (Sprague-Dawley (SD) vs Wistar (W)) and suppliers (Harlan, USA (H) vs Bantin-Kingman, UK (BK)) (Swerdlow et al, 2000). In one comparison, SDH rats were more sensitive to the PPI-disruptive effects of the D1/D2 agonist apomorphine (APO). Differences in APO sensitivity were ‘dose-dependent’—detected most prominently at relatively higher doses of APO (0.25–0.5 mg/kg)—and were evident at 18 days of age in rats reared under identical conditions, making it likely that these are ‘constitutional’ or genetic differences, rather than differences arising from environmental or later-developing influences.
In the present study, we examined the inheritance patterns for sensitivity to the PPI-disruptive effects of APO in SDH and WH rat pups, an F1 SDH×WH cross (‘F1’), and an F1×SDH cross (‘N2’). If clear inheritance patterns can be detected for sensitivity to the PPI-disruptive effects of APO, it should be possible to utilize our substantial understanding of the neural circuit regulation of PPI in rats to localize the neural substrates by which genes confer differences in the DAergic regulation of sensorimotor gating. In turn, these neural substrates would be promising candidates to assess their contribution to gating deficits in specific, heritable neuropsychiatric disorders.
METHODS
Experimental Animals
A total of 348 rats were used in these experiments. To closely match the rearing environments of SDH and WH pups, all timed pregnant female rats were housed individually, and pups were housed with their mothers until 3–5 days after birth. At that point, pups were assigned to same-strain litters and mother (‘in-fostered’) to produce litters of comparable size and sex distribution. Aside from the strain of the nursing female rat, rearing conditions for all pups were comparable; parental strains, F1 and N2 generations were raised in the same room, on the same cage racks. Adult male and nonpregnant female rats were housed in same-sex rooms (except for rats used for breeding), in groups of 2–4. Methods for housing and all behavioral testing were consistent with the substantial literature of startle measures in rodents (cf Geyer and Swerdlow, 1998). For example, a reversed 12 h light/dark cycle was used (lights on at 19:00 h, off at 07:00 h) for at least 7 days prior to testing. Rats were handled regularly prior to any procedures to minimize stress during behavioral testing, and were given ad libitum access to food and water except during behavioral testing. Throughout the studies, all efforts were made to minimize animal suffering and to reduce the number of animals used. All experiments conform to guidelines of the National Institute of Health for the use of animals in biomedical research and were approved by the Animal Subjects Committee at the University of California, San Diego (protocols 0224907 and 0224908).
Drugs
APO (0.1% ascorbate/saline vehicle, 0.1, 0.25 or 0.5 mg/kg) or phencyclidine (saline vehicle, 0.25, 0.75 or 1.25 mg/kg) was administered subcutaneously to rats immediately (APO) or 10 min (phencyclidine) prior to testing, in a volume of 1 ml/kg.
Apparatus
Startle experiments used four startle chambers (SR-LAB; San Diego Instruments, San Diego, CA) housed in a sound-attenuated room with a 60 dB ambient noise level. Each startle chamber consisted of a Plexiglas cylinder (8.7 cm internal diameter for adults; 3.75 cm internal diameter for pups) resting on a 12.5×25.5 cm Plexiglas stand. Acoustic stimuli and background noise were presented via a Radioshack Supertweeter mounted 24 cm above the Plexiglas cylinder. Startle magnitude was detected and recorded as transduced cylinder movement via a piezoelectric device mounted below the Plexiglas stand. Response sensitivities were calibrated (SR-LAB Startle Calibration System) to be nearly identical in each of the four startle chambers (maximum variability <1% of stimulus range and <5% of response ranges). Response sensitivities were calibrated for adult and pup chambers separately and recalibrated each time the chambers were changed, always within the <5% response range. Chambers were also balanced across all experimental groups. Sound levels were measured and calibrated with a sound level meter (Quest electronics: Oconomowoc, WI), A scale (relative to 20 μN/M2), with microphone placed inside the Plexiglas cylinder. Methodological details can be found in published material (Geyer and Swerdlow, 1998).
Startle Testing Procedures
In our testing apparatus, reliable measures of startle could first be obtained in pups at 14 days of age. At 14–19 days of age, different groups of rat pups were exposed to a brief ‘matching’ startle session, as reported previously (Geyer and Swerdlow, 1998; Martinez et al, 2000). Rat pups were placed in a startle chamber, and exposed to 5 min of 70 dB background noise followed by 17 ‘PULSE’ trials of 40 ms, 120 dB noise bursts and 5 ‘PREPULSE’ trials consisting of a 20 ms, 82 dB (12 dB above background) prepulse followed 100 ms by a 120 dB pulse (onset to onset). Adult rats were exposed to this matching session 2–4 days prior to testing. Data from this session were used to assign rat pups and adults to balanced dose groups according to their average PULSE startle magnitude.
Behavioral testing continued 2–4 days after the ‘matching’ session for pups (mean age at testing (day): SDH=18.0, WH=18.0, F1=17.13, N2=17.82). Pups were brought to the laboratory in their home cages with their mothers, weighed, and returned to a cage with their testing cohort, to minimize stress before and after testing. Adult rats were brought to the laboratory in individual cages. In most cases, test sessions were approximately 16 min long and consisted of 5 min of 70 dB background followed by five trial types: PULSE noise bursts, PREPULSE trials (20 ms noise bursts 5, 10, or 15 dB above background followed 100 ms by a PULSE) and NOSTIM trials (stabilimeter recordings obtained when no stimulus was presented). The session consisted of initial and final blocks of 3 PULSE trials, separated by two blocks that included 8 PULSE trials and 15 PREPULSE trials (the latter divided equally among 5, 10, and 15 dB prepulse intensities); NOSTIM trials were interspersed between startle trials. ‘NOSTIM’ trials were used to assess gross motor activity during the test session, but were not included in the calculation of intertrial intervals, which were variable and averaged 15 s. Reflex ‘habituation’ was determined based on the change in startle magnitude from the initial to the final block of PULSE trials. Using this design, PPI is measured during a portion of the session in which startle magnitude is relatively constant, that is, has already undergone the maximal rate of habituation during the initial three PULSE trials.
Breeding Procedures
For parental strains, pregnant dams were obtained from Harlan Laboratories, and litters were treated as described above (‘Experimental Animals’). Pups were ‘matched’ at day 14 and tested at day 18, as described above, with one of the 4 doses of APO (vehicle, 0.1, 0.25, or 0.5 mg/kg). Behavioral data from these parental rats were reported previously (Swerdlow et al, 2000), and served as the ‘parental phenotype’ in the present study. We have previously reported that sensitivity to the PPI-disruptive effects of APO is present and fully mature in SDH rats by day 18 (Martinez et al, 1999).
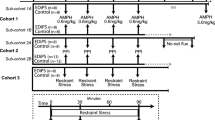
To produce an F1, SDH and WH rats were reciprocally crossed (with equal representation of both sexes from both strains) (Figure 1). F1 SDH×WH litters (n=95) were ‘matched’ on days 14–15, and subsequently tested with one of the 4 doses of APO (vehicle, 0.1, 0.25, or 0.5 mg/kg). This dose-response profile served as the ‘F1 phenotype’. Since this APO dose–response study resulted in only 25% of the entire population tested at each of the 4 different doses (including vehicle), it could not be used to estimate APO sensitivity across the entire F1 population. For this reason, all F1 SDH×WH rats (‘F1’) were tested on days 56–58 for their behavioral response to APO (0.1 mg/kg), using the test paradigm described above. Male and female rats with the highest and lowest 10% PPI were identified. F1 rats within each extreme decile were crossed with outbred SDH adults obtained from the supplier, with an equal number of male and female rats from each strain (F1, SDH) and sex of maternal F0 represented in this cross. SDH×F1 (‘N2’) litters (n=147) were treated as described above, ‘matched’, and subsequently tested at approximately 18 days of age, with one of the 4 doses of APO (vehicle, 0.1, 0.25, or 0.5 mg/kg). This dose-response profile served as the ‘N2 phenotype’.

Pedigree of SDH×WH cross and F1xSDH backcross to produce N2. An F1 was created by crossing parental SDH and WH rats, with an equal representation of sexes from either strain. The F1 was tested on day 18, and then stratified on day 56 based on its level of PPI in response to a low dose of apomorphine (0.1 mg/kg). Male and female rats with the highest (‘hi’) and lowest (‘lo’) 10% of PPI levels were then back-crossed to outbred SDH rats to produce an N2.
To examine whether strain differences in PPI APO sensitivity reflected generalized differences in PPI ‘disruptibility’, sensitivity to the PPI-disruptive effects of phencyclidine (PCP) was assessed in naive 18 days SDH and WH pups, using methods identical to those described above, except that instead of APO, rats were treated with PCP (saline vehicle, 0.25, 0.75, or 1.25 mg/kg) 10 min prior to testing.
Data Analysis
PPI was calculated as a percent reduction in startle magnitude on PREPULSE trials compared to PULSE trials. Any drug effects on %PPI prompted separate analyses to assess the relationship of these effects with drug-induced changes in startle magnitude on PULSE and PREPULSE trials. Because drug-induced changes in startle magnitude—independent of prepulse effects—can change the amount of %PPI, unequivocal changes in sensorimotor gating occur when the reflex-inhibiting effects of prepulses are modified, independent of changes in startle magnitude on PULSE trials. Thus, for each strain, data were assessed to determine whether drug-induced changes in the calculated amount of %PPI reflected actual changes in sensorimotor gating per se. Five pups (<1.5%) were excluded from analyses based on negligible startle magnitude (mean startle magnitude on pulse alone trials <10).
All startle data were analyzed using an ANOVA with strain, drug treatment and sex as between-subject factors, and trial block and trial type as within-subject repeated measures. Relevant ANOVA values are shown in Table 2. Post hoc comparisons of significant interaction effects and relevant main factor effects were conducted using Tukey–Kramer and one-factor ANOVA tests. Initial analyses of strain differences in sensitivity to the PPI-disruptive effects of APO included all four strains (SDH, WH, F1 and N2) and four doses of APO. However, given the known SDH>WH difference in APO sensitivity at the higher doses of APO (0.25 and 0.5 mg/kg), specific comparisons with F1 and N2 strains were planned a priori, with the simplest ‘additive’ model predictions that: (1) SDH and WH sensitivity would differ by the largest magnitude, (2) F1 sensitivity would be intermediate between parental strains, (3) N2 sensitivity would be intermediate between F1 and SDH and (4) these strain differences would be most evident at the higher doses of APO (0.25 and 0.5 mg/kg). Alpha was 0.05.
For ease of presentation, unless otherwise stated, several normal parametric effects can be assumed to be statistically significant in all startle analyses: effects of trial block on startle magnitude and effect of prepulse intensity on prepulse inhibition. Also, unless otherwise stated, reported values of mean %PPI can be assumed to be collapsed across all prepulse intensities and trial blocks. For most instances, only statistically significant effects, or those relevant to the critical comparisons, will be reported in detail.
RESULTS
The major dependent measure of these studies was PPI; all findings with this measure, in addition to startle magnitude, and the implications of these results in terms of changes in sensorimotor gating are summarized in the text and in Tables 1 and 2. Additional behavioral measures are also reported, as they may influence the interpretation of PPI results.
General Appearance
Litter size for all 4 populations was comparable, ranging from a mean of 14.21 pups (N2) to 16 pups (F1). The general appearance of F1 and N2 rats was comparable to parental SDH and WH rats, although F1 pups weighed significantly less at the time of testing, compared to other populations. ANOVA revealed that F1 pups at testing weighed significantly less than either SDH (p<0.05), WH (p<0.05) or N2 pups (p<0.05) at this age (F=23.90, df 3, 292, p<0.0001). F1 weight did not differ based on maternal strain (F<1).
Startle patterns across the four strains
‘Baseline’ levels of PPI in response to vehicle (0 mg/kg) in SDH, WH, F1 and N2 rats, are seen in Figure 2. ANOVA of %PPI revealed no significant effects of sex or strain, and no sex×strain interaction. There were significant effects of prepulse intensity and trial block, but no other 2-, 3- or 4-way interactions. Analysis of raw startle scores revealed relevant findings. The significant effect of trial type reflected reduced startle magnitude on prepulse trials compared to pulse-alone trials. The sex×trial type interaction narrowly missed statistical significance (p<0.052), reflecting a trend toward relatively greater startle-inhibiting effects of prepulses in male vs female rats across all strains, and there was a significant interaction of strain×trial type. This latter interaction reflected the proportionally greater startle-inhibiting effects of prepulses in SDH and N2 strains, compared to WH and F1 strains (Figure 2b). There were no other meaningful 2-, 3- or 4-way interactions.

(a) Percent PPI in SDH, N2, F1 and WH rats, in response to APO. Values are means collapsed across three prepulse intensities. ‘N’ are indicated above each bar. Values from parental SDH and WH strains, tested at day 18, were previously reported (Swerdlow et al, 2000). (b) Raw startle scores, on pulse-alone and prepulse+pulse trials, in SDH, F1, N2 and WH rats. PP5, PP10 and PP15.
Sensitivity to the PPI-disruptive effects of APO in SDH, WH, F1 and N2 rats is seen in Figure 3. Most generally, sensitivity to the PPI-disruptive effects of higher doses of APO (0.25 and 0.5 mg/kg) followed the predicted ‘additive’ gradient: SDH>N2>F1>WH. When all doses of APO (vehicle, 0.1, 0.25 and 0.5 mg/kg) were included in the analyses, ANOVA of %PPI revealed significant effects of APO, strain and intensity, and a significant interaction of APO×intensity (Figure 2a). The interaction of strain×APO did not reach significance. However, when ANOVA included only the predicted ‘sensitive’ dose range (vehicle, 0.25 and 0.5 mg/kg), ANOVA revealed the same significant effects of APO (p<0.0001) and strain (p<0.005), and a significant strain×APO interaction (p<0.02). A comparable pattern of results emerged when only the vehicle vs 0.5 mg/kg doses were included in the analyses (eg strain×APO interaction: p<0.001). Confirming that this pattern reflected differences in sensitivity to the sensorimotor gating-disruptive effects of APO, ANOVA of raw startle scores with vehicle, 0.25 and 0.5 mg/kg doses confirmed the critical interaction of trial type×APO×strain (p<0.04) (Figure 2b). Inspection of the data confirmed the predicted ‘additive’ gradient: mean % (SEM) disruption of PPI by 0.25 and 0.5 mg/kg APO (relative to ‘vehicle’ levels of PPI) for SDH, N2, F1 and WH rats was 45.61, 32.96, 21.78, and 13.93%, respectively (see Figure 3 for N2 distributions). Post hoc comparisons for 0.5 mg/kg APO confirmed significantly lower levels of PPI in SDH vs WH strains (p<0.0001), and in N2 vs F1 strains (p<0.001).

Magnitude of apomorphine effect on PPI, in SDH, N2, F1 and WH rats, calculated as mean % reduction compared to vehicle dose. Note relatively simple gradient of SDH>N2>F1>WH, corresponding to the genetic background: greater representation of SDH genes is accompanied by greater APO sensitivity. APO effect distribution among F1 and N2 rats (inset) revealed generally normal distributions, with a small number of ‘outliers’.
Startle magnitude in SDH, WH, F1 and N2 rats is seen in Figure 4. For vehicle-treated rats, ANOVA of startle magnitude on pulse-alone trials revealed a significant effect of strain, but not sex, and no significant strain×sex interaction. Post hoc comparison revealed F1<N2 startle magnitude (p<0.05), but no other group differences. When all doses of APO were considered, ANOVA revealed significant effects of APO and strain, but no significant interaction of APO×strain. There was no significant effect of sex, or other informative 2-, 3- or 4-way interactions.

Startle magnitude on pulse-alone trials in SDH, N2, F1 and WH rats, during blocks 2 and 3 (when PPI was assessed).
Habituation (change in startle magnitude in initial vs final blocks of pulse-alone trials) in SDH, WH, F1 and N2 rats is seen in Figure 5. For vehicle-treated rats, ANOVA of startle magnitude in the initial and final trial block revealed no significant effect of sex, a significant effect of strain and no significant sex×strain interaction. Habituation was reflected in a significant effect of trial block, and there was a significant interaction of strain×trial block. Inspection of the data revealed that this apparent group difference in habituation primarily reflected the substantially lower levels of startle magnitude in the initial trial block in F1's, compared to all other strains (p<0.05, all comparisons). Thus, this interaction more directly reflects reduced startle magnitude in F1 rats, rather than diminished habituation per se.

Startle magnitude on pulse-alone trials in SDH, N2, F1 and WH rats, during the initial and final trial blocks (blocks 1 and 4) which contained only three pulse-alone trials. Decline and startle magnitude in block 4 vs block 1 is a reflection of reflex habituation across the test session.
When all doses of APO were included, ANOVA revealed a significant effect of strain, no significant effect of APO or sex, and no significant 2- or 3-way interactions. There was a significant effect of trial block, and a significant interaction of strain×trial block, but the interaction of APO×trial block narrowly missed statistical significance (p<0.06). Inspection of the data revealed that APO reduced or eliminated habituation in SDH rats, and to a lesser degree in N2 and F1 rats. For SDH rats, APO (0.1 mg/kg) eliminated the reduction in startle magnitude between the first and last trial blocks, without significantly changing startle magnitude in the first trial block. Higher doses of APO reduced startle magnitude in the initial block, and no additional reflex diminution was observed across the session. In N2 and F1 rats, the highest dose (0.5 mg/kg) caused a small APO-induced reduction in startle magnitude in the first trial block, and a proportional APO-induced increase in startle magnitude in the last trial block. In contrast, APO had no evident effects on habituation in WH rats.
Levels of gross motor activity during startle testing (‘NOSTIM’ activity) in SDH, WH, F1 and N2 rats are seen in Table 1, and ANOVA values are seen in Table 2. As reported previously (Swerdlow et al, 2000), APO increased NOSTIM levels, and NOSTIM levels differed across strains. There were no effects of sex on NOSTIM activity, and no significant 2- or 3-way interactions. The main effect of rat strain on this measure appeared to reflect the relatively reduced impact of APO on NOSTIM activity in F1 rats, compared to all other strains, at all active doses of APO.
Startle patterns within F1 and N2 generations
While the main aim of these studies was to identify a pattern of inheritance of the APO-sensitivity ‘phenotype’ across these four strains, it was also possible to identify the impact of some specific lineage features on basal (vehicle) PPI levels, independent of the ‘APO sensitivity’ phenotype.
Each F1 rat had one SDH and one WH parent (‘F0’), and basal (vehicle) levels of PPI in F1 rats did not differ based on F0 maternal strain (F<1). Each N2 rat had one F1 parent and one SDH parent; in this case, litters differed in the sex of the F1 vs SDH parental strains, and in the sex of the F0 WH vs SDH strains. In the N2, stronger maternal WH background led to less baseline PPI. ANOVA of PPI in vehicle-treated rats revealed a significant effect of F1 maternal strain and a significant interaction of F1 maternal strain×F0 maternal strain. N2 rats derived from maternal WH F1s had relatively lower baseline PPI compared to those derived from maternal SDHs, and this effect was amplified if the F1 had been derived from maternal WH's.
It was also possible to assess within the N2 strain the impact of selectively crossing the extreme 10% of F1 rats (based on PPI level after 0.1 mg/kg APO) to SDH rats. Among N2 rats treated with vehicle, PPI was significantly greater for those whose F1 was from the higher decile of PPI (F1/high) compared to those whose F1 was from the lower decile of PPI (F1/low). This effect did not interact with any of the other aspects of lineage discussed above (F1 or F0 maternal strain).
Among F1 rats, the significant effect of APO on PPI did not differ based on the F0 maternal strain. Similarly, in N2 rats, the significant effect of APO did not differ based on either the F1 maternal strain the F0 maternal strain, or the F1×F0 interaction. There were also no interactions of this APO sensitivity with the F1 level of PPI, and there were no other relevant 2-, 3- or 4-way interactions.
Related phenotypes in parental SDH and WH rats
An unaddressed issue regarding the heritability of the PPI APO sensitivity phenotype is its neurochemical specificity, that is, does it simply reflect a more ‘disruptible’ PPI in the SDH rats, or does it have something more specific to do with the DAergic system? To address this issue, we compared sensitivity to the PPI-disruptive effects of the NMDA antagonist PCP in SDH and WH pups at 18 days of age (Figure 6). ANOVA of PPI revealed a dose-dependent PCP-induced disruption of PPI that did not differ across strains. Other strain differences were observed: in this particular comparison, startle magnitude in SDH rats was significantly greater than in WH rats, PCP increased startle magnitude in WH but not SDH rats and PCP increased NOSTIM activity in SDH rats more than in WH rats (data not shown).

PPI in 18 days SDH and WH rats 10 min after treatment with PCP (vehicle, 0.25, 0.75 or 1.25 mg/kg). Comparable sensitivity to the PPI-disruptive effects of PCP was observed across strains. ‘N’ are indicated above each bar.
DISCUSSION
Strain and supplier differences among outbred SD and W rats have been reported across several domains of neurobiological function (eg Luedtke et al, 1992; Gleason et al, 1999; Loscher et al, 1998; Oliff et al, 1997,1996; Turnbull and Rivier, 1999). Rigdon (1990) and others (Varty and Higgins, 1994; Hitchcock et al, 1999) have reported differences in sensitivity to the PPI-disruptive effects of APO across strains, and within strains, across suppliers. Consistent with this, our group (Swerdlow et al, 2000) reported opposite patterns of strain differences in APO sensitivity among rats obtained from two different suppliers. The major finding of the present study is that the sensitivity to the PPI-disruptive effects of APO in rats is a heritable trait that differs among outbred SDH and WH rats, and is apparently controlled by relatively simple genetic mechanisms. In two successive generations, the sensitivity to APO in this measure reached intermediate levels, first between parental SDH and WH strains, and then between F1and SDH strains. In more recent studies (Swerdlow et al, 2002a,2002b), we have observed a similar generational pattern in APO sensitivity among SDH, Long Evans (LEH) rats, their F1 offspring and an N2 cross (sensitivity: SDH>N2>F1>LEH). The physiological basis for the observed ‘gradients’ in sensitivity to the mixed D1/D2 agonist APO is not known, but previous studies have identified strain differences in D2 receptor gene polymorphisms between SD and W rats (Luedtke et al, 1992).
Studies conducted with SDH and LEH rats have demonstrated larger differences in PPI APO sensitivity (d=3.55) (Swerdlow et al, 2001a,2001b), compared to those detected in SDH vs WH rats (d=2.01) (Swerdlow et al, 2000). While it was not assessed in the present study, we previously reported that SDH vs LEH strain differences in PPI APO sensitivity do not reflect differences in brain regional levels of APO, and are reproduced by central (intracerebroventricular) administration of APO (Swerdlow et al, 2002a,2002b). Thus, at least in the comparison of SDH vs LEH strains, differences in this measure appear to reflect differences in the APO sensitivity of brain circuitry, rather than pharmacokinetic or pharmacodynamic differences that might influence the ability of APO to access brain substrates responsible for the regulation of PPI.
The present study compared the phenotype of PPI drug sensitivity—quantified by a dose–response function—across four different rat populations. The dose-sensitivity function for each population reflected the behavioral response to four doses of APO, each assessed with different sets of rats in a between-subject design. Since the response of any given rat in this design represented only a ‘fragment’ of the population phenotype, this design is not optimally suited for quantitative genetic analyses. For example, estimates of the genetic vs nongenetic contributions to the PPI APO sensitivity phenotype would be more easily obtained using a single ‘APO effect’ value for each rat (eg F1) and both of its parents (eg F0), based on within-subject comparisons of PPI after vehicle vs 1 active dose of APO (eg 0.5 mg/kg). Such information was not available from the present study.
Strain differences in the APO sensitivity ‘phenotype’ corresponded to differences in some, but not other, startle characteristics. For example, among the four populations in this study, PPI sensitivity to APO was lowest in WH rats, but WH rats were among the most sensitive to the impact of APO on NOSTIM levels. Reduced PPI sensitivity to APO also showed no obvious relationship to basal levels of PPI (which did not differ significantly among the four populations), startle magnitude (lowest in the F1 strain) or reflex habituation (not different between SDH and WH rats). Interestingly, APO appeared to reduce startle habituation in a manner that parallelled (albeit modestly) its impact on PPI across strains, disrupting habituation most potently in SDH and N2 rats. The conceptual and physiological relationships between PPI and startle habituation are areas of intense interest, in part because both of these forms of startle plasticity are impaired in schizophrenia (Braff et al, 1978; Geyer and Braff, 1982).
We previously reported SDH vs WH strain differences in other behavioral effects of DA agonists (Swerdlow et al, 2000). Compared to SDH rats, WH rats were relatively more sensitive to the locomotor-activating effects of the indirect DA agonist d-amphetamine, and to behaviorally activating effects of the D1 agonist SKF 82956. Identical patterns of sensitivity to the PPI-disruptive effects of PCP in SDH and WH rats suggest that the observed differences in PPI APO sensitivity do not reflect a ‘generalized’ physiological tendency for greater PPI ‘disruptibility’ in SDH vs WH rats. Heritable influences on basal levels of PPI (rather than sensitivity to APO per se) were also seen in the present study, both via selective breeding of F1 rats with extremes of PPI after challenge with a single low dose of APO, and via the maternal contribution of WH background. While this latter effect might be explained by differences in the in utero or preweaning environment, it did not relate in any simple way to litter size or weight difference among the litters of different lineage.
One major focus of our work over the past 20 years has been the neural circuit regulation of PPI in rats. The simplest interpretation of the present data is that properties of this PPI regulatory circuitry that determine the sensitivity to DA receptor stimulation are different in two outbred rat strains, and are regulated by genes in a manner that results in a relatively simple pattern of inheritance. Based on our past work and that of other groups (cf Swerdlow et al, 2001a,2001b), the most parsimonious predictions would be that the genes responsible for these inherited patterns are acting via DAergic substrates within the ventral striatum (nucleus accumbens and anteromedial caudate nucleus) or medial prefrontal cortex. Of course, it is equally possible that the differences reflect the impact of genes ‘beyond’ the DA receptor, within efferent projections from the ventral forebrain to pontine circuitry that mediates PPI. In the latter instance, SDH>WH APO sensitivity would reflect differences not in the immediate impact of APO on DA receptors, but instead differences in the translation of this impact through pallidal GABAergic synapses, and ultimately to the pontine tegmentum. Studies in progress are designed to assess these several likely candidate substrates. Knowing the neural circuit basis for genetically based population differences in the DAergic regulation of sensorimotor gating would be very useful for modeling the physiological basis for DA-linked gating deficits in clinical populations, like schizophrenia or Tourette Syndrome (cf Braff et al, 2001).
Several previous findings support the fact that basal level of PPI is a heritable trait. Perhaps the most direct evidence for simple, heritable patterns of basal PPI comes from the observation that PPI is reduced or eliminated in humans with an autosomal dominant genetic disorder—Huntington's disease (HD) (Swerdlow et al, 1995)—and in mice transgenic for the HD gene (Carter et al, 1999). Ellenbroek et al (1995) utilized pharmacogenetic inbreeding to produce strains of rats that were either sensitive (APO-SUS) or insensitive (APO-UNSUS) to the behavioral effects of APO. Male and female rats that exhibited the most (APO-SUS) or least (APO-UNSUS) gnawing in response to 1.5 mg/kg APO were identified from each generation. Within a single generation, APO-SUS rats exhibited significantly less basal PPI than did APO-UNSUS rats. The present observation of reduced PPI in N2 offspring of F1/low rats (F1 rats with relatively low post-APO levels of PPI) may reflect a similar impact of selective breeding. Differences in the levels of basal PPI among inbred Brown–Norway and Wistar/Kyoto strains have also been reported (Palmer et al, 2000).
Of relevance to a wide body of startle research is the degree to which findings in preclinical startle measures can be translated across species to humans (Swerdlow et al, 1999). In addition to genetically extreme conditions such as HD, there is only modest evidence that startle characteristics—and particularly their sensitivity to DA agonists—exhibit heritable patterns in humans. Startle magnitude, and its modulation by affective valence, is highly concordant among monozygotic, but not dizygotic, twins (Carlson et al, 1997). In another study, Hutchison et al (1999) reported that the PPI-disruptive effects of the indirect DA agonist amphetamine were most pronounced in individuals with a personality profile that may be genetically linked with specific D4 DA receptor functions (Paterson et al, 1999). Recent studies have demonstrated the PPI-disruptive effects of direct DA agonists in humans (Abduljawad et al, 1998), and based on the present findings, one might predict that such drug effects would exhibit some degree of genetically mediated variability in humans. The present studies suggest that a relatively simple animal model can be used to study brain mechanisms responsible for the expression of genetic differences in the dopaminergic regulation of sensorimotor gating—a phenotype with direct relevance to several inherited neuropsychiatric disorders.
References
Abduljawad KA, Langley RW, Bradshaw CM, Szabadi E (1998). Effects of bromocriptine and haloperidol on prepulse inhibition of the acoustic startle response in man. J Psychopharmacol 12: 239–245.
Braff DL, Geyer MA, Swerdlow NR (2001). Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharm 156: 234–258.
Braff D, Stone C, Callaway E, Geyer MA, Glick ID, Bali L (1978). Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15: 339–343.
Cadenhead KS, Geyer MA, Braff DL (1993). Impaired startle prepulse inhibition and habituation in schizotypal personality disordered patients. Am J Psychiatry 150: 1862–1867.
Cadenhead KS, Swerdlow NR, Shafer KM, Diaz M, Braff DL (2000). Modulation of the startle response and startle laterality in relatives of schizophrenia patients and schizotypal personality disordered subjects: evidence of inhibitory deficits. Am J Psychiatry 157: 1660–1668.
Carlson SR, Katsanis J, Lacono WG, McGue M (1997). Emotional modulation of the startle reflex in twins: preliminary findings. Biol Psychol 46: 235–246.
Carter RJ, Lione LA, Humby T, Mangiari L, Majal A, Bates GP et al. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington's Disease mutation. J Neurosci 19: 3248–3257.
Ellenbroek BA, Geyer MA, Cools MA (1995). The behavior of APO-SUS rats in animal models with construct validity for schizophrenia. J Neurosci 15: 7604–7611.
Geyer MA, Braff DL (1982). Habituation of the Blink reflex in normals and schizophrenic patients. Psychophysiology 19: 1–6.
Geyer MA, Swerdlow NR (1998). Measurement of startle response, prepulse inhibition, and habituation. In: Crawley JN, Skolnick P (eds). Current Protocols in Neuroscience. John Wiley & Sons: New York, pp 8.7.1–8.7.15.
Gleason TC, Dreiling JL, Crawley JN (1999). Rat strain differences in response to galanin on the Morris water task. Neuropeptides 33: 265–270.
Graham F (1975). The more or less startling effects of weak prestimuli. Psychophysiology 12: 238–248.
Hitchcock JM, Selk DE, Wettstein JG, Rush DK (1999). Intrastrain differences in the disruption of prepulse inhibition in rats by PCP, DOI, and 7-OH-DPAT. Schizophr Res 36: 115.
Hutchison KE, Wood MD, Swift R (1999). Personality factors moderate subjective and psychophysiological responses to d-amphetamine in humans. Exp Clin Psychopharmacol 7: 493–501.
Kinney GG, Wilkinson LO, Saywell KL, Tricklebank MD (1999). Rat strain differences in ability to disrupt sensorimotor gating are limited to the dopaminergic system, specific to prepulse inhibition, and unrelated to changes in startle amplitude or nucleus accumbens dopamine receptor sensitivity. J Neurosci 19: 5644–5653.
Koch M (1998). Sensorimotor gating changes across the estrous cycle in female rats. Physiol Behav 64: 625–628.
Koch M, Schnitzler HU (1997). The acoustic startle response in rats—circuits mediating evocation, inhibition and potentiation. Behav Brain Res 89: 35–49.
Kumari V, Soni W, Sharma T (1999). Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry 156: 1046–1051.
Lipska BK, Swerdlow NR, Geyer MA, Jaskiw GE, Braff DL, Weinberger DR (1995). Neonatal excitotoxic hippocampal damage in rats causes post-pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacology 122: 35–43.
Loscher W, Cramer S, Ebert U (1998). Differences in kindling development in seven outbred and inbred rat strains. Exp Neurol 154: 551–559.
Luedtke RR, Artymyshyn RP, Monks BR, Molinoff PB (1992). Comparison of the expression, transcription and genomic organization of D2 dopamine receptors in outbred and inbred strains of rat. Brain Res 584: 45–54.
Mansbach RS, Geyer MA, Braff DL (1988). Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology 94: 507–514.
Martinez Z, Oostwegel J, Geyer M, Swerdlow NR (2000). Ontogeny of phencyclidine and apomorphine effects on prepulse inhibition (PPI). Pharmacol Biochem Behav 65: 449–457.
Meloni EG, Davis M (1999). Enhancement of the acoustic startle response in rats by the dopamine D1 receptor agonist SKF 82958. Psychopharmacology 144: 373–380.
Oliff HS, Coyle P, Weber E (1997). Rat strain and vendor differences in collateral anastomoses. J Cereb Blood Flow Metab 17: 571–576.
Oliff HS, Marek P, Miyazaki B, Weber E (1996). The neuroprotective efficacy of MK-801 in focal cerebral ischemia varies with rat strain and vendor. Brain Res 731: 208–212.
Palmer AA, Dulawa SC, Mottiwala AA, Conti LH, Geyer MA, Printz MP (2000). Prepulse startle deficit in the Brown Norway rat: a potential genetic model. Behav Neurosci 114: 374–388.
Paterson AD, Sunohara GA, Kennedy JL (1999). Dopamine D4 receptor gene: novelty or nonsense? Neuropsychopharmacology 21: 3–16.
Rigdon G (1990). Differential effects of apomorphine on prepulse inhibition of acoustic startle reflex in two rat strains. Psychopharmacology 102: 419–421.
Swerdlow NR, Braff DL, Taaid N, Geyer MA (1994). Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry 51: 139–154.
Swerdlow NR, Braff DL, Geyer MA (1999). Cross-species studies of sensorimotor gating of the startle reflex. Ann N Y Acad Sci 877: 202–216.
Swerdlow NR, Geyer MA (1998). Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr Bull 24: 285–302.
Swerdlow NR, Geyer MA, Braff DL (2001a). Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenge. Psychopharmacology 156: 194–215.
Swerdlow NR, Martinez ZA, Hanlon FM, Platten A, Farid M, Auerbach P et al (2000). Toward understanding the biology of a complex phenotype: rat strain and substrain differences in the sensorimotor gating-disruptive effects of dopamine agonists. J Neurosci 20: 4325–4336.
Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR (1995). Impaired prepulse inhibition of acoustic and tactile startle in patients with Huntington's Disease. J Neurol Neurosurg Psychiatry 58: 192–200.
Swerdlow NR, Platten A, Kim YK, Gaudet I, Shoemaker J, Pitcher L et al (2001b). Sensitivity to the dopaminergic regulation of prepulse inhibition in rats: evidence for genetic, but not environmental determinants. Pharmacol Biochem Behav 70: 219–226.
Swerdlow NR, Shoemaker JM, Kuczenski R, Pitcher L, Platten A, Auerbach P (2002a). Strain differences in the gating-disruptive effects of apomorphine (APO) are innate and do not reflect differences in brain APO levels. Biol Psychiatry 51: 120S.
Swerdlow NR, Shoemaker JM, Pitcher L, Platten A, Kuczenski R, Eeley CC et al (2002b). Genetic differences in startle gating-disruptive effects of apomoprhine: evidence for central mediation. Behav Neurosci 116: 682–690.
Swerdlow NR, Varty GB, Geyer MA (1997). Discrepant findings of clozapine effects on prepulse inhibition of startle: is it the route or the rat? Neuropsychopharmacology 18: 50–56.
Turnbull AV, Rivier CL (1999). Sprague–Dawley rats obtained from different vendors exhibit distinct adrenocorticotropin responses to inflammatory stimuli. Neuroendocrinology 70: 186–195.
Varty GB, Higgins GA (1994). Differences between three rat strains in sensitivity to prepulse inhibition of acoustic startle response: influence of apomorphine and phencyclidine pretreatment. J Psychopharmacol 8: 148–156.
Weike AL, Bauer U, Hamm AO (2000). Effective neuroleptic medication removes prepulse inhibition deficits in schizophrenia patients. Biol Psychiatry 47: 61–70.
Acknowledgements
This research was supported in part by grants from the National Institute of Mental Health (MH-01436, MH-53484, MH-42228) and the Department of Veterans Affairs VISN 22 Mental Illness Research, Education and Clinical Centers (MIRECC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Swerdlow, N., Platten, A., Hanlon, F. et al. Sensitivity to Sensorimotor Gating-Disruptive Effects of Apomorphine in two Outbred Parental Rat Strains and their F1 and N2 Progeny. Neuropsychopharmacol 28, 226–234 (2003). https://doi.org/10.1038/sj.npp.1300035
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300035
Keywords
This article is cited by
-
Translational value of startle modulations
Cell and Tissue Research (2013)
-
Evaluating the antipsychotic profile of the preferential PDE10A inhibitor, papaverine
Psychopharmacology (2009)
-
Realistic expectations of prepulse inhibition in translational models for schizophrenia research
Psychopharmacology (2008)
-
Prepulse Inhibition Deficits in GAD65 Knockout Mice and the Effect of Antipsychotic Treatment
Neuropsychopharmacology (2004)
-
Heritable differences in the dopaminergic regulation of sensorimotor gating
Psychopharmacology (2004)
