
Abstract
Genetic neuromuscular diseases are clinically and genetically heterogeneous genetic disorders that primarily affect the peripheral nerves, muscles, and neuromuscular junctions. This study aimed to identify pathogenic variants, calculate carrier frequency, and predict the genetic prevalence of autosomal recessive neuromuscular diseases (AR-NMDs). We selected 268 AR-NMD genes and analyzed their genetic variants sourced from the gnomAD database. After identifying the pathogenic variants using an algorithm, we calculated the carrier frequency and predicted the genetic prevalence of AR-NMDs. In total, 10,887 pathogenic variants were identified, including 3848 literature verified and 7039 manually verified variants. In the global population, the carrier frequency of AR-NMDs is 32.9%, with variations across subpopulations ranging from 22.4% in the Finnish population to 36.2% in the non-Finnish European population. The predicted genetic prevalence of AR-NMDs was estimated to be 24.3 cases per 100,000 individuals worldwide, with variations across subpopulations ranging from 26.5 to 41.4 cases per 100,000 individuals in the Latino/Admixed American and the Ashkenazi Jewish populations, respectively. The AR-NMD gene with the highest carrier frequency was GAA (1.3%) and the variant with the highest allele frequency was c.-32-13 T>G in GAA with 0.0033 in the global population. Our study revealed a higher-than-expected frequency of AR-NMD carriers, constituting approximately one-third of the global population, highlighting ethnic heterogeneity in genetic susceptibility.
Similar content being viewed by others

Introduction
Genetic neuromuscular diseases are clinically and genetically heterogeneous genetic disorders that primarily affect the peripheral nerves, muscles, and neuromuscular junctions. Clinical symptoms include muscle weakness, muscle atrophy, joint contractures, cardiomyopathy, exercise intolerance, sensory deficits, fatigue, myalgia, tremors, ataxia, and dysarthria. These symptoms exhibit heterogeneity according to the subtypes and causative genes. The application of high-throughput analysis has revolutionized the diagnosis of genetic neuromuscular diseases, enabling a more accessible and cost-effective approach1. To date, approximately 600 causative genes have been identified2. The incidence and prevalence of genetic neuromuscular diseases have been explored in many studies, and it has been observed that variations in methodologies and ethnicities contribute to substantial discrepancies in reported values3,4,5. To achieve a comprehensive understanding of the epidemiological landscape of genetic neuromuscular diseases on a global scale, a large-scale study using uniform analytic methods is urgently required.
Since the completion of the Human Genome Project6 several databases of human exomes and genomes have been created. Notably, the Genome Aggregation Database (gnomAD) is the most representable resource7. This database serves as a comprehensive repository for information from diverse exomes and genomes, offering valuable insights into human genetic variation. These data suggest that a certain percentage of individuals in the general population carry pathogenic or likely pathogenic variants (PLPVs) of a specific gene. It could provide crucial genomic data for predicting the carrier frequency and genomic prevalence of autosomal recessive Mendelian disorders including inherited retinal diseases, Pompe disease, congenital hypothyroidism, and Upshaw-Schulman syndrome8,9,10,11. This genetic information provides simple epidemiological information and plays a crucial role in various fields, including genetic counseling and drug development.
Therefore, our study aimed to identify PLPVs using genetic data sourced from the gnomAD database. Additionally, our objectives were to the calculate the carrier frequency and predict genetic prevalence of autosomal recessive neuromuscular diseases (AR-NMDs) across major ethnicities worldwide.
Results
Identification of PLPVs of AR-NMD genes
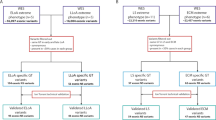
Figure 1 and Supplementary Table 2 illustrate the analytical scheme used to evaluate the AR-NMD gene variants. We identified a comprehensive set of 326,632 AR-NMD variants derived from the gnomAD database. We excluded two variants that were present in only one homozygous individual and were not found in heterozygotes, suggesting the potential unreliability of the reads. We also excluded two variants: c.5C>G in SMN1 and c.359-1G>T in TTPA). Despite being considered PLPVs, the total allele counts at the genomic positions of the two variants were < 1000 (86 and 526, respectively). Subsequently, 326,628 variants were categorized into two main subgroups: 25,917 truncating variants and 300,711 non-truncating variants. Among the truncating variants, most (25,659) exhibited an allele frequency < 0.005. This analysis identified 9658 truncating PLPVs (2,623 literature verified variants and 7035 manually verified variants). Among the 300,711 non-truncating variants, 45,047 variants had references in scientific literature, whereas 255,664 lacked references. This analysis identified 1229 non-truncating PLPVs (1225 literature verified variants and 4 manually verified variants). In summary, a total of 10,887 PLPVs were identified, including 3,848 (35.3%) literature verified and 7039 (64.7%) manually verified variants (Supplementary Tables 2 and 3). Among the manually verified variants, 7035 were truncating and classified as PLPVs based on two pieces of evidence: (1) a null variant in a gene where the loss of function is a known mechanism of the disease, and (2) absent or an extremely low frequency in controls in the gnomAD database. The remaining four manually verified variants were missense variants and were classified as PLPVs based on three pieces of evidence: (1) the same amino acid change as a previously established pathogenic variant, (2) absent or extremely low frequency in controls in the gnomAD database, and (3) multiple lines of computational evidence supporting a deleterious effect on the gene or gene product. There were 22 homozygous PLPVs of AR-NMDs in the gnomAD database (Supplementary Table 4).

Flowchart depicting the analytical scheme for the variants sourced from the gnomAD database.
The allele frequency ratio of the literature verified PLPVs to total PLPVs is 60.4% in the global population. This allele frequency ratio was the highest in the ASJ (83.3%) population, followed by the NFE (65.0%), FIN (64.7%), AMR (59.4%), SAS (52.7%), EAS (51.4%), and AFR (50.3%) populations (Supplementary Table 5).
Carrier frequency and predicted genetic prevalence of AR-NMDs
Figure 2 shows the carrier frequency and predicted genetic prevalence of AR-NMDs. The carriers of AR-NMDs are predicted to comprise 32.9% of the global population. Among subpopulations, the NFE population showed the highest carrier frequency at 36.2%, followed by AFR, EAS, AMR, ASJ, SAS, and FIN at 34.0%, 33.4%, 29.7%, 25.6%, 24.1%, and 22.4%, respectively. The predicted genetic prevalence of AR-NMD is estimated to be 24.3 cases per 100,000 individuals worldwide. The ASJ population had the highest predicted genetic prevalence of 41.4 cases per 100,000, followed by EAS, FIN, NFE, AFR, SAS, and AMR at 36.9, 35.2, 33.2, 30.3, 29.8, and 26.5 per 100,000 individuals, respectively.

Global carrier frequency and predicted genetic prevalence of autosomal recessive neuromuscular diseases represented per subpopulation worldwide. The gray bars measured along the left vertical axis indicate the carrier frequency. The blue bars measured along the right vertical axis indicate the predicted genetic prevalence.
Common AR-NMD genes according to subpopulation
Table 1 and Supplementary Table 6 show the carrier frequency and predicted genetic prevalence for each AR-NMD gene in the different subpopulations. The GAA (1.3%) was the only gene with a carrier frequency exceeding 1% in the global population. However, the number of genes with a carrier frequency exceeding 1% varied among subpopulations. ASJ had the highest count, with four genes exceeding this threshold (FKTN, 1.7%; GBE1, 1.4%; GAA, 1.3%; and PFKM, 1.0%), followed by EAS with three (GAA, 1.6%; SLC22A5, 1.4%; and NEB, 1.1%), FIN with three (GLE1, 2.6%; ANO5, 1.4%; and UBA5, 1.2%), NFE with two (GAA, 1.7% and ANO5, 1.1%), AFR with two (PGAM2, 1.4% and GAA, 1.3%), AMR with one (ANO5, 1.4%), and SAS with one (GNE, 2.7%).
Allele frequencies of PLPVs of AR-NMD genes
Table 2 and Supplementary Table 3 show the allele frequency of each PLPV associated with AR-NMDs in the major subpopulations. The variant with the highest allele frequency was c.-32-13T>G in GAA with 0.0033 in the global population. Within the subpopulations, the PLPV with the highest allele frequency was c.2179G>A in GNE, reaching 0.0133 in SAS, followed by c.433-10A>G in GLE1 at 0.0118 in FIN; c.1167dupA in FKTN at 0.0078 in ASJ; c.233G>A in PGAM2 at 0.0068 in AFR; c.-32-13T>G in GAA at 0.0053 in NFE; c.1385-42G>C in AGRN at 0.0028 in EAS; and c.-32-13T>G in GAA at 0.0027 in AMR.
gnomAD individuals with homozygous PLPVs in AR-NMD genes
To determine whether gnomAD included individuals with AR-NMDs, we compared the number of gnomAD individuals with homozygous PLPVs and the expected number of individuals with homozygous PLPVs calculated using the predicted genomic prevalence. We identified 28 gnomAD individuals with homozygous PLPVs in the AR-NMD genes (Supplementary Table 4). However, the expected number of individuals with homozygous variants using the predicted genomic prevalence was 5.8 (Supplementary Table 6).
Discussion
We conducted the first systematic analysis to estimate PLPVs using a global genomic database. One-third of the PLPVs were previously reported in databases and literature, and the remaining two-thirds were manually classified by assessing their pathogenicity according to the 2015 ACMG guidelines. Therefore, we believe that we accurately analyzed the pathogenicity of all variants and the selected PLPVs.
Our study revealed that the allele frequency ratio for the literature verified PLPVs compared to the total PLPVs is high in European populations, including ASJ, NFE, and FIN, compared with the non-European population including AMR, SAS, EAS, and AFR. This finding is consistent with previous observations, indicating the limited knowledge of genetic diversity outside European populations12,13.
Our study revealed that the predicted genetic prevalence of AR-NMDs was 24.3 per 100,000 individuals. This result is lower than the prevalence of genetic neuromuscular diseases ranging from 28.6 to 82.8 per 100,000 individuals in previous studies3,4. However, considering that these studies included other neuromuscular diseases3,4, our results showed that the prevalence of AR-NMDs was significantly higher than expected. This conclusion is based on the following evidence. First, the major common causative genes of genetic neuromuscular diseases are not inherited in an autosomal recessive manner. X-linked dystrophinopathy, autosomal dominant myotonic dystrophy, and autosomal dominant facioscapulohumeral muscular dystrophy account for more than 27% of all patients with genetic myopathy5. Additionally, autosomal dominant or X-linked hereditary motor and sensory neuropathy account for 96–98% of patients with genetically-confirmed cases14,15. Second, our analysis did not include both deletions or duplications of one or more exons. For example, homozygous absence of exon 7 in SMN1 is found in approximately 95% of patients with spinal muscular atrophy, the most common disease of genetic motor neuron disease16. Third, the diagnostic rates of exome sequencing and genome sequencing are only 30–40% in patients with Mendelian diseases including genetic neuromuscular diseases1. Conversely, two-thirds of patients with genetic neuromuscular diseases cannot be identified using exome and genome sequences sourced from the gnomAD database.
Our study revealed that carrier frequency is high in the NFE and AFR populations, but the predicted genetic prevalence is high in the ASJ and EAS populations. This is because the number of genes with carrier frequencies exceeding 1% in the ASJ and EAS populations is four and three, respectively, which is higher than that in other populations.
Our results revealed that the most common causative gene of AR-NMD is GAA in the global population. One previous study showed that SMN1 and GAA is the most common causative genes of AR-NMDs in 108 autosomal recessive Mendelian diseases17. However, we could not analyze large exonal deletions, the main alterations in SMN1 in this study. This is likely why GAA emerged as the most common AR-NMD gene. This previous study showed that the carrier frequency of individuals with PLPVs in GAA was 0.8%, which is slightly lower than our results17. This study also indicated that the carrier frequency of PLPVs in GAA was much lower in the EAS population (0.3%) than in the European population (0.9%)17. This contrasts our results, which found similar frequencies in both populations. This was because the [c.752C>T; c.761C>T] variant, the most common PLPV in the EAS population, was not found or was classified as a variant of uncertain significance in this study. This variant is currently considered the major PLPV in GAA, especially in the EAS population, but has been frequently classified as a variant of uncertain significance18. Our predicted genetic prevalence was somewhat consistent with the results from newborn screening programs, which reported a prevalence ranging from 3.6 to 11.5 per 100,000 individuals11,19,20,21,22. Additionally, one previous study used the same analytical method to assess the carrier frequency and genetic prevalence of individuals with PLPVs in GAA11. The results reported a carrier frequency of 1.3% and genetic prevalence of 4.3 per 100,000 persons in GAA, which is nearly identical to our results11. The most common PLPVs in GAA vary by ethnicity, with the c.-32-13T>G and [c.752C>T; c.761C>T] variants being most common in the NFE and EAS population, respectively. These findings are consistent with previous study11.
Our findings on the common causative genes in the EAS, NFE, ASJ, FIN, and SAS populations were consistent with previous results. In the NFE population, the most common causative gene is GAA and the most common PLPV is c.-32-13T>G in GAA, which is supported by several studies11,17. However, our predicted genetic prevalence was higher than previous prevalence data (1.7–2.5 per 100,000) for the United States and Dutch populations23,24. In the ASJ population, FKTN is well-known to be associated with the common founder variant, c.1167dupA25. The carrier frequency of this variant was 0.0160, which is consistent with our results (0.0156)26. In the EAS population, GAA was the most common AR-NMD gene, which is consistent with previous results in the Chinese population27. One study showed the carrier frequency of GAA ranged from 0.01 to 0.00528. Another recent study showed that the carrier frequency of individuals with PLPVs in GAA in Chinese population was 0.0145, which is similar to our result (0.0158) in the EAS population27. Lethal congenital contracture syndrome and lethal arthrogryposis with anterior horn cell disease associated with GLE1 were first reported in FIN population29. In particular, c.433-10A>G in GLE1, called FinMajor, is a representative pathogenic variant in GLE1 that is commonly observed in the Finnish population29,30. Additionally, the prevalence of individuals with PLPVs in GAA are relatively low in the FIN population compared with the NFE population31. In the Indian population, a representative group of the SAS population, large-scale genetic analysis showed that the most common causative gene of genetic myopathy was GNE, which is consistent with our result for the SAS population32. Additionally, c.2179 G>A in GNE is a founder and major PLPV in the Indian population, which is consistent with results from previous studies32,33.
Our findings on the common causative genes in the AFR and AMR populations differ from those of previous studies. However, there have been few large-scale genetic analyses of these populations compared to those in the European population. Our results showed that PGAM2 is the most common gene in the AFR population. However, no study has investigated the prevalence of individuals with alterations in PGAM2. Muscle phosphoglycerate mutase deficiency caused by alterations in PGAM2 are frequently found in African-Americans34. In particular, c.233G>A in PGAM2 is a founder variant in the African-American population, which is consistent with our result34. One epidemiological study in the Moroccan population showed that the carrier frequency of the c.525del pathogenic variant in SGCG was 4%, which contrasts our result (1%) in the AFR population35. However, because the Moroccan population is a small subset of the AFR population, the results of two studies cannot be directly compared. In the AMR population, our study showed that ANO5 was the most common causative gene, and the c.692G>T variant in ANO5 was the second most common PLPV. This variant in ANO5 is a common PLPV in the European population but is different from the c.191dupA and c.2272C>T variants, which are the most common variants in the Northern European and FIN populations, respectively36,37. Several molecular genetic studies have shown that DYSF and CAPN3 are more prevalent as causative genes than ANO5 in patients with limb-girdle muscular weakness in the Latino, Chilean, or Argentine populations38,39,40. Analysis of common causative genes of AR-NMD in the AFR and AMR populations requires additional large-scale studies.
The number of individuals from the gnomAD database with PLPVs was approximately five times higher than the expected number of individuals with homozygous PLPVs. This finding suggests that the gnomAD database includes patients with AR-NMD and other possible genetic diseases, which is consistent with the results of a previous study8. Additionally, this discrepancy might lead to an overestimation of the allele frequency of PLPVs, despite the small number of homozygous gnomAD individuals (28 of 141,456).
Our study had several limitations. First, we identified carriers and individuals by focusing on genes and PLPVs known to cause AR-NMDs. Therefore, our study could not analyze patients with alterations in unidentified causative genes. Furthermore, we posit that many disease-causing variants, particularly non-truncating variants that have not been documented in literature, are classified as variants of unknown significance. Second, we could not analyze large deletions or duplications of exons because of the nature of the gnomAD database. Third, we calculated the predicted genetic prevalence based on the Hardy–Weinberg equation. Therefore, the actual prevalence of AR-NMDs may be higher than the values obtained in the African and South Asian populations, which have high levels of consanguinity and intracommunity marriages41. Forth, this study analyzed gnomAD data, next-generation sequencing data that has not been verified through Sanger sequencing.
In summary, our study offers a comprehensive analysis of the carrier frequency and predicted genetic prevalence of AR-NMDs in the global population and six major subpopulations. These results provide crucial insights for the epidemiological analysis, genetic counseling, newborn screening, diagnostic approaches, and therapeutic development of AR-NMDs. We found that the carrier frequency of AR-NMDs was higher than expected, constituting approximately one-third of the entire human population. Furthermore, our findings highlight the heterogeneity of genetic susceptibility to AR-NMDs based on ethnicity.
Materials and methods
Selection of AR-NMD genes
Based on the gene table of neuromuscular diseases available at (https://www.musclegenetable.fr), we identified 584 genes associated with neuromuscular diseases, including muscular dystrophies, congenital muscular dystrophies, congenital myopathies, distal myopathies, other myopathies, myotonic syndromes, ion channel muscle diseases, metabolic myopathies, hereditary cardiomyopathies, congenital myasthenic syndromes, motor neuron diseases, hereditary ataxia, hereditary motor and sensory neuropathies, and hereditary paraplegias. We then selected genes linked to AR-NMDs. Among these, LAMA5 and LAMB2 were excluded for the following reasons: LAMA5 is associated with various multisystem syndromes including neuromuscular diseases, epilepsy, and nephropathy; LAMB2 is associated with congenital myasthenic and nephrotic syndromes. However, although CAPN3 is associated with autosomal dominant and recessive inheritance patterns, it was included because it is a common causative agent of AR-NMDs. A total of 268 genes linked to AR-NMDs were selected (Supplementary Table 1).
Analysis of pathogenicity of variants from the gnomAD database
We analyzed genetic variants derived from a dataset of 125,748 exome sequences and 15,708 genome sequences, sourced from the gnomAD database (version 2.1.1; https://gnomad.broadinstitute.org). This dataset comprised individuals from diverse populations: 12,487, 17,720, 5185, 9977, 12,562, 64,603, and 15,308 from the African/African-American (AFR), Latino/Admixed American (AMR), Ashkenazi Jewish (ASJ), East Asian (EAS), Finnish (FIN), non-Finnish European (NFE), South Asian (SAS) populations, as well as 3614 from individuals categorized as “other.”
We organized all the variants associated with AR-NMD genes, sourced from the gnomAD database, into distinct categories (Fig. 1). This classification involved two main subgroups: truncating variants, such as frameshift, splice-site, nonsense, and start-loss variants, and non-truncating variants, including missense, intron, in-frame-deletion/duplication, 5ʹUTR, and 3ʹUTR variants. For further analysis, we initially separated the truncating variants based on their allele frequencies, using a cutoff threshold of 0.005, and divided the non-truncating variants into two subgroups, those with and without references in the scientific literature. Subsequently, we stratified these non-truncating variants using an allele frequency threshold of 0.005.
For literature verified variants, we compiled relevant data from scientific literatures and pertinent databases, such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk/ac/), and LOVD (https://databases.lovd.nl/shared/genes). Variants lacking representation in scientific literature underwent a comprehensive manual assessment to ascertain their pathogenicity. The process of identifying PLPVs was performed in accordance with the 2015 guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology42. If the total allele counts at the genomic position of the variants were < 1000, they are not classified as PLPVs. This is because variants with very low allele numbers may have a large effect on overall allele frequency.
Analysis of allele frequency, carrier frequency, and predicted genetic prevalence
The gnomAD database provides specific values for each subpopulation, including allele count, allele number, and homozygote count. Using these values, we calculated the allele and carrier frequencies for a single variant. These calculations were based exclusively on heterozygous PLPVs, as previously described8:
Subsequently, we calculated the carrier frequency and predicted the genetic prevalence at the genetic level, as previously described8:
Finally, we determined whether gnomAD included individuals who might have been affected by AR-NMDs but were still asymptomatic, or whose condition was not immediately recognized. To do this, we compared the number of gnomAD individuals with homozygous PLPVs and the expected number of individuals with homozygous variants, calculated using a previously described method8.
Ethnical consideration
This study was approved by the Institutional Review Board of the Gangnam Severance Hospital, Korea (approval number: 3-2023-0065). The requirement for written informed consent was waived by the board because of all data were provided by the gnomAD with all personal information anonymously encrypted according to a strict confidentiality protocol.
Data availability
Public datasets used in the study are described in the Methods section. All data are available in the manuscript.
References
Chung, C. C. Y. et al. Meta-analysis of the diagnostic and clinical utility of exome and genome sequencing in pediatric and adult patients with rare diseases across diverse populations. Genet. Med. 25, 100896 (2023).
Benarroch, L., Bonne, G., Rivier, F. & Hamroun, D. The 2023 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 33, 76–117 (2023).
Carey, I. M. et al. Prevalence and incidence of neuromuscular conditions in the UK between 2000 and 2019: A retrospective study using primary care data. PLoS One 16, e0261983 (2021).
Emery, A. E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1, 19–29 (1991).
Theadom, A. et al. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 43, 259–268 (2015).
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Karczewski, K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Biorxiv. 531210 (2019).
Hanany, M., Rivolta, C. & Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 117, 2710–2716 (2020).
Zhao, T., Fan, S. & Sun, L. The global carrier frequency and genetic prevalence of Upshaw-Schulman syndrome. BMC Genom. Data 22, 1–8 (2021).
Park, K.-S. Analysis of worldwide carrier frequency and predicted genetic prevalence of autosomal recessive congenital hypothyroidism based on a general population database. Genes 12, 863 (2021).
Park, K. S. Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Mol. Genet. Metab. Rep. 27, 100734 (2021).
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 26–31 (2019).
Pereira, L., Mutesa, L., Tindana, P. & Ramsay, M. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genet. 22, 284–306 (2021).
Fridman, V. et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: A cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 86, 873–878 (2015).
Rudnik-Schöneborn, S. et al. Pregnancy outcome in Charcot–Marie–Tooth disease: Results of the CMT-NET cohort study in Germany. Eur. J. Neurol. 27, 1390–1396 (2020).
Cobben, J. et al. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet. 57, 805 (1995).
Lazarin, G. A. et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: Results from an ethnically diverse clinical sample of 23,453 individuals. Genet. Med. 15, 178–186 (2013).
Momosaki, K. et al. Newborn screening for Pompe disease in Japan: Report and literature review of mutations in the GAA gene in Japanese and Asian patients. J. Hum. Genet. 64, 741–755 (2019).
Chiang, S.-C., Hwu, W.-L., Lee, N.-C., Hsu, L.-W. & Chien, Y.-H. Algorithm for Pompe disease newborn screening: Results from the Taiwan screening program. Mol. Genet. Metab. 106, 281–286 (2012).
Mechtler, T. P. et al. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 379, 335–341 (2012).
Burton, B. K. et al. Newborn screening for Pompe disease in Illinois: Experience with 684,290 infants. Int. J. Neonatal Screen 6, 4 (2020).
Minter Baerg, M. M. et al. Precision newborn screening for lysosomal disorders. Genet. Med. 20, 847–854 (2018).
Ausems, M. et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur. J. Hum. Genet. 7, 713–716 (1999).
Martiniuk, F. et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am. J. Med. Genet. 79, 69–72 (1998).
Wallace, S. E. & Bean, L. J. GeneReviews®[Internet] (University of Washington, 2018).
Shi, L. et al. Comprehensive population screening in the Ashkenazi Jewish population for recurrent disease-causing variants. Clin. Genet. 91, 599–604 (2017).
Chan, S. H. et al. Analysis of clinically relevant variants from ancestrally diverse Asian genomes. Nat. Commun. 13, 6694 (2022).
Lin, C.-Y. & Shieh, J.-J. Molecular study on the infantile form of Pompe disease in Chinese in Taiwan. Zhonghua Min Xiao Er Ke Yi Xue Hui Za Zhi 37, 115–121 (1996).
Nousiainen, H. O. et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet. 40, 155–157 (2008).
Folkmann, A. W., Collier, S. E., Zhan, X., Ohi, M. D. & Wente, S. R. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 155, 582–593 (2013).
Palmio, J. et al. Screening for late-onset Pompe disease in Finland. Neuromuscul. Disord. 24, 982–985 (2014).
Chakravorty, S. et al. Clinical and genomic evaluation of 207 genetic myopathies in the Indian subcontinent. Front. Neurol. 11, 559327 (2020).
Attri, S., Sharma, V., Kumar, A., Verma, C. & Gahlawat, S. K. Dissecting role of founder mutation p. V727M in GNE in Indian HIBM cohort. Open Med. 16, 1733–1744 (2021).
Naini, A. et al. Muscle phosphoglycerate mutase deficiency revisited. Arch Neurol. 66, 394–398 (2009).
El Kerch, F. et al. Carrier frequency of the c.525delT mutation in the SGCG gene and estimated prevalence of limb girdle muscular dystrophy type 2C among the Moroccan population. Genet. Test Mol. Biomarkers 18, 253–256 (2014).
Hicks, D. et al. A founder mutation in Anoctamin 5 is a major cause of limb girdle muscular dystrophy. Brain 134, 171–182 (2011).
Penttilä, S. et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 78, 897–903 (2012).
Schiava, M. et al. Genetic characterization of limb girdle muscular dystrophies and pompe disease in a large argentine cohort. Neurol. Perspect. 2, 123–133 (2022).
Bevilacqua, J. A. et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet. J. Rare Dis. 15, 1–11 (2020).
Cerino, M. et al. Genetic profile of patients with limb-girdle muscle weakness in the Chilean population. Genes 13, 1076 (2022).
Hamamy, H. Consanguineous marriages: Preconception consultation in primary health care settings. J. Community Genet. 3, 185–192 (2012).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Acknowledgements
We would like to thank the many individuals whose sequence data were aggregated in gnomAD for their contributions to the research, and the users of gnomAD for their collaborative feedback. We would also like to thank the gnomAD steering committee for their contribution to the development of the gnomAD resources.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education [Grant no. RS-2023-00242210].
Author information
Authors and Affiliations
Contributions
H.P. conceived the idea and all the authors performed literature search, wrote the manuscript, and proofread the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, WJ., Kim, SH., Lee, S.R. et al. Global carrier frequency and predicted genetic prevalence of patients with pathogenic sequence variants in autosomal recessive genetic neuromuscular diseases. Sci Rep 14, 3806 (2024). https://doi.org/10.1038/s41598-024-54413-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54413-1
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
