
Abstract
Neglected tropical diseases, such as leishmaniasis, lead to serious limitations to the affected societies. In this work, a structure–activity relationship (SAR) study was developed with a series of quinoxaline derivatives, active against the promastigote forms of Leishmania amazonensis. As a result, a new quinoxaline derivative was designed and synthesized. In addition, a quantitative structure–activity relationship (QSAR) model was obtained [pIC50 = − 1.51 − 0.96 (EHOMO) + 0.02 (PSA); N = 17, R2 = 0.980, R2Adj = 0.977, s = 0.103, and LOO-cv-R2 (Q2) = 0.971]. The activity of the new synthesized compound was estimated (pIC50 = 5.88) and compared with the experimental result (pIC50 = 5.70), which allowed to evaluate the good predictive capacity of the model.
Similar content being viewed by others

Introduction
According to the World Health Organization (WHO), the neglected tropical diseases (NTDs) are a diverse group of 20 conditions that are mainly prevalent in the tropical and subtropical regions of the world, such as Latin America, Africa, and Asia, predominantly in developing countries. The NTDs are considered endemic in low-income populations, as they affect vulnerable people, who in most cases have limited access to clean and potable water, and poor hygiene and sanitation conditions1.
Among the 20 NTDs, tuberculosis, Chagas disease, leprosy, malaria, dengue, schistosomiasis, and leishmaniasis are included in the Brazilian National Agenda of Priorities in Health Research2. Although they have been present on our planet for thousands of years, they continue without being eradicated, and impose serious limitations on the affected societies, leading to a panorama of illness, suffering, disability, and death, with serious social, economic, and psychological consequences, affecting more than 1 billion people worldwide, according to the WHO data3.
In January 2021, WHO launched its new roadmap for tackling NTDs from 2021 to 2030. Targets include achieving prevention, control, elimination, and eradication of a diverse set of diseases until 2030. The goal includes a 90% decrease in the number of people in need of treatment, that at least one hundred countries eliminate at least one neglected disease present in their nation, and a 75% reduction in years of life lost due to disability caused by these diseases3.
The treatments available for the NTDs are very limited and insufficient, in addition to presenting a series of problems, such as low efficacy, high toxicity, and the emergence of resistant strains4.
Leishmaniasis are a set of diseases caused by protozoan of the Leishmania genus and the Trypanosomatidae family, and they are transmitted to humans by the bite of infected female phlebotomine sandflies. Leishmaniasis can occur in three different clinical forms: (i) visceral leishmaniasis (VL), which is generally fatal without treatment; (ii) cutaneous leishmaniasis (CL) that causes skin ulcers; and (iii) mucocutaneous leishmaniasis (MCL), affecting nose, mouth, and throat. The WHO estimates that 700,000–1 million new cases occur each year worldwide5.
The antileishmanial treatment is performed, as a first choice, with pentavalent antimonial agents, such as sodium stibogluconate. Meanwhile, amphotericin B, pentamidine, miltefosine, and paromomycin have also been used with varying results. However, the current drugs have undesirable side effects and important toxicities. For example, amphotericin and pentavalent antimonial drugs cause high nephrotoxicity and cardiac effects, respectively, in addition to being administered intravenously, which sometimes hinders the patient’s adherence to the treatment, resulting in therapeutic failures and favoring the parasite’s resistance to the drugs5,6.
Some N-heterocyclic nucleus are considered privileged scaffolds due to their broad spectrum of biological activity highlighted in the literature7. Privileged scaffolds generally exhibit physicochemical properties that allow a single class of molecules to provide potent and selective ligands for different biological targets8. The N-heterocycle quinoxaline (1,4-naphthyridine) (Fig. 1) is considered a privileged scaffold.

Chemical structure and atom numbering of the quinoxaline nucleus (1).
Quinoxaline derivatives represent a class of biologically active compounds, showing anti-inflammatory, anticancer, antibacterial, antimicrobial, antifungal, antiviral, and antileishmanial activities, among others8,9,10,11,12. The quinoxaline heteroaromatic scaffold is found in more than 30 drugs available in the DrugBank (DB, https://go.drugbank.com/), including approved, nutraceuticals, investigational or experimental ones, such as brimonidine (DB00484), chlorsulfaquinoxaline (DB12921), erdafitinib (DB12147), rabeximod (DB05772), riboflavin (DB00140), and varenicline (DB01273).
On the search of new drug leads, there is a need for efficient and robust procedures that can be used to screen chemical databases against molecules with known activities. To this end, quantitative structure–activity relationships (QSAR) studies provide a mean for rationalizing the relationship between chemical structure and its biological action towards the development of new drug candidates13.
Cherkasov et al.14 have described several QSAR studies where computational and medicinal chemists worked together to discover novel molecules with unique biological activities.
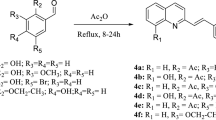
In this context, this work aims to evaluate by an in silico approach which physicochemical properties of the quinoxaline derivatives (2a–2i, 3a–3i, and 4a–4d) (Table 1)11 contribute to their in vitro inhibitory activity against the promastigote forms of Leishmania amazonensis, to propose and to synthesize a new potential antileishmanial agent, and to build a QSAR model able to predict its activity.
Methods
Computational chemistry
The three-dimensional (3D) structures of the quinoxaline derivatives (2a–2i, 3a–3i, and 4a–4d) (Table 1) were constructed using the Spartan’10 software (Wavefunction, Inc.)15. Each structure was submitted to a full geometry optimization step by a molecular mechanics model, using the Merck molecular force field (MMFF), available in the Spartan software. Then, each optimized structure was submitted to the default systematic conformational analysis at Spartan, using the same molecular mechanics force field. The lowest-energy conformer for each quinoxaline derivative was submitted to a full geometry optimization (energy minimization) step by a semi-empirical model, using the Austin Method 1 (AM1) Hamiltonian at Spartan. Then, each optimized conformer was submitted to a single-point energy calculation by a density functional theory (DFT) model, using the B3LYP hybrid DFT method at Spartan, considering the 6-311 + + G(d,p) basis set. For each energy minimized DFT structure, the following thirteen physicochemical properties were obtained: total energy (ET, au), energy of the highest occupied molecular orbital (EHOMO, eV), energy of the lowest unoccupied molecular orbital (ELUMO, eV), HOMO–LUMO energy gap (GAP, eV), dipole moment (μ, Debye), base-10 logarithm of the partition coefficient (LogP), surface area (SA, Å2), molecular volume (MV, Å3), molecular weight (MW, amu), polarizability (P, 10−30 m3), number of hydrogen bond donors (HBD), number of hydrogen bond acceptors (HBA), and polar surface area (PSA, Å2).
A linear cross-correlation matrix was constructed with the calculated thirteen physicochemical properties as a criterion to exclude at least one from the two highly correlated pair of properties and generate a subset of properties to be used in the QSAR equations construction. Therefore, the calculated values of a set of selected properties were set as the independent variables (X) used to calculate the QSAR equations along with the values of the dependent variable (Y), i.e., the biological activity values, which were converted from IC50 (μM) (Table 1) to the corresponding pIC50 (M) values, before the QSAR equations generation. Then, the QSAR equations were obtained by multiple linear regression (MLR) analysis, using the Microsoft Excel® program (Microsoft Inc.).
In addition, using the OSIRIS Property Explorer server16, the toxicity risks of the quinoxaline derivatives were evaluated in silico and fragment-based drug-likeness score was calculated in the same server.
Organic synthesis
The proposed quinoxaline derivative (5) was synthesized following the route used by Cogo et al.11 in the synthesis of 2-amino-3-sulfonylquinoxalines. This synthetic route consists in four steps: (i) vinylic substitution of 1,1-bis(methylsulfanyl)-2-nitroethene using 4-chloroaniline as nucleophile and ethanol as solvent to obtain the 4-chloro-N-(1-methylsulfanyl-2-nitroethenyl)aniline intermediate; (ii) cyclization of the intermediate with phosphoryl chloride (POCl3) to produce the pyrazine ring of the quinoxaline nucleus of the 3,6-dichloro-2-methylsulfanylquinoxaline intermediate, using acetonitrile as solvent; (iii) microwave assisted nucleophilic substitution, using ethanol as solvent to install the methylamino substituent in the 2-position of the quinoxaline nucleus; and (iv) oxidation of the methylsulfanyl group with 3-chloroperbenzoic acid (mCPBA) to obtain the sulfone (5) in dichloromethane as solvent (Fig. 2).

Synthetic route of 7-chloro-N-methyl-3-(methylsulfonyl)quinoxalin-2-amine (5).
The 1H NMR spectra of all intermediates and final product were obtained by using a Bruker ARX-400 equipment (400 MHz).
In vitro growth inhibition assay
Promastigote (1 × 106 cells/mL) cultures were inoculated in a 24-well plate in the absence or presence of different concentrations of the quinoxaline derivatives (0.1 and 100 μM). The inhibitory activity was evaluated after 72 h. The cell density for each concentration was determined by counting in a hemocytometer (Improved Double Neubauer). The concentration that inhibited cell growth in 50% (IC50) was determined by nonlinear regression analysis11.
Results and discussion
SAR analysis of the quinoxaline derivatives and design of a new derivative
Many descriptors reflect simple molecular properties give an insight referent to physicochemical nature of the observed biological activity17.
Table 2 shows the physicochemical descriptor values calculated at the DFT(B3LYP)/6-311 + + G(d,p) level of theory for the quinoxaline derivatives (2a–2i, 3a–3i, and 4a–4d). All the most active quinoline derivatives (IC50 < 3 μM, i.e., pIC50 from 5.54 to 6.70 M, compounds 3a-3i, see Table 1) presented the number of hydrogen bond acceptors (HBA) ranging from 5 to 7, the polar surface area (PSA) values ranging from 46 to 74 Å2, and the LUMO energy (ELUMO) values more negative than − 2.5 eV. In addition, the LogP values range from 1.6 to 3.5, and the HOMO energy (EHOMO) values are more negative than − 5.9 eV.
Unfortunately, the fragment-based drug-likeness values predicted by the OSIRIS server for these compounds are negative like most of the Fluka chemicals that have negative values, whereas 80% of the commercial drugs have a positive drug-likeness value. Toxicity was also predicted by the OSIRIS, and compounds 3g and 4a–d showed alerts of mutagenic risks. On the other hand, 3d showed the highest drug-score value (0.63). The drug-score index combines drug-likeness, cLogP (lipophilicity), LogS (water solubility), MW, and toxicity risks in one value used to predict the compound's overall potential as a drug.
Lipinski’s rule-of-five18 proposes that poor absorption or cell permeability of a drug occurs when its chemical structure fulfils more than one of the following criteria: the molecular weight (MW) is greater than 500 Daltons; the calculated LogP is greater than 5; the number of hydrogen bond donors (NH + OH) are more than 5; and the number of hydrogen bond acceptors (N + O) are more than 10. According to the Veber’s rule19, for good oral availability, the PSA value must be less than or equal to 140 Å2. The physicochemical properties calculated for the studied compounds fit these parameters, except the LogP values for 4a–d (Table 2).
In order to improve these parameters, structural modifications on the studied compounds were proposed to design an antileishmanial agent with higher chances to become a drug.
Cogo and co-workers11 noticed that hydrogen replacement at R1 position (Table 1) by halogen elements (Cl or Br) increases the activity, and substitution at R2 position (Table 1) did not show great interference on the activity. The methylsulfonyl group is present in all the most active compounds studied in this work (Table 1) and literature data also indicates that it is one of the main groups at 3-position of quinoxaline derivatives, which are responsible for the observed activity against Trypanosoma cruzi and Leishmania amazonensis11.
Based on this SAR analysis, several structural modifications were proposed and their synthetic viability as well as the OSIRIS Property Explorer’s risk alerts were evaluated. After that, some of the designed compounds were selected for structural optimization and calculation of the corresponding physicochemical properties. Considering the properties related to the biological activity, compound 5 was proposed as a potential antileishmanial agent (Fig. 3).

Toxicity risks and physicochemical properties predicted by the OSIRIS Property Explorer server for compound (5), 7-chloro-N-methyl-3-(methylsulfonyl)quinoxalin-2-amine, proposed as a potential antileishmanial agent.
It fulfilled all the requirements, presenting the physicochemical descriptors according to the most active compounds of the studied series: LUMO energy of − 2.79 eV, five H-bond acceptors, polar surface area of 53.18 Å2, LogP equal to 1.74, and HOMO energy of − 6.52 eV.
Besides, according to the OSIRIS Property Explorer server, compound 5 (Fig. 3) seems to have low toxicity risks (green color) and the drug-likeness and drug-score indexes were improved to 0.88 and 0.82, respectively, when compared to the other compounds of the series. It is also important to mention that compound 5 follows Lipinski’s rule-of-five and Veber’s rule related to PSA range of drug candidates.
QSAR model construction and validation
A QSAR model was built to predict the activity value of compound 5. Firstly, the degree of correlation between all pairs among the thirteen descriptors (Table 1) was verified by constructing a cross-correlation matrix. After removing multicollinear descriptors, seven of them were selected (ET, EHOMO, ELUMO, dipole moment, LogP, MW, and PSA), and equations that describe the dependency relationship between the independent (X, properties or descriptors) and dependent (Y, biological activity) variables were obtained based on Hansh and Unger’s work20, who suggest that, in a selection of independent variables, for each independent variable included in the QSAR model, there must be no more than five observations (i.e., compounds), thus avoiding chance correlation21.
Therefore, the calculated values of those seven descriptors (Table 1) were set as the independent (X) variables used to calculate the QSAR equations along with the values of the dependent (Y) variable (i.e., biological activities), which were converted from IC50 (μM) (Table 1) to the corresponding pIC50 (M) values, before the QSAR equations generation.
Among the main methods used in the selection of the independent variables in QSAR, we applied the systematic search method, which consists in combining the available independent variables to build and analyze all possible linear regression equations. In the QSAR method, compounds are generally divided into training set and test set, compounds from the training set are used in the construction of QSAR equations and compounds from the test set are used in validation. Since there are 22 compounds (Table 1) and that part of them (~ 20% from the total number of compounds) should be removed from the model as a test group, we used a maximum of three independent variables to be included in each equation, considering N = 18 for the training set and N = 4 for the test set (namely, compounds 2i, 3g, 3h, and 4d).
The systematic search generated 63 regression equations: seven equations with one independent variable, 20 equations with two independent variables, and 34 equations with three independent variables. Tables 3, 4 and 5 list the previously selected independent variables included in the linear equations and the following statistical parameters of each equation calculated by the Microsoft Office Excel® program (Microsoft Inc.): correlation coefficient (R), coefficient of determination (R2), adjusted coefficient of determination (R2Adj), standard error (s) and F-test.
Comparing the best equations (highlighted in bold on Tables 3, 4 and 5) of the three groups containing one (Eqs. 1–7), two (Eqs. 8–28) and three (Eqs. 29–63) theoretical physicochemical descriptors (independent variables or terms), i.e., Eq. 2 (pIC50 = − 0.84 (EHOMO), R2Adj = 0.936), Eq. 18 (pIC50 = − 1.58–0.97 (EHOMO) + 0.02 (PSA), R2Adj = 0.967), and Eq. 33 (pIC50 = − 1.85 + 4.31 × 10−5 (ET) − 1.02 (EHOMO) + 0.02 (PSA), R2Adj = 0.967), respectively, the EHOMO term is present in all of them. However, Eq. 2 should be excluded because it has the lowest R2Adj value (a normalized R2 value, used to compare equations containing a different number of terms).
Therefore, considering only Eqs. 18 and 33, we can observe that the inclusion of the ET term in Eq. 33 does not alter the R2Adj value, which makes these two equations to be equivalent. Nevertheless, since the parsimony principle advises the choice of the simplest model, Eq. 18 was used to calculate the antileishmanial activity value for the 18 quinoxaline derivatives (Table 6), using as descriptors the EHOMO and PSA independent variables (Table 4).
The HOMO and LUMO energies are important properties in chemical and pharmacological processes because these properties give information on the electron-donating and electron-accepting character of a compound. It is possible to notice that the EHOMO for the most active studied compounds are the more negative ones (Table 2). This means that the more active compounds are not so good electron-donor molecules when compared to the less active ones.
The PSA is a molecular descriptor extensively used to characterizing the transport properties of drugs, related to its intestinal absorption and the penetration of the blood–brain barrier. According to the model (Eq. 18), together with EHOMO, it is a key descriptor to explain the biological activity of the quinoxaline derivatives.
These descriptors shows that not only steric but also electronic properties are important to understand the interaction between quinoxaline derivatives that present antileishmanial activity and the biological receptor. The steric properties are related to the positioning of the molecule when interacting with the receptor, while the electronic properties are related to the intensity of the molecular association due to electronic interaction.
Among the 18 compounds, only one (4b) presents a deviation greater than 5% from the experimental activity value, characterizing it as an outlier. Compound 4b has bulky substituents, altering its physicochemical properties (such as a much larger area and volume values—see Table 2) when compared to the other compounds of the series, and consequently making discrepant the relationship between structure and biological activity through the proposed equation.
Excluding the outlier 4b, coefficients were recalculated for Eq. 18 providing Eq. 1 (N = 17, R2 = 0.980, R2Adj = 0.977, s = 0.103, and R2 from the leave-one-out-cross-validation (Q2) = 0.971), in which analysis of residues (Table 7) and plot of pIC50 (calculated) versus pIC50 (observed) (Fig. 4) did not show any outlier.

Experimentally observed antileishmanial activity values (pIC50 (observed)) versus calculated activity values (pIC50 (calculated)), using Eq. 1, for the 17 quinoxaline derivatives in the training set (after removing outlier 4b).
Since literature data indicates that there is evidence that only models validated externally, after internal validation, can be considered reliable and applicable for external prediction and regulatory purposes22,23, the model was applied for external molecules.
Carrying out an external validation, it was possible to confirm the robustness of the proposed model (Eq. 1). A set of four compounds (2i, 3g, 3h, and 4d) was used as external test, representing about 20% of the quantity of observations (N = 22). The test group with its values of observed and calculated pIC50, residuals, and percentual deviation are shown in Table 8, where is possible to verify that all of them present a deviation smaller than or equal to 5% of the biological activity value observed experimentally.
Synthesis of the new derivative and activity prediction by the QSAR model
Unpublished compound 5 was synthesized, characterized by NMR, and its biological activity in the promastigote form of Leishmania amazonensis was evaluated. The built and validated QSAR model, corresponding to Eq. 1, was used to predict the activity of this new derivative.
Therefore, the descriptors present in Eq. 1 were calculated for the new compound 5 (EHOMO = − 6.52 eV and PSA = 53.19 Å2) and a value of 5.81 was predicted for biological activity (pIC50) against Leishmania amazonensis. Comparison with the experimental result (IC50 = 2.0 ± 1.2 μM and pIC50 = 5.70) shows that the QSAR model (Eq. 1) proposed here, presented a good predictive capacity with a deviation of 1.93%, being useful to drive the synthesis of new quinoxaline derivatives, saving time and resources that would be spent on synthesis and testing of biological activity.
NMR data
7-chloro-N-methyl-3-(methylsulfonyl)quinoxalin-2-amine (5).
61% yield. 1H NMR (300 MHz, CDCl3) δ: 7.77 (d, J = 8.9 Hz, 1H), 7.72 (d, J = 2.3 Hz, 1H), 7.35 (dd, J = 8.9, 2.3 Hz, 1H), 6.93 (m, 1H), 3.41 (s, 3H), 3.12 (d, J = 4.8 Hz, 3H). 13C NMR (300 MHz, CDCl3) δ: 148.68, 144.21, 141.19, 138.69, 132.96, 130.47, 126.38, 125.43, 40.45, 27.92.
Conclusions
SAR studies of a series of quinoxaline derivatives were carried out and a new quinoxaline derivative was proposed as a potential antileishmanial agent. The unpublished compound was synthesized and tested against Leishmania amazonensis promastigotes. A new QSAR model was built, and it was capable to predict the activity of the new compound being useful to drive the synthesis of other ones.
Data availability
The authors confirm all data generated and analyzed during this study are available in the article.
References
World Health Organization. Global report on neglected tropical diseases 2023; Retrieved Aug 2023 from WHO/CED/PHE/18.10; World Health Organization: Geneva, Switzerland, 2023.
Fonseca, B. P., Albuquerque, P. C. & Zicker, F. Neglected tropical diseases in Brazil: Lack of correlation between disease burden, research funding and output. Trop. Med. Int. Health 25, 1373–1384. https://doi.org/10.1111/tmi.13478 (2020).
World Health Organization. Ending the neglect to attain the Sustainable Development Goals: A road map for neglected tropical diseases 2021–2030; World Health Organization, 2021. Retrieved Feb 2023.
Varikuti, S. et al. Host-directed drug therapies for neglected tropical diseases caused by protozoan parasites. Front. Microbiol. 9, 2655. https://doi.org/10.3389/fmicb.2018.02655 (2018).
World Health Organization. Leishmaniasis; World Health Organization, 2023. Retrieved Feb 2023.
Tuon, F. F., Amato, V. S., Zequinao, T. & Cruz, J. A. W. Emerging computational technologies in human leishmaniasis: Where are we?. Trans. R. Soc. Trop. Med. Hyg. 116, 981–985. https://doi.org/10.1093/trstmh/trac047 (2022).
Kerru, N., Gummidi, L., Maddila, S., Gangu, K. K. & Jonnalagadda, S. B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 25, 1909. https://doi.org/10.3390/molecules25081909 (2020).
Rivera, G. Quinoxaline 1,4-di-N-oxide derivatives: Are they unselective or selective inhibitors?. Mini-Rev. Med. Chem. 22, 15–25. https://doi.org/10.2174/1389557521666210126142541 (2022).
Montana, M., Montero, V., Khoumeri, O. & Vanelle, P. Quinoxaline derivatives as antiviral agents: A systematic review. Molecules 25, 2784. https://doi.org/10.3390/molecules25122784 (2020).
Aakasha, V. B., Ramalakshmia, N., Bhuvaneswaria, S., Sankaria, E. & Arunkumarb, S. Comprehensive review on versatile pharmacology of quinoxaline derivative. Russ. J. Bioorg. Chem. 48, 657–677. https://doi.org/10.1134/S1068162022040069 (2022).
Cogo, J. et al. Synthesis and biological evaluation of novel 2,3-disubstituted quinoxaline derivatives as antileishmanial and antitrypanosomal agents. Eur. J. Med. Chem. 90, 107–123. https://doi.org/10.1016/j.ejmech.2014.11.018 (2015).
Cogo, J. et al. Quinoxaline derivatives as potential antitrypanosomal and antileishmanial agentes. Bioorg. Med. Chem. 26, 4065–4072. https://doi.org/10.1016/j.bmc.2018.06.033 (2018).
Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 29, 476–488. https://doi.org/10.1002/minf.201000061 (2010).
Cherkasov, A. et al. QSAR modeling: Where have you been? Where are you going to?. J. Med. Chem. 57, 4977–5010. https://doi.org/10.1021/jm4004285 (2014).
SPARTAN’10, Wavefunction Inc. Irvine, CA 92612, U.S.A., 2011.
Sander, T. OSIRIS Property Explorer. Organic Chemistry Portal (2001). https://www.organic-chemistry.org/prog/peo
Karelson, M., Lobanov, V. S. & Katrizky, A. R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 96, 1027–1044. https://doi.org/10.1021/cr950202r (1996).
Lipinski, C. A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today: Technol. 1, 337–341. https://doi.org/10.1016/j.ddtec.2004.11.007 (2004).
Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. https://doi.org/10.1021/jm020017n (2002).
Hansh, C. & Unger, S. H. Strategy in drug design. Cluster analysis as an aid in the selection of substituents. J. Med. Chem. 26, 1217–1221 (1973).
Gaudio, A. C. & Zonade, E. Proposição, validação e análise de modelos que correlacionam estrutura química e atividade biológica. Quím. Nova. 24, 658–671 (2001).
Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 26(5), 694–701. https://doi.org/10.1002/qsar.200610151 (2007).
Golbraikh, A. & Tropsha, A. Beware of q2!. J. Mol. Graph. Model. 20, 269–276. https://doi.org/10.1016/S1093-3263(01)00123-1 (2002).
Acknowledgements
To the Brazilian governmental agencies: FAPERJ (“Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro”), CAPES (“Coordenação de Aperfeiçoamento de Pessoal de Nível Superior”), and CNPq (“Conselho Nacional de Desenvolvimento Científico e Tecnológico”).
Author information
Authors and Affiliations
Contributions
Project design, students supervision, writing the manuscript and approval of the final version of the manuscript were the responsibility of J.Y. Suggestion of research idea was performed by D.P.S. He and J.A.D were responsible for the organic synthesis. J.A.D. also proposed the structural modifications on the studied compounds. C.V.N. realized the biological tests. F.L.L. worked on theoretical calculations for the SAR study. A.C.S.J.P. developed the QSAR study together with M.G.A., which reviewed the manuscript and also approved the final version. The authors confirm that the individuals named are the authors of this work, and are satisfied to publish their research work in Journal of Scientific Reports.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Silva de Jesus Passaes, A.C., Arantes Dantas, J., Landim Lopes, F. et al. Quinoxalines against Leishmania amazonensis: SAR study, proposition of a new derivative, QSAR prediction, synthesis, and biological evaluation. Sci Rep 13, 18136 (2023). https://doi.org/10.1038/s41598-023-45436-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45436-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
