
Abstract
The synthesis, spectral properties, thermal analysis, structural characterization and in silico prediction of pharmacokinetic parameters of tetramethylammonium (compound 1) and tetraethylammonium (compound 2) salt of nimesulide were described in this article. Both compounds crystallize in the monoclinic P21/n space group, with one tetraalkylammonium cation and one nimesulide anion in the asymmetric unit and their crystal structures are stabilized by C–H···O hydrogen bonds between ions. Additionally, structures of title compounds are stabilized by π–π interactions (compound 1), or C–H···π interactions (compound 2) between nimesulide anions. The TG and DSC measurements show that compound 1 melts at a temperature higher than nimesulide, whereas the compound 2 melts at a temperature lower than nimesulide. The MALDI-TOF, 1H NMR, 13C NMR and ATR-FTIR analyses confirm the SCXRD study, that in compounds 1 and 2 nimesulide exists in an ionized form. Studies performed by SWISS ADME and ProTOX II tools, predict to be oral bioavailability of both salts obtained, and one of them (compound 1) is predicted to be well-absorbed by digestive system, while both compounds obtained are classified into toxicity class 4.
Similar content being viewed by others

Introduction
The development of crystal engineering makes it possible to obtain new forms of drugs1,2,3,4,5,6,7,8. This is possible, among other things, due to the understanding of intermolecular interactions between the molecules or ions, which enables the design of multi-component crystals involving Active Pharmaceutical Ingredients (API). Obtaining multi-component crystals using APIs makes it possible to improve the physicochemical properties of drugs such as thermal stability, lipophilicity and solubility, which allows for, among others, various possibilities of drug administration, reducing the dose of the drug substance and extending the time interval between drug dosing9,10,11,12,13. Determining the structures of such crystals using XRD methods makes it possible to determine the correlation between their structure and properties such as melting point, absorption, or reactivity14,15,16,17,18. Crystallization of multi-component crystals can also improve the narrow therapeutic index of drugs9,10,11,12,13,14,15,16,17,18. What is more, pharmacodynamic interactions between two or more bioactive substances can change properties and pharmaceutical action of different drugs which is called synergism19,20,21,22. Such a combination causes that properties one or both drugs may be greatly strengthened, or substances may have an antagonistic action19,20,21,22.
An interesting example of multicomponent crystals of pharmaceutical interest are crystals containing two or more bioactive substances, such as nimesulide and quaternary ammonium cations. Nimesulide (4-nitro-2-phenoxymethanesulfonanilide) is an Active Pharmaceutical Ingredient belongs to the non-steroidal anti-inflammatory drugs (NSAIDs) (Fig. 1). It has antipyretic, analgesic and anti-inflammatory properties and it is use in acute pain treatment and primary dysmenorrhea. Like most NSAIDs, the mechanism of action of substances is based on the inhibition of cyclooxygenase—an enzyme that is involved in the synthesis of prostaglandins from lipids in cell membranes23,24,25,26. In turn, tetramethylammonium cation is a cholinomimetic that imitate effects as acetylcholine and causes stimulation and then block of neurotransport nicotinic and muscarinic acetylcholine receptors27,28,29,30, whereas tetraethylammonium anion is ganglionic blocker and is inhibitor at nicotinic acetylcholine receptors31,32.

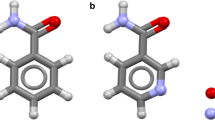
The molecular structures of nimesulide and tetramethylammonium (compound 1) and tetraethylammonium (compound 2) salts of nimesulide.
The structures of crystals involving nimesulide are poorly known—in the Cambridge Structural Database (CSD ver. 5.41, update 01.2023) there are only 11 such structures33. Two of them are the structures of two nimesulide polymorphs determined at room and low temperatures (100 K)26,34,35, five structures are complexes of silver with nimesulide and various ligands35, whereas four structures are cocrystals of nimesulide with pyridine analogues36. In the CCDC there are also information about inclusion complex of nimesulide with cyclodextrin, however the crystal structure of this compound was not determined37,38.
Considering the above, in this article we describe the synthesis, spectral and structural characterization of tetramethylammonium (compound 1) and tetraethylammonium (compound 2) salts of nimesulide (Fig. 1). The spectral characterization of compounds obtained were studied by MALDI-TOF, 1H NMR, 13C NMR and ATR-FTIR methods, whereas the crystal structures were determined using the Single-Crystal X-Ray Diffraction method. Physicochemical features of compounds obtained were predicted in silico using SWISS ADME and Protox II tools.
Experimental
Materials
Tetramethylammonium hydroxide pentahydrate and tetraethylammonium hydroxide solution were purchased from Sigma-Aldrich, whereas nimesulide was received from Ambeed and they were used without further purification. The purity of nimesulide and compounds 1 and 2 and were controlled by TG/DSC methods.
Synthesis of compound 1
Nimesulide (0.05 g, 0.162 mmol) and tetramethylammonium hydroxide pentahydrate (0.026 g, 0.162 mmol) were dissolved in the mixture of solvents: 5 cm3 of ethanol and 5 cm3 of a methanol. After this, the mixture was stirred. The solution was allowed to evaporate in place without sunlight for a few days to give yellow crystals (m.p. = 179 °C).
MALDI-TOF MS (m/z): calc. for [C13H11N2O5S]-: 307.303; found: 307.045 (in negative-ion mode, Fig. 2a); calc. for [C4H12N]+: 74.144; found: 74.218 (in positive-ion mode, Fig. 2b).

MALDI-TOF mass spectra for compound 1 in negative-ion (a) and positive-ion (b) mode.
1H NMR (500 MHz, DMSO-d6) δ (ppm): 7.83 (dd, J = 9.3, 2.8 Hz, 1H), 7.50 (d, J = 2.8 Hz, 1H), 7.35 (d, J = 9.3 Hz, 1H), 7.30 (dd, J = 8.6, 7.3 Hz, 2H), 7.03 (tt, J = 7.3, 1.2 Hz, 1H), 6.87 (dd, J = 8.6, 1.2 Hz, 2H), 3.09 (s, 12H), 2.62 (s, 3H) (Fig. 4a).
13C NMR (126 MHz, DMSO-d6) δ (ppm): 158.23, 152.15, 145.56, 134.05, 129.93, 122.34, 122.29, 117.40, 117.33, 116.25, 54.86 (1:1:1 triplet by coupling with 14N), 40.80 (Fig. 4b).
ATR-FTIR (/cm): 3098 and 3059 (νC–Haromatic), 2993 (νC–Haliphatic) 1582–1467 (νC=C and νasNO2), 1320 and 1165 (shoulder low-intensity bands, νas and νsymSO2), 1280–1070 (νC–N and νC–O) (Fig. 5).
Synthesis of compound 2
Nimesulide (0.05 g, 0.162 mmol) was dissolved in 0.12 ml tetraethylammonium hydroxide solution (20 wt. % in H2O, d = 1.01 g/cm3 in 20 °C, 0.162 mmol). After this, the mixture was stirred. The solution was allowed to evaporate in place without sunlight for a few days to give yellow crystals (m.p. = 113 °C).
MALDI-TOF MS (m/z): calc. for [C13H11N2O5S]−: 307.303; found: 307.008 (in negative-ion mode, Fig. 3a); calc. for [C8H20N]+: 130.250; found: 130.198 (in positive-ion mode, Fig. 3b).

MALDI-TOF mass spectra for compound 2 in negative-ion (a) and positive-ion (b) mode.
1H NMR (500 MHz, DMSO-d6) δ (ppm): 7.84 (dd, J = 9.3, 2.8 Hz, 1H), 7.50 (d, J = 2.8 Hz, 1H), 7.37 (d, J = 9.3 Hz, 1H), 7.31 (dd, J = 8.3, 7.2 Hz, 2H), 7.03 (t, J = 7.2 Hz, 1H), 6.88 (d, J = 8.3 Hz, 2H), 3.21 (q, J = 7.3 Hz, 8H), 2.64 (s, 4H), 1.16 (m, 3JHH = 7.3, J14N,CH3 = 1.8 Hz, 12H) (Fig. 4a).

1H NMR (a) and 13C NMR spectra (b) of nimesulide and compounds 1 and 2 in DMSO-d6 at 298 K.
13C NMR (126 MHz, DMSO-d6) δ (ppm): 158.27, 153.43, 145.52, 133.92, 129.92, 122.32, 122.29, 117.34, 117.29, 116.30, 51.84 (1:1:1 triplet by coupling with 14N), 40.80, 7.54 (Fig. 4b).
ATR-FTIR (/cm): 3032 (νC–Haromatic), 1577–1468 (νC=C and νasNO2), 1318 and 1163 (shoulder low-intensity bands, νas and νsymSO2), 1280–1073 (νC–N and νC–O) (Fig. 5).

ATR-FTIR spectra of studied compounds in the range of 4000–500/cm (left panel) with spectra enlargement in the range of 1800–1000/cm (right panel).
Methods
MALDI-TOF mass spectra
Positive-ion and negative-ion mode MALDI-TOF mass spectra for compounds 1 and 2 were obtained using a Bruker Biflex III spectrometer with α-cyano-4-hydroxycinnamic acid matrix in positive- and negative-ion mode.
Nuclear magnetic resonance (NMR)
The 1H NMR and 13C NMR spectra were recorded on a Bruker III Avance 500 MHz spectrometer (1H frequency 500.13 Hz) operated at magnetic fields of 11.7 T, using standard experimental conditions in DMSO-d6 solution.
Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR–FTIR)
The ATR-FTIR spectra were acquired using a Perkin Elmer Spectrum 2™ instrument (Perkin Elmer, Waltham, USA) equipped with attenuated total reflectance (ATR) accessory. The spectra were recorded at room temperature in the spectral range from 4000 to 500/cm at a resolution of 4/cm averaging 16 scans for each measurement.
Thermogravimetry (TG) and differential scanning calorimetry (DSC)
Thermogravimetric measurements (TG) were performed with a Netzsch 209 thermobalance: samples weighing ~ 4.0 mg were placed in a platinum crucible and heated at 10.0 K/min in a dynamic Ar atmosphere. Differential scanning calorimetry (DSC) measurements were performed with a Perkin Elmer TGA 8000 instrument: samples weighing ~ 4.0 mg were placed in an aluminium crucible and heated at 10.0 K/min in a dynamic N atmosphere.
Single-Crystal X-Ray Diffraction (SCXRD) measurements and structure refinement
SCXRD data were collected on an Oxford Diffraction Gemini R ULTRA Ruby CCD diffractometer MoKα (λMo = 0.71073 Å, T = 293(2) K) (Table 1). The lattice parameters were obtained using CrysAlis CCD, while data were reduced using CrysAlis RED software (multi-scan absorption corrections were applied)39. Crystal structures were solved by direct methods using SHELXS-97 and refined by full-matrix least-squares on F2 with anisotropic displacement parameters for non-H atoms using SHELXL-2017/140. All H–atoms were placed geometrically and refined using a riding model with C–H = 0.93 ÷ 0.97 Å and Uiso(H) = 1.2Ueq(C) (C–H = 0.96 Å and Uiso(H) = 1.5Ueq(C) for the methyl groups). The phenoxy group in compound 1 has disordered orientations with refined site-occupancy factors of the disordered parts of 0.533(6) and 0.467(6) (the disordered benzene rings were refined as rigid ideal hexagons with C–C = 1.38 Å and constrained with isotropic displacement parameters). In all figures, the disordered phenoxy group is omitted for clarity. All interactions were found using the PLATON program41. The molecular graphics were prepared using ORTEPII42, PLUTO-7843, and Mercury44 software.
ADME and Protox II analyses
The web-service SWISS-ADME tool by the Swiss Institute of Bioinformatics (http://www.swissadme.ch/) was used to calculate physicochemical descriptors, important for drug discovery45. Compounds were analyzed to predict ADME (absorption, distribution, metabolism, and excretion) parameters, pharmacokinetic properties, or drug-like nature in comparison to pure nimesulide. The web-service ProTOX II was used for the prediction of the toxicity of the title compounds46.
Results and discussion
Spectral characterization
Negative-ion and positive-ion mode MALDI-TOF mass spectra for compounds 1 and 2 are shown in Figs. 2 and 3. For compounds 1 and 2, the intensity for nimesulide anion were found at m/z 307.045, and 307.008, respectively (calculated for [C13H11N2O5S]−: 307.303), whereas the intensity for tetramethylammonium and tetraethylammonium cations appeared at m/z 74.218 and m/z 130.198, respectively (calculated for [C4H12N]+: 74.144 and calculated for [C8H20N]+: 130.250). The differences between intensities found and calculated are insignificant.
The 1H and 13C NMR spectra of pure nimesulide, and compounds 1 and 2 are shown in Fig. 4. The 1H and 13C NMR data are presented in the “Experimental” section.
Chemical shifts, multiplicities and the observed proton-proton coupling constants are in accordance with the structure of the studied compounds. The number of carbon signals corresponds to the expected number of carbon atoms of the studied compounds. A singlet at 10.2 ppm in the 1H NMR spectra of pure nimesulide indicates N–H proton of sulfonamide group47. This resonance disappeared in the 1H NMR spectra of compounds 1 and 2 due to the amide proton detachment (Fig. 4a). Resonances between 6.5 and 8.5 ppm correspond to aromatic protons. NMR resonances with chemical shifts in the range of 1–3.5 ppm correspond to aliphatic protons of the methylene and methyl groups. The signals in the range of 110–160 ppm in the 13C NMR spectra indicate aromatic carbons, whereas those in the range of 0–55 ppm correspond to saturated functional groups (Fig. 4b).
ATR-FTIR spectra of pure nimesulide and compounds 1 and 2 are presented in Fig. 5. The ATR-IR spectrum of the pure nimesulide shows characteristic bands at 3278/cm (N–H stretch), 1587/cm (aromatic rings), 1335/cm and 1150/cm (asymmetric and symmetric stretching vibrations of SO2 group), 1515/cm and 1315/cm (asymmetric and symmetric stretching vibrations of aryl NO2 group). These values are in agreement with literature results 26,34,48,49,50.
Formation of the salts results in a disappearance of N–H band, which is related to proton detachment from N-amine atom. The changes are also observed within the stretching vibrations of SO2 and NO2 groups. The asymmetric and symmetric stretching vibrations of SO2 group are observed at slightly lower wavenumbers as shoulder low-intensity bands for both salts, whereas those of nitro group remain strong but are shifted to lower wavenumbers by 25–35/cm.
TG/DSC studies
The thermal behaviour of pure nimesulide and compounds 1 and 2 are shown in Fig. 6. The TG-DSC diagrams for the nimesulide show a sharp endothermic peak at 149 °C, which represent the melting point, whereas the first decomposition step occurs between 220 and 390 °C with a mass loss of 71.9% associated with an exothermic peak at 330 °C attributed to the decomposition of the compound. These temperatures are in agreement with literature results 26,34,48,49,50. The TG-DSC curves for compounds 1 and 2 exhibit sharp endothermic peaks at 179 and 113 °C, which represent the melting points of both compounds, respectively. For the compound 1, the first decomposition step occurs between 219 °C and 377 °C with a mass loss of 64.7% associated with exothermic peaks at 269 and 297 °C. For the compound 2, the first decomposition step is observed in range of 205–360 °C with a mass loss of 75.4% associated with exothermic peaks at 258 and 297 °C.

DSC-TG curves of nimesulide and compounds 1 and 2.
Single-Crystal X-Ray Diffraction (SCXRD) studies
Compounds 1 and 2 crystallize in the monoclinic P21/n space group with one tetraalkylammonium cation and one nimesulide anion in the asymmetric unit (Fig. 7, Table 1).

Crystal structures of compounds 1 and 2 with the atom-labelling scheme (hydrogen bonds are represented by dashed lines).
In both crystal structures, the proton detachment from N7-atom of nimesulide is observed which is confirmed by d(C–N) and d(N–S) bond length and ∠(C–N–S) angle within the sulfonamide group. In the crystals of title compounds, the values of d(C–N) bond length are 1.37 and 1.35 Å, whereas the d(N–S) bond length are 1.56 and 1.58 Å for compounds 1 and 2, respectively. For comparison, in the crystal structures of nimesulide (form I and II) the d(C-N) bond length is 1.40 and 1.41 Å, whereas the d(N–S) bond length are 1.64 and 1.65 Å, respectively for form I and II26,34,35,36. In turn, in the crystals of complexes of silver with nimesulide the d(C–N) bond length is in the range 1.38 ÷ 1.40 Å, while the d(N–S) bond length is in the range 1.38 ÷ 1.62 Å35. For the structures of cocrystals of nimesulide with pyridine analogues, the values of these bond length are 1.42 and 1.64 Å, respectively37. In the crystals of title compounds, the values of ∠(C–N–S) angle are 122.2° and 119.2°, for compounds 1 and 2, respectively, which indicates the sp2 hybridization of the nitrogen atom. For comparison, in the crystal structures of nimesulide the ∠(C–N–S) angle is 124.6 and 124.1°, for form I and II, respectively26,34,35,36. In turn, in the crystals of complexes of silver with nimesulide the ∠(C–N–S) angle is in the range 118.7 ÷ 122.3°35, whereas for the structures of cocrystals of nimesulide with pyridine analogues, this angle is in the range 119.9 ÷ 124.0°37.
In the crystal of compound 1, the nimesulide anions are linked by π–π interactions between aromatic ring of phenoxy groups [d(Cg···Cg) = 3.374(6) Å and offset = 1.496 Å] (Fig. 8, Table S3) to form homodimer. The neighbouring homodimers are linked through C(methyl)–H···O(nitro) hydrogen bond, building blocks along [1 0 1] direction (Fig. 8, Table S2). The adjacent blocks of nimesulide anion are connected by C(methyl)–H···O(sulfo) hydrogen bond to create 3D framework with channels (Fig. 8, Table S2). In these channels the tetramethylammonium cations are located and interact with the nimesulide anions by weak C(methyl)–H···O(nitro)/(sulfo) hydrogen bonds.

Crystal packing of compound 1 viewed along b-axis (interactions between nimesulide anions are highlighted by green, whereas hydrogen bonds between nimesulide anion and tetramethylammonium cation are highlighted by orange).
In the crystal of compound 2, the homodimer of anions is formed by C–H···π interactions (d(H···Cg) = 2.97 Å) between both aromatic rings of nimesulide (Fig. 9, Table S4). The neighbouring dimers are linked by C(methyl)–H···O(nitro) hydrogen bond, building columns along b-axis (Fig. 9, Table S2) in voids of which tetraethylammonium cations are located and linked with nimesulide anions via C(methyl)–H···O(nitro) hydrogen bonds. The adjacent columns are connected through C(methyl)–H···O(sulfo) hydrogen bonds between nimesulide anions to produce 3D framework (Fig. 9, Table S2).

Crystal packing of compound 2 viewed along b-axis (interactions between nimesulide anions are highlighted by green, whereas hydrogen bonds between nimesulide anion and tetraethylammonium cation are highlighted by orange).
ADME analysis
Bioavailability Radar is one of results, which the Swiss ADME web tool is giving. It is prediction on six physicochemical properties, such as lipophilicity, size, polarity, insolubility, insaturation, flexibility51. Descriptors used for it, in comparison to pure nimesulide, are presented in Table 2.
The pink area illustrates optimal range for six parameters. Nimesulide represents proper values for 5 properties. Lipophilicity, calculated by XLOGP3 is between − 7 and + 5 and size (MW) is between 150 and 500 g/mol. Other properties, such as polarity (TPSA) and solubility (logS), are in range 20–130 Å2 and not higher than 6, respectively. One of properties of nimesulide, saturation, which is described as a number of carbons in the sp3 hybridization—is out of range. The value of the parameter should be more than 0.25. All parameters for compounds 1 and 2 are in the range of the pink area, which means that substances are predicted to be orally bioavailable. Another parameter, referring to drug-likeness, is described by Lipinski’s rule of five55 from Pfizer and Bioavailability Score56 by Abbott. Both criteria, for all structures are in the acceptable range—Lipinski’s filter causing 0 violation and Bioavailability score is 0.55. Amount PAINS #alarm57 for all compounds is 0 alarm and the value of Synthetic accessibility58 for all structures is in an optimal range.
Method to predict at the same time two properties: the passive gastrointestinal absorption (HIA), brain access (BBB) is called BOILED-Egg. This model is based on two physicochemical parameters—WLOGP and TPSA. The BOILED-Egg plot (Fig. 10) contains 3 areas: the yellow one is assigned for most likely BBB permeation, the white one is a space for most likely HIA absorption molecules and the grey one stands for structures predicted to have weak absorption and low brain penetration. Yellow and white compartments are not mutually exclusive59. Occurred points are colored. Molecules, which are not effluated from the central nervous system by P-glycoprotein substrates (PGP-), are marked as red dots. Blue dots are for substances predicted to dispose from the central nervous system by P-glycoprotein substrates (PGP+)60. Nimesulide and compound 1 are predicted to be well-absorbed and not crossing the blood–brain barrier, which is confirmed in the literature for the nimesulide61,62, whereas compound 2 is predicted not to be absorbed and not accessing the brain. Compounds 1 and 2 are rid of the nervous system (PGP+), while nimesulide is not pumped-out by P-glycoprotein (PGP-). The Servis ProTox II has classified all molecules (nimesulide, compound 1 and 2) into toxicity class 4 (predicted LD50: 997 mg/kg).

BOILED-Egg diagram for the nimesulide (Molecule 1) and compounds 1 (Molecule 2) and 2 (Molecule 3).
Conclusion
The synthesis, spectral properties, thermal analysis, and structural characterization of tetramethylammonium (compound 1) and tetraethylammonium (compound 2) salts of nimesulide were described in this article. The MALDI-TOF, 1H NMR, 13C NMR and ATR-FTIR analyses confirm SCXRD study, that nimesulide exists in an ionized form in both studied compounds. MALDI-TOF mass spectra of the compounds 1 and 2 showed the peaks of nimesulide anion (found at m/z 307.045, and 307.008, respectively), and tetramethylammonium and tetraethylammonium cations (found at m/z 74.218 and m/z 130.198, respectively). A singlet at 10.2 ppm in the 1H NMR spectra of pure nimesulide corresponding to H–N proton of sulfonamide group, disappeared in the 1H NMR spectra of both title compounds due to the amide proton detachment. Likewise, formation of the salts resulted in a disappearance of N–H stretching band at 3278/cm in the ATR-FTIR spectra. The TG and DSC measurements confirmed that compound 1 melts at a temperature higher than nimesulide (179 °C), whereas the compound 2 melts at a temperature lower than nimesulide (113 °C). Compounds 1 and 2 crystallize in the monoclinic P21/n space group, with one tetraalkylammonium cation and one nimesulide anion in the asymmetric unit, and their crystal structures are stabilized by C–H···O hydrogen bonds between ions. Additionally, structure of compound 1 is stabilized through π–π interactions, whereas in the crystal packing of compound 2 the C–H···π interactions between nimesulide anions playing an important role. Studies performed by SWISS ADME and ProTOX II tools, predict to be oral bioavailability of both salts obtained, and one of them (compound 1) was predicted to be well-absorbed by digestive system. Both compounds were predicted to be classified into toxicity class 4 (predicted LD50: < 1000 mg/kg).
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers No. CCDC 2281374 (1) and CCDC 2281375 (2). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Aitipamula, S. et al. Polymorphs, salts, and cocrystals: What’s in a name?. Cryst. Growth Des. 12(5), 2147–2152 (2012).
El-Zhry El-Yafi, A. K. & El-Zein, H. Technical crystallization for application in pharmaceutical material engineering: Review article. Asian J. Pharm. Sci. 10(4), 283–291 (2015).
Thakuria, R. & Sarma, B. Drug-drug and drug-nutraceutical cocrystal/salt as alternative medicine for combination therapy: A crystal engineering approach. Crystals 8(2), 1–39 (2018).
Datta, S. & Grant, D. J. W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Discov. 3(1), 42–57 (2004).
Friscic, T. & Jones, W. Benefits of cocrystalisation in pharmaceutical materials science: An update. J. Pharm. 62(11), 1547–1559 (2010).
Qiao, N., Schlindwein, W., Malek, N., Davis, A. & Trappitt, G. Pharmaceutical cocrystals: An overview. Int. J. Pharm. 419, 1–11 (2011).
Braga, D. & Grepioni, F. Making Crystal by Design (Wiley-VCH Verlag GmbH & Co, 2006).
Desiraju, G. R. Crystal engineering. From molecules to materials. J. Am. Chem. Soc. 135, 9952–9967 (2013).
Chaurasia, G. A review on pharmaceutical preformulation studies in formulation and development of new drug molecules. Int. J. Pharm. Sci. Res. 7(6), 23132–32320 (2016).
Shekunov, B. Y. & York, P. Crystallization processes in pharmaceutical technology and drug delivery design. J. Cryst. Growth 211(1–4), 122–136 (2000).
Bergström, C. A. et al. Early pharmaceutical profiling to predict oral drug absorption: Current status and unmet needs. Eur. J. Pharm. Sci. 57, 173–199 (2014).
Singhal, D. & Curatolo, W. Drug polymorphism and dosage form design: A practical perspective. Adv. Drug Deliv. Rev. 56(3), 335–347 (2004).
Dressman, J. B. & Reppas, C. In vitro–in vivo correlations for lipophilic, poorly water-soluble drugs. Eur. J. Pharm. Sci. 2000(11), S73–S80 (2000).
Van de Walle, A. A complete representation of structure–property relationships in crystals. Nat. Mat. 7(6), 455–458 (2008).
Bernstein, J. Crystal growth, polymorphism and structure-property relationships in organic crystals. J. Phys. D: Appl. Phys. 26(8B), B66–B76 (1993).
Gessner, V. H., Däschlein, C. & Strohmann, C. Structure formation principles and reactivity of organolithium compounds. Chem. A Eur. J. 15(14), 3320–3334 (2009).
Aakeröy, C. B., Forbes, S. & Desper, J. Using cocrystals to systematically modulate aqueous solubility and melting behaviour of an anticancer drug. J. Am. Chem. Soc. 131(47), 17048–17049 (2009).
Kesisoglou, F. & Wu, Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 10, 516–525 (2008).
Griffin, J. P. & D’Arcy, P. F. Synergistic Drug Interactions. Handb. Exp. Pharmacol. 1996, 235–248 (1996).
Weinstein, Z. B., Bender, A. & Cokol, M. Prediction of synergistic drug combinations. Curr. Opin. Syst. Biol. 4, 24–28 (2017).
Lehár, J. et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 27(7), 659–666 (2009).
Robertson, D. Agonistic and antagonistic drug action. Nurse Prescrib. 15(7), 356–358 (2017).
Bennett, A. & Villa, G. Nimesulide: An NSAID that preferentially inhibits COX-2 and has various unique pharmacological activities. Expert Opin. Pharmacother. 1(2), 277–286 (2000).
Vane, J. R. & Botting, R. M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 104, 2–8 (1998).
Kress, H. G. et al. Acute pain: A multifaceted challenge—the role of nimesulide. Curr. Med. Res. 32(1), 23–36 (2016).
Dupont, L., Pirotte, B., Masereel, B., Delarge, J. & Geczy, J. Nimesulide. Acta Cryst. 51(3), 507–509 (1995).
Anthoni, U. et al. Tetramine: Occurrence in marine organisms and pharmacology. Toxicon 27(7), 707–716 (1989).
DiPalma, J. R., McGraw-Hill, E. & Drill’s, E. D. Pharmacology in Medicine (McGraw-Hill, 1971).
Burn, J. H. & Dale, H. H. The action of certain quaternary ammonium bases. J. Pharmacol. Exp. Ther. 6, 417–438 (1915).
Neef, C., Oosting, R. & Meijer, D. K. F. Structure-pharmacokinetics relationship of quaternary ammonium compounds. Naunyn-Schmiedeberg’s Arch. Pharmacol. 328(2), 103–110 (1984).
Kleinhaus, A. L. & Prichard, J. W. Close relation between TEA responses and Ca-dependent membrane phenomena of four identified leech neurons. J. Physiol. 270(1), 181–194 (1977).
Akk, G. & Steinbach, J. H. Activation and block of mouse muscle-type nicotinic receptors by tetraethylammonium. J. Physiol. 551, 155–168 (2003).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 72(2), 171–179 (2016).
Sanphui, P., Sarma, B. & Nangia, A. Phase transformation in conformational polymorphs of nimesulide. J. Pharm. Sci. 100(6), 2287–2299 (2011).
Banti, C. N., Papatriantafyllopoulou, C., Manoli, M., Tasiopoulos, A. J. & Hadjikakou, S. K. Nimesulide silver metallodrugs containing the mitochondriotropic triaryl derivatives of pnictogen; anticancer activity against human breast cancer cells. Inorg. Chem. 55(17), 8681–8696 (2016).
Wang, M. et al. Similar but not the same: Difference in the ability to form cocrystals between nimesulide and the pyridine analogues. Cryst. Growth Des. 21(1), 287–296 (2021).
Geczy, J. Nimesulide salt cyclodextrin inclusion complexes. U.S. Patent no. 5744165 (1998).
Braga, S. S. et al. Encapsulation of sodium nimesulide and precursors in β-cyclodextrin. Org. Biomol. Chem. 1(5), 873–878 (2003).
CrysAlis CCD and CrysAlis RED. Version 1.171.36.24. Oxford Diffraction Ltd. (Yarnton, 2012).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. 71(1), 3–8 (2015).
Spek, A. L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 36, 7–13 (2003).
Johnson, C. K. ORTEP II. Report ORNL-5138. ORNL (Oak Ridge, 1976).
Motherwell S. & Clegg. S. PLUTO-78. Program for Drawing and Molecular Structure. UC (Cambridge, 1978).
Macrae, C. F. et al. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 41(2), 466–470 (2008).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Tetko, I. V., Bruneau, P., Mewes, H. W., Rohrer, D. C. & Poda, G. I. Can we estimate the accuracy of ADME–Tox predictions?. Drug Discov. Today 11, 700–707 (2006).
Jian, Y.-S. et al. Hyaluronic acid–nimesulide conjugates as anticancer drugs against CD44-overexpressing HT-29 colorectal cancer in vitro and in vivo. Int. J. Nanomed. 12, 2315–2333 (2017).
Swamy, P. V. et al. Dissolution rate enhancement of Nimesulide using electro-spinning and cogrinding techniques: A comparative study. Am. J. Pharm. Tech. Res. 5(4), 609–624 (2015).
Georgescu, M., Meltzer, V., Stănculescu, I. & Pincu, E. Thermal behavior of the nimesulide-salicylic acid eutectic mixtures prepared by mechanosynthesis and recrystallization. Materials 14, 7715 (2021).
Gálico, D. A. et al. Thermoanalytical study of nimesulide and their recrystallization products obtained from solutions of several alcohols. J. Therm. Anal. Calorim. 115(3), 2385–2390 (2013).
Ritchie, T. J., Ertl, P. & Lewis, R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov. Today 16, 65–72 (2011).
Cheng, T. et al. Computation of octanol−water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 47(6), 2140–2148 (2007).
Ertl, P., Rohde, B. & Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 43, 3714–3717 (2000).
Delaney, J. S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. 44(3), 1000–1005 (2004).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 46, 3–26 (2001).
Martin, Y. C. A bioavailability score. J. Med. Chem. 48, 3164–3170 (2005).
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740 (2010).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 1, 8 (2009).
Daina, A. & Zoete, V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 11, 1117–1121 (2016).
Montanari, F. & Ecker, G. F. Prediction of drug-ABC-transporter interaction–Recent advances and future challenges. Adv. Drug Deliv. Rev. 86, 17–26 (2015).
Candelario-Jalil, E. et al. Post-ischaemic treatment with the cyclooxygenase-2 inhibitor nimesulide reduces blood-brain barrier disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats. J. Neurochem. 100(4), 1108–1120 (2007).
Candelariojalil, E. Nimesulide as a promising neuroprotectant in brain ischemia: New experimental evidence. Pharmacol. Res. 57(4), 266–273 (2008).
Acknowledgements
The authors acknowledge the Research of Young Scientists grant (BMN) no. 539-T080-B063-23 (University of Gdansk) and DS no. 530-8228-D738-23 (University of Gdańsk). The article was financed by the project “Innovation Incubator 4.0” established by the announcement of the Minister of Science and Higher Education on 5 June 2020.
Author information
Authors and Affiliations
Contributions
Conceptualization, M.R. and A.S.; methodology, M.R. and A.S.; software, M.R. and A.S.; formal analysis, M.R. and A.S.; investigation, M.R. and A.S.; writing-original draft preparation, M.R. and A.S.; visualization, M.R. and A.S.; project administration, M.R. and A.S. All authors reviewed and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Title compounds are protected by European patent application no. EP22208962 submitted by the authors of this paper.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rybczyńska, M., Sikorski, A. The synthesis, thermal behaviour, spectral and structural characterization, and in silico prediction of pharmacokinetic parameters of tetraalkylammonium salts of non-steroidal anti-inflammatory drug nimesulide. Sci Rep 13, 17268 (2023). https://doi.org/10.1038/s41598-023-44557-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44557-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
