
Abstract
In the current study, transcriptome profiles of mare endometrium, classified into categories I, IIA, and IIB according to Kenney and Doig, were compared using RNA sequencing, analyzed, and functionally annotated using in silico analysis. In the mild stage (IIA) of endometrosis compared to category I endometrium, differentially expressed genes (DEGs) were annotated to inflammation, abnormal metabolism, wound healing, and quantity of connective tissue. In the moderate stage (IIB) of endometrosis compared to category I endometrium, DEGs were annotated to inflammation, fibrosis, cellular homeostasis, mitochondrial dysfunction, and pregnancy disorders. Ingenuity pathway analysis (IPA) identified cytokines such as transforming growth factor (TGF)-β1, interleukin (IL)-4, IL-13, and IL-17 as upstream regulators of DEGs associated with cellular homeostasis, metabolism, and fibrosis signaling pathways. In vitro studies showed the effect of these cytokines on DEGs such as ADAMTS1, -4, -5, -9, and HK2 in endometrial fibroblasts at different stages of endometrosis. The effect of cytokines on ADAMTS members’ gene transcription in fibroblasts differs according to the severity of endometrosis. The identified transcriptomic changes associated with endometrosis suggest that inflammation and metabolic changes are features of mild and moderate stages of endometrosis. The changes of ADAMTS-1, -4, -5, -9, in fibrotic endometrium as well as in endometrial fibroblast in response to TGF-β1, IL-4, IL-13, and IL-17 suggest the important role of these factors in the development of endometrosis.
Similar content being viewed by others


Introduction
Equine endometrosis is a degenerative chronic condition of the uterus, defined as fibrosis that develops around the endometrial glands and in the stroma1,2,3. Endometrial fibrosis in mares is associated with excessive deposition of extracellular matrix (ECM) components and pathological changes in the endometrial glands, such as cystic dilation and atrophy or hypertrophy of the epithelium1,2,3. This condition affects the endometrium structure as well as the uterine microenvironment and leads to early pregnancy loss3, 4. Depending on the degree of structural changes, the endometria are divided into four categories: I (without fibrosis), and IIA, IIB, III which correspond to mild, moderate, and severe fibrosis, inflammatory infiltrates, and degree of dilation of both endometrial glands and lymphatic vessels, respectively2. In endometrosis, the expected foaling rate is 80–90%, 50–80%, 10–50%, and 10% for categories I, IIA, IIB, and III endometria, respectively2. Therefore, endometrosis is a serious problem in the horse-breeding industry.
The mechanisms underlying tissue fibrosis are very complex with several overlapping stages including inflammation, proliferation, and remodeling. It is worth emphasizing that fibrosis is not only limited to mare endometrium. According to Zeisberg and Kalluri5, chronic loss of function of most organs, including the heart, intestine, kidney, liver, lung, bone marrow, and skin is associated with fibrosis, contributing to an estimated one-third of premature deaths worldwide. Fibrosis across the organs shows many histomorphological similarities and shares pathways and mechanisms contributing to its development5. Despite many similarities, there are organ-specific mechanisms responsible for fibrosis development.
In mare endometrosis, research has mainly focused on the changes in the endometrial expression of inflammatory markers, and components of ECM including synthases of prostaglandin, cytokines, and metalloproteinases (MMP) at the different stages of endometrosis3, 6,7,8,9,10,11. Our previous results described the effects of mediators of inflammation on cell proliferation, fibroblast to myofibroblast differentiation, and the expression of MMPs and their tissue inhibitors (TIMPs)10,11,12,13. Recently, transcriptomic analysis of mare endometrium with severe endometrosis in anestrus was examined14. In endometrium with severe fibrotic changes, the expression of genes was annotated to processes such as to collagen catabolic process, lipid metabolic process, one-carbon metabolic process, defense response to the virus, metalloendopeptidase activity, and extracellular matrix organization. However, to the best of our knowledge, the transcriptomic analysis of endometrial tissue in categories IIA and IIB of endometrosis in the estrous cycle was not described so far.
The application of transcriptomic analysis allows for filling gaps in understanding the nature of endometrial fibrosis as well as identifying the novel pathways and regulators that participate in the development of endometrosis. Therefore, we compared the transcriptome profiles of mare endometrium at different stages of endometrosis using RNA sequencing (RNA-seq). Furthermore, in in vitro study, the effect of transforming growth factor (TGF)-β1 and interleukins (IL)-4, -13, -17 on the expression of genes selected based on global transcriptomic analysis was investigated in endometrial fibroblasts.
Results
Experiment 1. Transcriptomic analysis of endometrial tissue at different stages of endometrosis at the follicular phase of the estrous cycle
In this experiment, we generated RNA-Seq data for 9 pools of 27 samples belonging to all three studied groups. After rejecting low-quality reads, we used from 14.6 to 20.1 M high-quality reads per sample (17.7 M on average). Of the reads, 89.06% on average were successfully mapped against the reference genome (Supplementary Table 1).
Differential changes in endometrial transcriptome at different stages of endometrosis
A comparison of the transcriptomes of endometria collected from categories IIA and IIB with that of category I revealed significant differences in gene expression between healthy and fibrotic endometrium. Most changes were observed in the category IIB endometrium with moderate changes due to endometrial fibrosis. We observed significant differences in the expression of 230 (58 up-regulated and 172 down-regulated), and 1101 (598 up-regulated and 503 down-regulated) in categories IIA and IIB compared to category I endometria, respectively. Additionally, significant differences in the expression of 2111 (1152 up-regulated and 959 down-regulated) transcripts in categories IIB compared to category IIA endometria were observed. In each comparison, besides known genes, several novel transcripts were identified as differentially expressed. A complete list of differentially expressed transcripts in endometrial tissue in categories IIA and IIB vs I as well as IIB vs IIA is reported in Supplementary Table 2.
Comparison of changes in endometrial transcriptome at different stages of endometrosis
The lists of up-and down-regulated DEGs across the categories IIA vs I and IIB vs I endometria were uploaded to jvenn software (http://jvenn.toulouse.inra.fr/app/example.html) to visualize genes whose expression changed across more than one endometrosis categories or only in a specific category as compared to the healthy endometrium (Fig. 1A and B). Change in 23 genes was common to categories IIA vs I and IIB vs I endometria of which seven were up-regulated and 16 were down-regulated (Fig. 1A and B, listed in Supplementary Table 2).

Shared and unique DEGs in endometria at different stages of endometrosis. Venn diagram showing up-regulated (A) and down-regulated (B) shared and unique DEGs in categories IIA and IIB in comparison to category I endometria in the follicular phase of the estrous cycle.
Analysis of biological processes, pathways, and upstream regulators of identified DEGs
Biological processes
To understand the functional significance of gene expression changes in endometrial tissue in categories IIA vs I, IIB vs I, as well as IIB vs IIA core analysis available in the IPA was used to identify pathways that were significantly enriched by the DEGs. Important enriched pathways and functional categories relevant to the process of endometrosis along with their associated DEGs in categories IIA vs I, IIB vs I, and IIB vs IIA endometria are presented in Table 1. A complete list of comparison analyses, comparing disease and biological functions, canonical pathways, and upstream regulators in categories IIA vs I, IIB vs I as well as IIB vs IIA endometria are presented in Supplementary Table 3.
The functional annotation of DEGs identified in category IIA vs I endometria was associated with functions such as inflammation of the organ, organismal death, the quantity of interleukin, cellular infiltration by macrophages, leukocyte migration, cell movement, the quantity of macrophages, abnormal metabolism. Infiltration of macrophages in endometria was confirmed by immunofluorescence staining where a significantly higher number of macrophages was observed in category IIA endometrium (Fig. 2A–D). Additionally, in category IIA vs I endometria, DEGs led to the enrichment of canonical pathways that can be linked to pathological processes such as fibrosis, ferroptosis signaling pathway, inhibition of matrix metalloproteases, wound healing signaling pathway, HIF-1α signaling pathway, IL-13 pathway, and IL-17 signaling.

Endometrial infiltration of macrophages. Representative immunostaining of CD68 in categories I (A) and IIA (B) endometria in the follicular phase of the estrous cycle. (C) negative control; scale bar, 50 µM. The white arrows indicate the macrophages. (D) The number of positive CD68+ cells counted in endometrial regions; Asterisks denote statistical differences (*P < 0.05).
In category IIB vs I endometria, many functional categories and pathways that can lead to the pathology of fibrosis were enriched. The DEGs in this category were annotated to processes such as leukocyte migration, organization of cytoskeleton, apoptosis, organismal injury and abnormalities, growth of connective tissue, cellular homeostasis, and proliferation of fibroblasts and epithelial cells.
Canonical pathways that were enriched among DEGs associated with category IIB vs I endometria included mitochondrial dysfunction, pulmonary fibrosis idiopathic signaling pathway, oxidative phosphorylation, endothelin-1 signaling, HIF-1α signaling pathway, IL-13 pathway, inhibition of matrix metalloproteases, Th1 and Th2 activation pathway.
Furthermore, in category IIB vs IIA endometria, DEGs were annotated to decrease the activation of processes such as recruitment of phagocytes, transport of molecules, organismal death. Canonical pathways that were enriched among DEGs associated with category IIB vs IIA endometria included inhibition of matrix metalloproteases, mitochondrial dysfunction, pulmonary fibrosis idiopathic signaling pathway, epithelial adherent junction signaling, production of nitric oxide and reactive oxygen species in macrophages, DNA methylation and transcriptional repression signaling, phagosome formation, oxidative phosphorylation, and mitochondrial dysfunction.
GO biological processes were analyzed using ShinyGO version 0.741 (FDR ≤ 0.05; hypergeometric test followed by FDR correction)15, and the top—10 GO terms or related biological processes in categories IIA vs I, IIB vs I, IIB vs IIA endometria are shown in Fig. 3.

Top 10 significant GO biological processes in endometria at different stages of endometrosis. (A) category IIA vs I endometria, (B) category IIB vs I endometria, (C) category IIB vs IIA endometria visualized using ShinyGO software (FDR ≤ 0.05; hypergeometric test followed by FDR correction).
Upstream regulators
The upstream regulator analysis tool in IPA was used to predict the potential upstream regulators of DEGs in categories IIA and IIB vs I endometria. Several factors including cytokines that are known to be associated with fibrosis were predicated to be activated or inhibited. A complete list of upstream regulators in categories IIA and IIB vs I as well as IIB vs IIA endometria are presented in Supplementary Table 3. The selected upstream regulators including cytokines and growth factors associated with DEGs between categories: IIA and IIB vs I as well as IIB vs IIA endometria are presented in Table 2. In category IIA vs I endometria, FLCN, CEBPA, GLI1, miR-16-5p, LAMA4, KLF3, MMP9, lipoteichoic acid, LDL, and FGF2 were shown as activated upstream regulators, (Table 2, Supplementary Table 3). In category IIB vs I endometria, growth factors such as BMP10, LEP, IGF2, and cytokines including IL-1β, LIF, IL-1α, TNFSF13B, C5, TNF, CXCL12, CD40LG, IL-33, EPO, IL-6, IL-15, TIMP1 were shown as activated upstream regulators (Table 2, Supplementary Table 3). In category IIB vs IIA endometria, collagenase, TNF, STAT3, IL-13, PDGD, LH was shown as activated upstream regulators, (Table 2, Supplementary Table 3).
qPCR validation of transcriptome sequencing
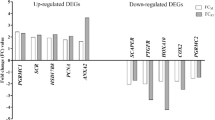
For qPCR validation of transcriptome sequencing results, 4 DEGs (HK2, CXCR4, ADAMTS9, SLC25A29) were investigated. A comparison of log2change and p-value of DEGs obtained after qPCR and data analysis of RNA-seq is presented in Supplementary Table 4. The qPCR analysis showed the direction of changes in the expression of all the evaluated DEGs.
Experiment 2. The effect of cytokines on the differentially expressed genes in endometrial fibroblasts
Transforming growth factor-β1
Transforming growth factor-β1 treatment did not affect the expression of ADAMTS1 in fibroblasts derived from categories I and IIB (Fig. 4A and C; P > 0.05), but it increased the expression of ADAMTS1 in fibroblasts derived from category IIA endometrium (Fig. 4B; P < 0.05). Additionally, TGF-β1 treatment induced an increase in ADAMTS4 in fibroblasts derived from categories I, IIA, and IIB endometria (Fig. 4D–F; P < 0.05). In contrast, treatment with TGF-β1 caused a decrease in ADAMTS5 mRNA transcription in the fibroblasts derived from categories I (Fig. 4G; P < 0.001), IIA and IIB endometria (Fig. 4H–I; P < 0.01). Transforming growth factor-β1 treatment did not affect the expression of ADAMTS9 in fibroblasts derived from categories I and IIA (Fig. 4J and K; P > 0.05) but it decreased the expression of ADAMTS9 in the fibroblasts derived from category III endometrium (Fig. 4L; P < 0.001).

The effect of TGF-β1 on expression of ADAMTS and HK2 mRNA expression in equine endometrial fibroblasts. TGF-β1 treatment (10 ng/ml) on (A–C) ADAMTS1, (D–F) ADAMTS4, (G–I) ADAMTS5, (J–L) ADAMTS9, (M–O) HK2 mRNA transcription in in vitro cultured fibroblast derived from categories I, IIA and IIB endometria. Results are presented as a fold change. Asterisks denote statistical differences (*P < 0.05; **P < 0.01; ***P < 0.001) as determined by the nonparametric Mann–Whitney U test.
Transforming growth factor-β1 treatment increased HK2 mRNA transcription in cultured fibroblasts derived from category I (Fig. 4M; P < 0.05) and category IIA (Fig. 4N; P < 0.05), but decreased HK2 mRNA transcription in cultured fibroblasts derived from category IIB endometria (Fig. 4O; P < 0.05).
IL-4, IL-13, and IL-17
Interleukin 4 treatment did not affect the expression of ADAMTS1 and ADAMTS4 in fibroblasts derived from categories I and IIA endometria (Fig. 5A–D; P > 0.05). However, IL-4 treatment decreased the expression of ADAMTS5 (Fig. 5E,F; P < 0.01, P < 0.05) and increased the expression of ADAMTS9 in cultured fibroblasts derived from categories I and IIA endometria (Fig. 5G,H; P < 0.001; P < 0.05).

The effect of IL-4 on expression of ADAMTS mRNA expression in equine endometrial fibroblasts. IL-4 treatment (10 ng/ml) on (A,B) ADAMTS1, (C,D) ADAMTS4, (E,F) ADAMTS5, (G,H) ADAMTS9 mRNA transcription in in vitro cultured fibroblast derived from (A,C,E,G) categories I and (B,D,F,H) IIA endometria. Results are presented as a fold change. Asterisks denote statistical differences (*P < 0.05; **P < 0.01) as determined by the nonparametric Mann–Whitney U test.
Interleukin 13 treatment did not affect the expression of ADAMTS1 and ADAMTS4 in fibroblasts derived from categories I and IIB (Fig. 6A–D, P > 0.05) but decreased ADAMTS5 mRNA transcription in cultured fibroblasts derived from category I and IIB endometria (Fig. 6E–F; P < 0.01). In turn, IL-13 treatment did not affect the expression of ADAMTS9 in fibroblasts derived from category I (Fig. 6G; P < 0.05) but increased the expression of ADAMTS9 in fibroblasts derived from category IIB endometrium (Fig. 6H; P < 0.01).

The effect of IL-13 on expression of ADAMTS mRNA expression in equine endometrial fibroblasts. IL-13 treatment (10 ng/ml) on (A,B) ADAMTS1, (C,D) ADAMTS4, (E,F) ADAMTS5, (G,H) ADAMTS9 mRNA transcription in in vitro cultured fibroblast derived from (A,C,E,G) categories I and (B,D,F,H) IIB endometria. Results are presented as a fold change. Asterisks denote statistical differences (**P < 0.01) as determined by the nonparametric Mann–Whitney U test.
Interleukin 17 treatment increased ADAMTS1, ADAMTS4, and ADAMTS9 mRNA transcription in cultured fibroblasts derived from category I endometrium (Fig. 7A,C,G; P < 0.05). However, IL-17 treatment decreased ADAMTS1 mRNA transcription in cultured fibroblasts derived from category IIB endometrium (Fig. 7B; P < 0.05). Interleukin 17 treatment did not affect the expression of ADAMTS4, ADAMTS5, ADAMTS9 in fibroblasts derived from category IIB (Fig. 7D,F,H; P > 0.05) and the expression of ADAMTS5 in fibroblasts derived from category I endometria (Fig. 7E > 0.05).

The effect of IL-17 on expression of ADAMTS mRNA expression in equine endometrial fibroblasts. IL-17 treatment (10 ng/ml) on (A,B) ADAMTS1, (C,D) ADAMTS4, (E,F) ADAMTS5, (G,H) ADAMTS9 mRNA transcription in in vitro cultured fibroblast derived from (A,C,E,G) categories I and (B,D,F,H) IIB endometria. Results are presented as a fold change. Asterisks denote statistical differences (*P < 0.05) as determined by the nonparametric Mann–Whitney U test.
Discussion
In the present study, the identified transcriptomic changes associated with endometrosis suggest that inflammation and metabolic changes are features of mild and moderate stages of endometrosis. In addition, the results of this study suggest that cytokines secreted, possibly by macrophages and T helper cells affect the expression of genes associated with ECM remodeling.
Fibrosis development and progression
The analysis of selected molecular and cellular functions and canonical pathways predicted to be associated with DEGs in endometria at different stages of endometrosis reflect events in the development and progression of tissue fibrosis described in other tissue.
In the present study, in the mild (IIA) and moderate (IIB) categories vs category I endometrium, DEGs were annotated to inflammation of organs (e.g. CXCR4, CD68, CD36, F8, NLRP3, TLR7), cellular infiltration by leukocytes, macrophages, and phagocytes as well as cell movement of phagocytes and leukocytes (e.g. ALOX5), and cytokine quantity (e.g. ALOX5, CD36, IL12RB2, NLRP3, PLD4, RNF128). The importance of macrophages in fibrosis development is well established16, 17. In our study, an increase in the expression of CD36, CD68, and CXCR4 in category IIA vs I endometria possibly points towards the role of macrophages in endometrosis development. Macrophages are known to drive fibrosis by producing profibrotic mediators including TGF-β1 and platelet-derived growth factor (PDGF) that directly activate fibroblasts and control extracellular matrix turnover by regulating the balance of MMPs and TIMPs16. The expression of CD68 was increased in rat models of liver fibrosis18 and in patients with intermediate and advanced stages of oral submucous fibrosis19 and idiopathic pulmonary fibrosis (IPF)20. The infiltration of macrophages (CD68+ cells) in category I and IIA endometria using immunofluorescence staining was confirmed. These results suggest the potential role of macrophages in the development of endometrosis in mares. However, the results from the transcriptomic analysis are not clear which population of macrophages is predominant in the endometrial tissue at different stages of endometrosis.
Chemokine CXCR4, a transmembrane protein receptor, expressed by most cell types, including macrophages is involved in processes such as cell proliferation and tissue regeneration21. Results of many studies revealed dysregulated CXCR4 expression in fibrotic tissue. An increased number of CXCR4+ cells were observed in the lung tissue of patients with IPF22 and, recently, it was suggested as a biomarker for IPF in humans23. Additionally, CXCR4 expression increased after renal injury and its sustained activation enhanced the fibrotic response24.
In a more advanced stage of endometrosis, in category IIB vs I, DEGs were annotated to the pulmonary fibrosis idiopathic signaling pathway (e.g. ACTA2, AREG, ADAMTS1, WNT4, WNT11), growth of connective tissue (e.g. AGER, TIMP1) and cell proliferation of fibroblasts (e.g. AGER, FADD, FTH1, MED31, NUPR1, PDGFD, SERPINE1, TGFB1I1). Moreover, our data showed the changes in the expression of genes annotated to apoptosis (e.g. AAMDC, AATK, ACER2, AGER, CASP2, NUPR1), necrosis (e.g. AGER, ATF3) and cell homeostasis (e.g. AGER, AQP1, ATP13A4, HK2, HKDC1). According to our results, NUPR1, ATF3 AGER, and CASP2, which are positive regulators of apoptotic processes, were up-regulated in category IIB endometria. Though the role of apoptosis in tissue fibrosis is not very well studied, apoptosis was shown to be related to the cell type in fibrotic tissue25,26,27. It was demonstrated that increased apoptosis of alveolar epithelial cells and decreased apoptosis of fibroblasts may play an important role in the pathogenesis of lung fibrosis25. Increased expression of apoptotic factors, NUPR1 and ATF3 has been reported to activate renal fibrosis in rats26 and liver fibrosis in humans and mice27, respectively. However, it has to be underlined that apoptotic factors were found to have roles different than only induction of apoptosis and their role need to be investigated in endometrosis.
Additionally, the results of our study demonstrated the changes in the expression of genes associated with mitochondrial dysfunction (e.g. COX17, COX6B1, CPT1B, FIS1, GLRX2) and oxidative phosphorylation (e.g. MT-ATP6, MT-CO1, MT-CO2, NDUFA3, NDUFS6, UQCRB) in category IIB endometria. There is a growing body of evidence that mitochondrial dysfunction contributes to the development and progression of fibrosis28. Results of our study demonstrated alteration in the expression of genes connected to the respiratory chain (COX6B1), proton transmembrane transporter activity (MT-ATP6, MT-CO1) mitochondria fission (FIS1), and redox signal transduction (GLRX2) in category IIB endometria. According to other study, the expression of COX17 was up-regulated in ischemia–reperfusion injury AKI mice which suggests that COX17 plays a role in the development of renal fibrosis29. Furthermore, Zhou et al.30 showed that the protein abundance of FIS1, involved in the fragmentation of the mitochondrial network and its perinuclear clustering, was up-regulated in the human fibrotic liver which suggests that FIS1 can promote HSC activation.
Metabolic changes
The presented study points to potential novel factors and pathways which has to be taken into consideration as important players in the development of fibrosis. Cellular metabolism is a critical regulator of changes in cellular function. The altered metabolism of carbohydrates, lipids, proteins, and hormones has been documented in lung, liver, and kidney fibrosis31, 32. The changes in cellular metabolism described in other fibrotic tissue have not been pointed out so far in equine endometrosis. In our study, the possible metabolic changes, especially in category IIA vs I endometria, were connected to metabolism and molecular transport of lipids (e.g. ABHD3, ALOX5, FABP5, KDSR, PLPP6), carbohydrates (e.g. ALDOB, FABP5, FOXO1, FUT2, HK2), and amino acids (e.g. SLC1A1, SLC25A29, SLC38A9, SLC6A20, SLC7A2). Results of our research showed that HK2 mRNA transcription declined in all stages of endometrosis. Accumulating evidence points to the role of HK2 in fibrosis development. The role of HK2 in fibrosis seems to be dependent on the affected organ. In the heart, Wu et al.33 demonstrated that a reduction in HK2 levels caused altered remodeling of the heart in ischemia/reperfusion by increasing cell death, fibrosis and reducing angiogenesis. The knockdown of HK2 resulted in exaggerated cardiac hypertrophy via increased reactive oxygen species production34. However, Yin et al.35 indicated that HK2 coupled glycolysis with the profibrotic actions of TGF-β in pulmonary fibrosis. In equine fibroblasts derived from category I and IIA endometria, TGF-β1 up-regulated mRNA transcription of HK2 and down-regulated it in fibroblasts derived from category IIB endometria. Probably, the level of HK2 is down-regulated in the course of fibrosis, but at the beginning of its development, TGF-β1 stimulates its transcription.
Inflammatory mediators and their effect on tissue remodeling
Our results link fibrosis to the inflammatory response which can be a result of cellular infiltration by macrophages, Th1 and Th2 activation pathways (e.g. CXCR4, IL12RB2), IL-13 (e.g. AGR2, CD36, DEFB1, SPDEF), IL-17 signaling pathways (e.g. CXCL1, DEFB1, LIF, MMP9, RASD1), and fMLP signaling in neutrophils (e.g. RASD1). Moreover, a huge number of inflammatory mediators such as TGF-β1, IL-4, IL-13, and IL-17 were determined as regulators of DEGs. Immune cells play an important role in the development of fibrosis by their effect on surrounding cells through secreted cytokines36. As suggested previously, the profibrotic action of TGF-β1, IL-1β, and IL-6 in the development of mare endometrial fibrosis can be connected to myofibroblast differentiation, fibroblast proliferation, MMP expression, and gelatinolytic activation, as well as synthesis of ECM10,11,12,13, 37.
In the current study, we were interested in the action of TGF-β1, which is the most potent profibrotic growth factor, on mRNA transcription of ADAMTS family members in endometrial fibroblasts. In general, ADAMTS are multi-domain matrix-associated zinc metalloendopeptidases that play multiple roles in tissue morphogenesis and pathophysiological remodeling, inflammation, and vascular biology38. These protein family members are strongly related to the TGF-β signaling and are involved in the ECM remodeling process39, 40. Recent studies suggest the role of ADAMTS members in the development of fibrosis as the important players in the turnover of ECM in various tissues and their altered regulation has been implicated in diseases such as cancer, osteoarthritis, and cardiovascular disease41,42,43,44. Results of our study showed that the expression of ADAMTS1, 4, and 5 were up-regulated, while ADAMTS9, 12, and 19 were down-regulated in endometrial tissue in category IIB. It suggests their important role in the development of endometrosis. Although the biological functions of the majority of the distinct ADAMTS subtypes remain poorly characterized, there is a growing interest in their role in the pathogenesis of tissue fibrosis. Dysregulated expression of members of ADAMTS family genes/proteins was found to be associated with fibrosis in the kidney and the lung42, 43, 45, 46. The expression and activation of ADAMTS-4 are augmented by several molecules, such as IL-1, tumor necrosis factor (TNF), TGF-β1, IL-17 as well as fibronectin44. These suggest their important function in endometrial fibrotic processes in mares. Thus, we further explored the effect of select upstream regulators on ADAMTS genes in equine endometrial fibroblasts derived from different stages of endometrosis. The effect of TGF-β1 on ADAMTS1 and -9 mRNA transcription differed in equine endometrial fibroblasts depending on endometrosis stage. Moreover, TGF-β1 treatment increased ADAMTS4 but decreased ADAMTS5 mRNA transcription in endometrial fibroblast.
We also determined the effect of IL-4, IL-13, and IL-17 on ADAMTS mRNA transcription in equine endometrial fibroblasts. The knowledge about the role of these cytokines in the development of endometrosis in mare remains unclear. However, in recent years, the role of T helper cells in the development of fibrosis in different organs has been intensively investigated47. Results of the study showed that DEGs were annotated to Th1 and Th2 activation pathways as well as IL-13 and IL-17 signaling pathways. In general, it was demonstrated that IL-17 exhibits both profibrotic and antifibrotic functions in different organs in a disease-specific manner. The cytokines produced by Th2 (IL-4 and IL-13) act as a profibrotic factor in skin fibrosis48. However, to the best of our knowledge, the effect of IL-4, IL-13, and IL-17 on the mRNA transcription of ADAMTS family members in fibroblasts has not studied so far. Their importance in fibrosis can be attributed to a narrow substrate specificity that makes them potentially better pharmaceutical targets.
Conclusion
The changes in gene expression appear to reflect the changes occurring in endometria at various stages of endometrosis. However, our results only suggest a possible mechanism of action and these findings need to be investigated by designing functional studies. In the mild stage of endometrosis compared to category I endometria, DEGs are annotated to inflammation, abnormal metabolism, wound healing, and quantity of connective tissue. In the moderate stage of endometrosis compared to category I, DEGs are annotated among others to inflammation, fibrosis, cell death, cellular homeostasis, and mitochondrial dysfunction. The changes of ADAMTS-1, -4, -5, -9, in fibrotic endometrium as well as in endometrial fibroblast in response to TGF-β1, IL-4, IL-13, and IL-17 suggest the important role of these factors in the development of endometrosis.
Material and methods
Uteri used in the experiments (42 in total; 27 in experiment 1 and 15 in experiment 2) were obtained post-mortem from cyclic mares at a local abattoir (Rawicz, Poland). Procedures were reviewed and accepted by the Local Ethics Committee for Experiments on Animals in Olsztyn, Poland (Approval No. 51/2011). The collection of material was conducted between April and June. Normally cycling cold-blooded mares (weighing from 400 to 600 kg) at age from 2 to 20 were used in this study. The mares were clinically healthy, as declared by official government veterinary inspection and individual veterinary histories of animal health. Immediately before death, peripheral blood samples were collected into heparinized tubes (Monovettes-Sarstedt, Numbrecht, Germany). The material was collected within 5 min after slaughter. The follicular phases of the estrous cycle were identified based on progesterone (P4) analysis of blood plasma and the macroscopic observation of ovaries. The follicular phase was characterized by the absence of an active corpus luteum (CL) and the presence of follicles of various sizes, but always at least one follicle > 35 mm in diameter, with a concentration of P4 < 1 ng/ml as described previously9. All endometria were confirmed to be free from inflammation, by microscopic evaluation of endometrial smears collected with sterile swabs before the endometrium was excised from the myometrium and stained with Diff-Quick49. The presence of bacteria detected by cytological examination was the criterion for the exclusion of tissue samples from the experiments. The presence of more than two neutrophils per four microscopic fields (mag = 400X) indicates acute endometritis50.
Endometrial tissue was divided into two parts, one part was placed in 4% paraformaldehyde for histological analysis after hematoxylin–eosin staining and the other part was stored in RNAlater (#AM7021; Invitrogen, Burlington, ON, Canada) for RNA-seq. The horns of uteri (n = 15) for cell culture were kept in cold sterile physiological saline with 0.01% of antibiotic antimycotic (AA) solution containing penicillin, streptomycin, amphotericin (A5955, Sigma-Aldrich) and transported to the laboratory on ice. After hematoxylin–eosin staining, endometria were retrospectively assigned to categories I, IIA, or IIB according to the Kenney and Doig classification2.
The isolation and culture of fibroblasts
The fibroblasts were isolated from mare endometrium, cultured, and passaged, as previously described with modifications10. Briefly, the uterine lumen was washed three times with 10 ml of sterile Hanks’ balanced salts (HBSS; H1387; Sigma-Aldrich) containing 0.01% of AA solution. A uterine horn was slit open with scissors to expose the endometrial surface. Endometrial strips were excised from the myometrium layer with a scalpel, washed once with sterile HBSS containing 0.01% of AA solution, and cut into very small pieces (1–3 mm3) with a scalpel. The minced tissues were digested once by stirring for 45 min in 100 ml of sterile HBSS containing 0.05%, (w/v) collagenase I (C2674, Sigma-Aldrich), 0.005% (w/v) DNase I (11,284,932,001; Roche), 0.01% AA, and 0.1% (w/v) bovine serum albumin (BSA; A9418, Sigma-Aldrich). Then, the cell suspension was filtered through the 100 μm, 70 μm, and 40 µm strainers to remove undigested tissue fragments and centrifuged at 100×g for 10 min. To lyse red blood cells, the cell pellet was resuspended and gently mixed with 1 ml of Red Blood Cell Lysing Buffer Hybri-Max™ (R7757; Sigma-Aldrich), then washed three times by centrifugation (4 °C, 100×g, 10 min) in HBSS supplemented with antibiotics and 0.1% (w/v) BSA. The final pellet of endometrial cells was resuspended in FBM™ Basal Medium (CC-3131, LONZA) supplemented with FGM™-2 SingleQuots™ supplements. The cells were counted using a Countess™ 3 FL Automated Cell Counter. The viability of endometrial cells was higher than 95% as assessed by the trypan blue exclusion test.
The dispersed cells were seeded separately at a density of 5 × 105 viable cells/ml and cultured at 38.0 °C in a humidified atmosphere of 5% CO2 in the air. To purify the fibroblast population, the medium was changed 18 h after plating, by which time selective attachment of fibroblasts had occurred, and other types of endometrial cells were eliminated (i.e. epithelial and endothelial cells). The medium was changed every second day until the cells reached confluence. The fibroblast homogeneity was confirmed using immunofluorescent staining for vimentin based on the protocol described recently [51; data not shown]. The purity of fibroblast after isolation was around 96%. After reaching 90% of confluency, the cells were cryopreserved as described previously52.
Experiment 1. Transcriptomic analysis of endometrial tissue at different stages of endometrosis at the follicular phase of the estrous cycle
RNA extraction, purification, and degradation analysis
The endometrial tissues were divided into three groups (n = 9 for each) according to Kenney and Doig’s categories: I, IIA, and IIB2. Total RNA was isolated from around 60 mg of endometrial tissue and homogenized with the use of TriReagent® (T9424; Sigma-Aldrich) and controlled in terms of quality using the Agilent TapeStation2200 system. Within each group, every three RNA isolates were pooled in equimolar ratios into one sample to obtain three replicates (pools). The pooling of samples was performed to reduce the experimental costs and the variability among individual samples and at the same time have more individuals within an experiment.
RNA integrity number (RIN) was assessed for each RNA isolate using Agilent 2100 system and Expert software (Agilent Technologies, Inc., Santa Clara, CA, USA). Only samples with a RIN above 8.0 were processed further. In total, 500 ng of RNA was used for library construction with the TruSeq RNA Sample Prep v2 kit (Illumina, San Diego, CA).
Library preparation
Standard library preparation steps including mRNA selection, fragmentation, cDNA synthesis, end repair, adenylation, indexed adapters ligation, and amplification were followed by a qualitative evaluation (Agilent TapeStation 2200) and quantitation (Qubit, Thermo Fisher Scientific). The libraries were eventually sequenced in a single 50 bp run (1 × 50 bp) on the HiScanSQ system using the TruSeq SBSv3 Sequencing kit, to obtain 15–20 million reads/sample.
Data analysis
Raw reads were controlled for quality using FastQC software and filtered with Flexbar software to remove adapters and those with a Phred quality score below 20 and read length below 30 nt. Filtered reads were mapped against Equus caballus reference genome assembly (EquCab3) with a TopHat2 software set to single-end reads and the “bowtie2 sensitive” option. The reads mapped to individual genes were counted using HTSeq software with ‘Union’ mode and GTF annotation file (Ensembl annotation version 104). Differential expression analysis was performed with DESeq2 software in pairwise comparisons. The quality of RNA-Seq read mappings was controlled using RseQC software. Only genes with adjusted P-value (after FDR correction using the Benjamini–Hochberg procedure, q-value) < 0.05 were considered as differentially expressed (DE). The characteristics of a given RNA-seq data set among the groups were evaluated using principal component analysis (PCA) (Supplementary Data 5).
To determine the gene network, the obtained data were analyzed using Ingenuity Pathways Analysis (IPA) tools (Ingenuity Systems, Mountain View, CA), a web-delivered application that enables the identification, visualization, and exploration of molecular interaction networks in gene expression data.
Raw sequencing reads obtained from all analyzed samples were deposited in the publicly available SRA (Sequence Read Archive) NCBI database under accession number PRJNA880454.
Experiment 2. The effect of cytokines on the differentially expressed genes in endometrial fibroblasts
In Experiment 1, DEGs in categories IIA vs I and IIB vs I endometria were observed to be regulated by TGF-β1, IL-4, IL-13, and IL-17. Thus, in Experiment 2, we aimed to determine how chosen cytokines (TGF-β1, IL-4, IL-13, IL-17) affect the expression of selected DEGs connected to extracellular matrix remodeling (ADAMTS family members) and cell metabolism (HK). In addition, using endometrial fibroblasts isolated from different endometrosis categories, we evaluated whether the response of fibroblasts to a given factor differs depending on the stage of endometrosis.
For in vitro experiments, thawed fibroblasts isolated previously from mare endometria (n = 5) were seeded on T75 cm2 in FBM™ Basal Medium supplemented with FGM™-2 SingleQuots™ supplements and ascorbic acid (100 ng/ml; A4544; Sigma-Aldrich). After reaching 90% confluence, fibroblasts were trypsinized and seeded on 24-well plates. When fibroblasts from passage 1 reached desired 80% confluence, the culture medium was replaced with starvation medium: Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham (DMEM/Ham’s F-12; D2906; Sigma-Aldrich) supplemented with 0.01% of AA solution, ascorbic acid (100 ng/ml) and 0.1% (w/v) BSA, and the cells were incubated at 38.0 °C in 5% CO2. After the starvation, cells were treated with TGF-β1 (10 ng/ml; 100–21; human recombinant; PeproTech), IL-4 (10 ng/ml; RP0003E, equine recombinant, Kingfisher Biotech), IL-13 (10 ng/ml; RP0102E, equine recombinant, Kingfisher Biotech) or IL-17A (10 ng/ml; RP0078E, equine recombinant, Kingfisher Biotech) for 48 h. All doses of cytokine were selected based on previous studies: TGF-β110, IL-453, IL-1354, and IL-1755. After 48 h of the treatments, the cells were dispersed with 1 mL TRI Reagent® and stored at − 80 °C for subsequent RNA extraction and qPCR.
RNA extraction and cDNA synthesis
Total RNA was extracted using TRI Reagent® according to the manufacturer’s instructions. The concentration and quality of RNA were determined spectrophotometrically and by agarose gel electrophoresis. The ratio of absorbance at 260 and 280 nm (A260/280) was approximately 1.8–2. The RNA samples were kept at − 80 °C. Dnase I (AMPD-1; Sigma-Aldrich) was used according to the manufacturer’s directions for eliminating DNA from RNA samples before qPCR. Total RNA (1 μg) was reverse transcribed using a ThermoScript RT-PCR System according to the manufacturer’s directions (no. 11146-016; Invitrogen). The cDNA was stored at − 20 °C until qPCR was carried out.
qPCR
To validate the RNA-Seq results, the expression level of selected DEGs was confirmed by qPCR. The same samples were used for both, RNA-Seq and qPCR analysis. However, to validate the results of RNA-seq the samples were not pooled but all individual samples were used. Additionally, the qPCR was used to determine the effect of selected cytokines on gene expression in endometrial cells in vitro. qPCR was performed in a 7900HT Fast Real-Time PCR System using TaqMan Universal Master Mix II (4,440,049; Applied Biosystems, Foster City, CA, USA) with 384-well plates. All samples were run in duplicates. For measurements of mRNA transcription of ADAMTS1 (cat. no. Ec03469970_m1), ADAMTS4 (cat. no. Ec03469176_m1), ADAMTS5 (cat.no. Ec03470669_m1), ADAMTS9 (cat.no. Ec06952347_m1), CXCR4 (cat. no. Ec07070263_s1), HK2 (cat.no. Ec03467576_m1), SLC25A29 (cat. no. Ec06979775_m1), HPRT (cat. no. Ec03470217_m1), SDHA (cat. no. Ec03470487_m1), GAPDH (cat. no. Ec03210916_gH), B2M (cat. no. Ec03468699_m1), RPL32 (cat. no. Ec06951800_m1), RPS18 (cat.no. Ec06969343_g1), RPS28 (cat. no. Ec06997545_g1), Single Tube TaqMan Gene Expression Assays (Life Technologies Thermo Fisher Scientific) were used. The selection of the most adequate reference genes was done using NormFinder software56. As SDHA and HPRT were found to have the most stable expression across the endometrosis categories, gene expression data were normalized to the average geometric mean of these two genes.
The reaction mixture for the qPCR assay comprised 5 μL TaqMan Universal PCR Master Mix, 0.5 μL TaqMan probe, 3 μL DNA (10 ng), and 1.5 μL nuclease-free water for a final volume of 10 μL. As a negative control, nuclease-free water instead of template cDNA was used. cDNA amplification was performed under the following conditions: initial denaturation for 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The data were analyzed using the method described previously57. The relative concentration of mRNA (R0) for each target and reference gene (SDHA; HPRT) was calculated using the equation R0 = 1/(1 + E)Ct, where, E is the average gene efficiency and Ct is the cycle number at the threshold. The relative gene expression was calculated as R0target gene/R0reference gene and was expressed in arbitrary units.
Immunofluorescence staining
The immunofluorescence staining was performed to determine the infiltration of macrophages in the categories I (n = 3) and IIA (n = 3) endometria to support results obtained from the transcriptomic analysis. The immunofluorescence staining was performed as described previously51. Briefly, formalin-fixed endometrial samples were processed, embedded in paraffin, and sectioned at 5 μm. Sections were deparaffinized (xylene I for 10 min, xylene II for 10 min) and rehydrated through a graded alcohol series (100% ethanol for 2 min, 96% ethanol for 2 min, and 70% ethanol for 2 min). Rehydration was followed by antigen retrieval in sodium citrate buffer (10 mM Sodium citrate, 0.05% Tween 20, pH = 6) at 95 °C for 25 min. Subsequently, the endometrial sections were permeabilized with PBS containing 0.3% TritonX-100 for 10 min, washed one time for 5 min in PBS, and blocked with 1% BSA and 5% normal goat serum in PBS for 1 h at room temperature. The endometrial sections were incubated overnight with CD68 + primary antibody (1:100, cat. no. 14–0681-82; Invitrogen) at 4 °C. Afterward, the sections were washed three times for 5 min with PBS and incubated with secondary antibodies labeled with Alexa Fluor 594 (1:400, cat no. A-11007, Thermo Fisher Scientific). The slides were washed three times for 5 min with PBS and mounted in Fluoroshield with DAPI (Sigma-Aldrich; F6057).
The number of CD68+ cells was counted manually in 15 regions that were randomly selected from 15 images at 20 × magnification from each endometrial section. The scoring results were shown as mean ± SD number of immunopositive cells per µm2 of the tissue section.
Radioimmunoassay
Plasma concentrations of P4 were assayed by RIA (Diasource; KIP1458). All samples were run in duplicate. The P4 standard curve ranged from 0.12 to 36 ng/ml. The intra- and inter-assay CVs were on average 8% and 10%, respectively. The sensitivity of the assay was 0.05 ng/ml. The validation of RIA in horse blood plasma was done as described previously58.
Statistical analysis
Data are shown as the mean ± SD. For each analysis, the Gaussian distribution of results was tested using the D'Agostino & Pearson normality test (GraphPad Software version 9; GraphPad, San Diego, CA). Whenever the assumptions of normal distribution were not met, nonparametric statistical analyses were done. In Experiments 1 and 2, the significant differences were determined by the nonparametric Mann–Whitney U test. The endometrial HK2 mRNA transcription in categories I, IIA, and IIB was determined by using parametric one-way ANOVA followed by a Newman-Keuls comparison test. The results were considered significantly different when P < 0.05.
Ethics approval and consent to participate
Procedures were reviewed and accepted by the Local Ethics Committee for Experiments on Animals in Olsztyn, Poland (Approval No. 51/2011). All methods were carried out in accordance with relevant guidelines and regulations. All methods are reported in accordance with ARRIVE guidelines for the reporting of animal experiments.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request. NGS results were deposed in the publicly available SRA (Sequence Read Archive) NCBI database under accession number PRJNA880454.
References
Kenney, R. M. Cyclic and pathologic changes of the mare endometrium as detected by biopsy, with a note on early embryonic death. J. Am. Vet. Med. Assoc. 172, 241–262 (1978).
Kenney, R. M. & Doig, P. A. Equine endometrial biopsy. In Current Therapy in Theriogenology Vol. 2 (ed. Morrow, D. A.) 726–29 (Saunders WB, 1986).
Hoffmann, C. et al. The equine endometrosis: New insights into the pathogenesis. Anim. Reprod. Sci. 111, 261–78 (2009).
Lehmann, J. et al. Morpho-functional studies regarding the fertility prognosis of mares suffering from equine endometrosis. Theriogenology 76, 1326–1336 (2011).
Zeisberg, M. & Kalluri, R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 304, C216-25 (2013).
Flores, J. M., Rodriguez, A., Sánchez, J., Gómez-Cuétara, C. & Ramiro, F. Endometrosis in mares: Incidence of histopathological alterations. Reprod. Dom. Anim. 30, 60–65 (1995).
Hanada, M., Maeda, Y. & Oikawa, M. A. Histopathological characteristics of endometrosis in thoroughbred mares in Japan: Results from 50 necropsy cases. J. Equine Sci. 25, 45–52 (2014).
Szóstek, A. Z. et al. mRNA transcription of prostaglandin synthases and their products in the equine endometrium in the course of fibrosis. Theriogenology 78, 768–776 (2012).
Szóstek, A. Z., Lukasik, K., Galvão, A. M., Ferreira-Dias, G. M. & Skarzynski, D. J. Impairment of the interleukin system in equine endometrium during the course of endometrosis. Biol. Reprod. 89, 79 (2013).
Szóstek-Mioduchowska, A. Z., Lukasik, K., Skarzynski, D. J. & Okuda, K. Effect of transforming growth factor-β1 on α-smooth muscle actin and collagen expression in equine endometrial fibroblasts. Theriogenology 124, 9–17 (2019).
Szóstek-Mioduchowska, A., Słowińska, M., Pacewicz, J., Skarzynski, D. J. & Okuda, K. Matrix metallopeptidase expression and modulation by transforming growth factor-β1 in equine endometrosis. Sci. Rep. 10, 1119 (2020).
Szóstek-Mioduchowska, A. Z., Baclawska, A., Okuda, K. & Skarzynski, D. J. Effect of proinflammatory cytokines on endometrial collagen and metallopeptidase expression during the course of equine endometrosis. Cytokine 123, 154767 (2019).
Szóstek-Mioduchowska, A. Z., Baclawska, A., Rebordão, M. R., Ferreira-Dias, G. & Skarzynski, D. J. Prostaglandins effect on matrix metallopeptidases and collagen in mare endometrial fibroblasts. Theriogenology 153, 74–84 (2020).
Witkowski, M. et al. Next-generation sequencing analysis discloses genes implicated in equine endometrosis that may lead to tumorigenesis. Theriogenology 189, 158–166 (2022).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020).
Wynn, T. A. & Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257 (2010).
Wynn, T. A. & Vannella, K. M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016).
Liu, C. et al. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab Investig. 90, 1805–1816 (2010).
Pereira, T., Naik, S. & Tamgadge, A. Quantitative evaluation of macrophage expression using CD68 in oral submucous fibrosis: An immunohistochemical study. Ann. Med. Health Sci. Res. 5, 435–441 (2015).
Nouno, T. et al. Elevation of pulmonary CD163+ and CD204+ macrophages is associated with the clinical course of idiopathic pulmonary fibrosis patients. J. Thorac. Dis. 11, 4005–4017 (2019).
Bianchi, M. E. & Mezzapelle, R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front. Immunol. 11, 2109 (2020).
Jaffar, J. et al. CXCR4+ cells are increased in lung tissue of patients with idiopathic pulmonary fibrosis. Respir. Res. 21, 221 (2020).
Derlin, T. et al. Clinical molecular imaging of pulmonary cxcr4 expression to predict outcome of pirfenidone treatment in idiopathic pulmonary fibrosis. Chest 159, 1094–1106 (2021).
Yuan, A., Lee, Y., Choi, U., Moeckel, G. & Karihaloo, A. Chemokine receptor Cxcr4 contributes to kidney fibrosis via multiple effectors. Am. J. Physiol. Ren. Physiol. 308, F459–F472 (2015).
Drakopanagiotakis, F., Xifteri, A., Polychronopoulos, V. & Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 32, 1631–1638 (2008).
Zhou, R. et al. Nupr1 mediates renal fibrosis via activating fibroblast and promoting epithelial-mesenchymal transition. FASEB J. 35, e21381 (2021).
Shi, Z. et al. Transcriptional factor ATF3 promotes liver fibrosis via activating hepatic stellate cells. Cell Death Dis. 11, 1066 (2020).
Li, X. et al. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 6, 80 (2020).
Zhu S, Ch, Y. COX17-Mediated abnormal mitochondrial copper metabolism promotes renal fibrosis. Am. Soc. Nephrol., SU—OR14 (2020).
Zhou, Y. et al. Oxidative stress-mediated mitochondrial fission promotes hepatic stellate cell activation via stimulating oxidative phosphorylation. Cell Death Dis. 13, 689 (2022).
Henderson, J. & O’Reilly, S. The emerging role of metabolism in fibrosis. Trends Endocrinol. Metab. 32, 639–653 (2021).
Ung, C. Y., Onoufriadis, A., Parsons, M., McGrath, J. A. & Shaw, T. J. Metabolic perturbations in fibrosis disease. Int. J. Biochem. Cell Biol. 139, 106073 (2021).
Wu, R. et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ. Res. 108, 60–69 (2011).
Wu, R. et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol. Med. 4, 633–646 (2012).
Yin, X. et al. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci. Signal. 12, eaax4067 (2019).
Borthwick, L. A., Wynn, T. A. & Fisher, A. J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta 1832, 1049–1060 (2013).
Szóstek, A. Z., Galvão, A. M., Ferreira-Dias, G. M. & Skarzynski, D. J. Ovarian steroids affect prostaglandin production in equine endometrial cells in vitro. J. Endocrinol. 220, 263–276 (2014).
Kelwick, R., Desanlis, I., Wheeler, G. N. & Edwards, D. R. The ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 16, 113 (2015).
Chen, C., Li, R., Ross, R. S. & Manso, A. M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 93, 162–174 (2016).
Margadant, C. & Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 11, 97–105 (2010).
Mead, T. J. & Apte, S. S. ADAMTS proteins in human disorders. Matrix Biol. 71–72, 225–239 (2018).
Taylor, S., Whitfield, M., Barratt, J. & Didangelos, A. The metalloproteinase ADAMTS5 is expressed by interstitial inflammatory cells in IgA nephropathy and is proteolytically active on the kidney matrix. J. Immunol. 205, 2243–2254 (2020).
Vojtusek, I. K. et al. First characterization of ADAMTS-4 in kidney tissue and plasma of patients with chronic kidney disease-a potential novel diagnostic indicator. Diagnostics (Basel) 12, 648 (2022).
Novak, R. et al. The role of ADAMTS-4 in atherosclerosis and vessel wall abnormalities. J. Vasc. Res. 59, 69–77 (2022).
Nakamura, A., Sakai, Y., Ohata, C. & Komurasaki, T. Expression and significance of a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-1 in an animal model of renal interstitial fibrosis induced by unilateral ureteral obstruction. Exp. Toxicol. Pathol. 59, 1–7 (2007).
Kreus, M., Lehtonen, S., Skarp, S. & Kaarteenaho, R. Extracellular matrix proteins produced by stromal cells in idiopathic pulmonary fibrosis and lung adenocarcinoma. PLoS One 16, e0250109 (2021).
Zhang, M. & Zhang, S. T cells in fibrosis and fibrotic diseases. Front. Immunol. 11, 1142 (2020).
Nguyen, J. K., Austin, E., Huang, A., Mamalis, A. & Jagdeo, J. The IL-4/IL-13 axis in skin fibrosis and scarring: Mechanistic concepts and therapeutic targets. Arch. Dermatol. Res. 312, 81–92 (2020).
Rebordão, M. R. et al. Impairment of anti-fibrotic PGE2 pathway might influence neutrophil extracellular traps-induced fibrosis in mare endometrium. Domest. Anim. Endocrinol. 67, 1–10 (2019).
Riddle, W. T., LeBlanc, M. M. & Stromberg, A. J. Relationships between uterine culture, cytology and pregnancy rates in a thoroughbred practice. Theriogenology 68, 395–402 (2007).
Szóstek-Mioduchowska, A. Z. et al. Effects of cortisol on prostaglandin F2α secretion and expression of genes involved in the arachidonic acid metabolic pathway in equine endometrium - in vitro study. Theriogenology 173, 221–229 (2021).
Szóstek, A. Z. et al. Effects of cell storage and passage on basal and oxytocin-regulated prostaglandin secretion by equine endometrial epithelial and stromal cells. Theriogenology 77, 1698–1708 (2012).
OuYang, Z. et al. Interleukin-4 stimulates proliferation of endometriotic stromal cells. Am. J. Pathol. 173, 463–469 (2008).
Nasu, K., Fukuda, J., Sun, B., Nishida, M. & Miyakawa, I. Interleukin-13 and tumor necrosis factor-beta differentially regulate the production of cytokines by cultured human endometrial stromal cells. Fertil. Steril. 79, 821–827 (2003).
Ahn, S. H. et al. IL-17A contributes to the pathogenesis of endometriosis by triggering proinflammatory cytokines and angiogenic growth factors. J. Immunol. 195, 2591–2600 (2015).
Andersen, C. L., Ledet-Jensen, J. & Ørntoft, T. Normalization of real-time quantitative RT-PCR data: A model-based variance estimation approach to identify genes suited for normalization - applied to bladder- and colon-cancer data-sets. Cancer Res. 64, 5245–5250 (2004).
Zhao, H. & Fernald, R. D. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 12, 1045–1062 (2005).
Mondal, M., Prakash, B., Rajkhowa, C. & Prakash, B. S. Development and validation of a sensitive radioimmunoassay for progesterone estimation in unextracted mithun (Bos frontalis) plasma. J. Immunoass. Immunochem. 26, 273–283 (2005).
Acknowledgements
Authors would like to thank Agnieszka Bacławska and Witold Krzywiec from the Institute of Animal Reproduction and Food Research, PAS, Olsztyn for their technical support.
Funding
Supported by the project Maestro no. 2011/02/A/NZ5/00338 to DJS (Exp.1.) and project Sonata 15 no. 2019/35/D/NZ9/02989 (Exp.2.) financed by the National Science Centre, Poland.
Author information
Authors and Affiliations
Contributions
A.Sz.-M.—writing-original draft, conception, and design of the study, methodology, funding acquisition, formal analysis, visualization; A.W.—investigation; A.S.—investigation; B.J.—formal analysis; M.S.—investigation, K.L.—investigation; A.G.—formal analysis; T.Sz.—formal analysis, M.B.-P.—formal analysis; G.F.-D editing the original draft; D.S.—conception, funding acquisition, editing the original draft. All authors edited, revised, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szóstek-Mioduchowska, A., Wójtowicz, A., Sadowska, A. et al. Transcriptomic profiling of mare endometrium at different stages of endometrosis. Sci Rep 13, 16263 (2023). https://doi.org/10.1038/s41598-023-43359-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43359-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
