
Abstract
Mare endometrial fibrosis (endometrosis), is one of the main causes of equine infertility. Despite the high prevalence, both ethology, pathogenesis and the nature of its progression remain poorly understood. Recent studies have shown that microRNAs (miRNAs) are important regulators in multiple cellular processes and functions under physiological and pathological circumstances. In this article, we reported changes in miRNA expression at different stages of endometrosis and the effect of transforming growth factor (TGF)-β1 on the expression of the most dysregulated miRNAs. We identified 1, 26, and 5 differentially expressed miRNAs (DEmiRs), in categories IIA (mild fibrosis), IIB (moderate fibrosis), and III (severe fibrosis) groups compared to category I (no fibrosis) endometria group, respectively (Padjusted < 0.05, log2FC ≥ 1.0/log2FC ≤ − 1.0). This study indicated the potential involvement of miRNAs in the regulation of the process associated to the development and progression of endometrosis. The functional enrichment analysis revealed, that DEmiRs target genes involved in the mitogen-activated protein kinases, Hippo, and phosphoinositide-3-kinase (PI3K)-Akt signalling pathways, focal adhesion, and extracellular matrix-receptor interaction. Moreover, we demonstrated that the most potent profibrotic cytokine—TGF-β1—downregulated novel-eca-miR-42 (P < 0.05) expression in fibroblasts derived from endometria at early-stage endometrosis (category IIA).
Similar content being viewed by others

Introduction
Mare endometrial fibrosis (endometrosis) is one of the major reasons for equine infertility1,2,3. Endometrosis leads to alterations in endometrial structure and function, causing changes in the uterine environment and early pregnancy loss1,4. It is a degenerative chronic condition of the mare uterus, described mainly as a fibrotic process developing around the endometrial glands and in the stroma. Moreover, endometrosis is associated with inflammatory infiltrates as well as the dilation of endometrial glands and lymphatic vessels in equine endometrium. There are two terms: endometrosis and endometriosis used in the medical literature, which is defined differently and should not be used interchangeably. Endometriosis means extrauterine implantation of endometrial tissue and refers to women5, whereas the term endometrosis means changes in the mare’s uterus previously referred to as chronic degenerative endometritis1.
Despite the increasing number of studies focusing on endometrosis, its pathogenesis remains not completely understood6. Its root cause seems to be repeated and chronic inflammation, associated with the age, numbers of foaling and breeding1,3,7. Studies on endometrosis focused mainly on the role of proinflammatory cytokines, neutrophils, and prostaglandins8,9,10,11,12,13,14. There are multiple signalling pathways associated with the development of fibrotic diseases, including transforming growth factor (TGF)-β1 signalling. TGF-β1 is a main profibrotic cytokine15, important in processes associated with the development of mare endometrosis11. Previously, TGF-β1 was shown to increase expression of extracellular matrix (ECM) components in mare endometrial fibroblasts cultured in vitro as well as to stimulate fibroblast proliferation8. The profibrotic mechanisms of TGF-β1 action are regulated by cross-talk with other signalling pathways, and epigenetics mechanisms, including microRNAs (miRNAs)15.
Nowadays, epigenetic determinants start to evoke great interest. Body of evidence indicates a meaningful role of miRNAs at the site of injury and fibrosis and disturbed miRNA expression along with the progression of liver, heart, skin, kidney, and lung fibrosis16. However, to the best of our knowledge, there is no information concerning the regulatory role of miRNAs and their regulators in the development of endometrosis in the mare. Recent studies revealed alteration in miRNAs expression in mare endometrial fibroblasts cultured in vitro treated with lipopolysaccharides (LPS)17. The stimulation with LPS imitates bacterial infection, thus LPS induced changes in miRNAs expression indicates possible regulatory role of miRNAs in inflammation, which is one of the major causes of endometrosis. Moreover, another study showed, that LPS stimulation changes of TGF-β signaling in human keratinocytes18.
miRNAs belong to a group of small (~ 22 nucleotides long) non-coding RNA molecules, that regulate gene expression at the post-transcriptional level, leading to mRNA degradation and translational inhibition. One miRNA can regulate multiple gene transcripts, which is why they are so significant expression regulators19. Recent studies identified many novel miRNAs associated with multiple organs fibrosis20,21,22. Moreover, miRNAs expression dysregulated with the progression of fibrosis seem to be susceptible to therapeutic modulation. Thus, miRNAs are in the area of interest for targeted therapies. The miRNA families can be described as either pro- or antifibrotic, depending on their target mRNA16.
Through understanding of the core molecular signalling pathways of fibrosis among multiple organs is the essence of further development of novel diagnostics methods and therapeutics. The main aim of the present study was to describe the miRNAs expression signature and thus elucidate the potential molecular processes regulated by differentially expressed miRNAs (DEmiRs) at every stage of endometrosis of mares being in the follicular phase of the estrous cycle. The follicular phase of the estrous cycle was selected based on our previous study, which showed the most changes in the transcriptome associated with fibrosis-related processes14. Further, we investigated, if the most potent profibrotic cytokine—TGF-β1 affects the expression of the most dysregulated in endometrial fibroblasts.
This study aims at describing mare endometrial miRNA profile at each stage of endometrosis and finding potential regulation of their expression by the most potent profibrotic cytokine—TGF-β1.
Results
We conducted next-generation sequencing (NGS) to characterize the miRNAs expression profile in the endometrium of mares in the follicular phase of the estrous cycle at every stage of endometrosis. The miRNA profiling results were submitted to the NCBI BioProject under the accession number PRJNA880660. From 26,304,055 to 2,650,974 reads in individual samples were obtained. Subsequent filtering resulted in on average 5,748,485 sequences, which were further mapped to the Equus caballus (genome EquCab3.0, Ensembl release 106) mature miRNA sequence from miRBase database 22.1. Most expressed miRNAs were 22 nt long (Supplementary Data 1). In this study, we identified 423 known and 90 novel miRNAs (Supplementary Data 2). Principal component analysis (PCA) performed on the basis of miRNA expression results revealed distinctly separated category IIB samples from category I-derived samples (Supplementary Data 3). The distribution of transcripts, including DEmiRs, identified in the examined samples is depicted in Supplementary Data 4.
Mare endometrial miRNA expression profile changes along with the severity of endometrosis
The full list of identified DEmiRs between categories IIA, IIB, and III vs. I endometria including their mature sequence, log2FC, and P-adjusted is provided in Supplementary Data 5.
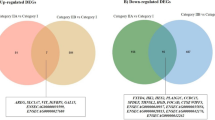
In category IIA vs. category I endometria, we identified one DEmiR—novel-eca-miR-42 (Fig. 1a). We found most DEmiRs in category IIB endometria compared to category I endometria. Briefly, we identified 26 DEmiRs (Fig. 1a) of which 12 were downregulated (including eca-miR-146b-5p, novel-eca-miR-58, novel-eca-miR-77, eca-miR-370, and eca-miR-409-3p), while 14 were upregulated (including eca-miR-34a, eca-miR-29b, eca-miR-34c, eca-miR-34b-5p, eca-miR-34b-3p, eca-miR-29c, eca-miR-190a, eca-miR-205, and novel eca-miR-25). In category III vs. I endometria, five DEmiRs were identified, three of which were downregulated (eca-miR-146b-5p, eca-miR-495 and eca-miR-1), and two were upregulated (eca-miR-151-5p and eca-miR-25; Fig. 1a).

Differentially expressed miRNA (DEmiRs) identified between different categories of mare endometrium with endometrosis. (a) Category IIA (mild fibrosis), IIB (moderate fibrosis), and III (severe fibrosis) vs. I (no fibrosis) pairs and (b) IIA, IIB, and III pairs presented as Venn diagram. Arrows indicate downregulation or upregulation of identified DEmiRs in particular comparisons.
In categories IIB vs. IIA, we identified 29 DEmiRs; 11 of them were downregulated and 19 were upregulated (Fig. 1b). In categories III vs. IIA endometria, two DEmiRs were identified; one downregulated (eca-miR-151-5p) and one upregulated (novel-eca-miR-42). In comparison between categories III and IIB endometria, we identified seven DEmiRs: three down- (eca-miR-145, eca-miR-205, eca-miR34b-3p) and four upregulated (eca-miR-370, eca-miR-77, eca-miR-58, eca-miR-9055).
The circos plot illustrates the expression profile of DEmiRs in endometrial samples in both comparisons between fibrous endometria (categories IIA, IIB, and III) vs. non-fibrous (category I) as well as between tissue with different stages of endometrosis (Fig. 2).

Circos plot illustrating the expression profile of differentially expressed miRNA (41 DEmiRs; P-adjusted ≤ 0.05 and log2FC ≥ 1.0 or log2FC ≤ − 1.0) in mare endometrium category I (no fibrosis), IIA (mild fibrosis), IIB (moderate fibrosis), and III (severe fibrosis). Track A (blue) represents log-transformed mean of Reads Per Kilobase Million (RPKM) values of control samples (maximum value = 7.384); Track B (violet) represents log-transformed mean of RPKM values of treatment samples (maximum value = 7.217); Track C (green–red) depicts fold changes in the miRNA expression level between two respective comparison (IIA compared to I, IIB compared to I, III compared to I, IIB compared to IIA, III compared to IIA or III compared to IIB), where the brightest green depicts the − 27.175-fold change and the brightest red the 27.103-fold change; Links in the middle of the circular plot connect these miRNA, expression of with significantly differed between any two sets of compared condition; Gene names originated from miRBase (v. 22.1).
Among all DEmiRs, the most downregulated were: novel-eca-miR-42, eca-miR-77, eca-miR-58, and eca-miR-146b-5p, while among most upregulated were: eca-miR-25, eca-miR-205, eca-miR-190a, eca-miR-29c, and eca-miR-34b-3p (Fig. 3a, Supplementary Data 5). Therefore, for the next steps of the study four miRNAs with the most changed expression and available TaqMan probes (novel-eca-miR-42, eca-miR-29c, eca-miR-34b-3p, and eca-miR-205) were selected.

Differentially expressed miRNAs (DEMiRs) in mare endometrium with I (no fibrosis), IIA (mild fibrosis), IIB (moderate fibrosis), and III (severe fibrosis) category, KEGG, and GO pathway analysis concerning DEmiRs target genes. (a) Differentially expressed miRNAs and the corresponding fold changes. (b) KEGG pathways of miRNA target genes. The y axis is pathway categories. The x axis refers to pathway enrichment. (c) Gene Ontology (GO) analysis of miRNA target genes. The y axis is GO categories. The x axis refers to GO enrichment. CC cellular components, MF molecular functions.
DEmiRs enriched molecular and cellular functions important during the development of fibrosis
The analysis of target genes of the identified DEmiRs enabled the prediction of signalling pathways through KEGG pathway analysis23,24,25. The number of target genes identified for particular DEmiRs in selected comparisons among different endometrosis categories is presented in Supplementary Data 6, while the full list of DEmiRs target genes is included in Supplementary Data 7. We identified seven (IIA vs. I), 12 (IIB vs. IIA), 58 (IIB vs. I), 65 (IIB vs. IIA), two (III vs. IIA), and nine (III vs. IIB) KEGG pathways enriched by DEmiRs identified in the indicated comparisons.
The enriched KEGG pathways shared by categories IIA vs. I and IIB vs. I endometria, include focal adhesion, extracellular matrix (ECM)-receptor interaction, MAPK and PI3-Akt signalling pathways, and regulation of actin cytoskeleton (Fig. 3b). The unique KEGG pathways enriched by IIB vs. I DEmiRs are, among others: Hippo, calcium, oxytocin, AMPK, Wnt, Hedgehog, mTOR, and JAK-STAT signalling pathways. The full list of KEGG enriched pathways with indicated DEmiRs and their target genes is provided in Supplementary Data 8. Selected KEGG pathways in category IIA vs. I and IIB vs. I as well as III vs. IIB endometria enriched by the identified DEmiRs in depicted comparisons are presented in Fig. 3b.
Gene ontology (GO) analysis was used to predict the biological processes, cellular components (CC) and molecular functions (MF) that were the gene product enrichments of identified DEmiRs (Fig. 3c). In categories IIB vs. I and IIB vs. IIA endometria, we indicated nine and 12 GO categories, respectively. Our results showed the following most significantly enriched GO terms in category IIB vs. I endometria among CC: intrinsic and integral component of plasma membrane and cell surface, while cell surface, transmembrane, transporter, and ion transmembrane transporter activity, ATP, adenyl ribonucleotide and nucleotide binding among MF category (Supplementary Data 9). In category IIB vs. IIA endometria, the analysis revealed plasma membrane region (CC) as well as anion transmembrane and ATPase-coupled transporter activity (MF; Supplementary Data 9).
Validation of selected DEmiRs by qRT-PCR
To validate the miRNA-Seq results, four miRNAs (novel-eca-miR-42, eca-miR-29c, eca-miR-34b-3p, and eca-miR-205) were selected for qRT-PCR. A comparison of DEmiRs log2fold change and P-value obtained after qPCR and P-adjusted after NGS analyses is presented in Table 1. Analysis confirmed decreased novel-eca-miR-42 expression in endometria category IIA compared to category I (P < 0.05) and increased eca-miR-205 expression in category IIB tissue compared to category I endometria (P < 0.05) (Table 1).
We also compared the expression level of selected DEmiRs in cultured endometrial fibroblasts derived from categories IIA and IIB compared to category I endometria samples. In cultured fibroblasts derived from category IIB endometria, eca-miR-205 expression was upregulated compared to fibroblasts derived from category I endometria (P < 0.05; Fig. 4d). In the case of the expression levels of novel-eca-miR-42 (P > 0.05; Fig. 4a), eca-miR-29c (Fig. 4b), and eca-miR-34b-3p (P > 0.05; Fig. 4c) there were no statistically significant differences in mare endometrial fibroblasts isolated from endometria category IIA (P > 0.05; Fig. 4a) and IIB (P > 0.05; Fig. 4b,c) cultured in vitro.

The comparison of the expression of selected for validation DEmiRs (a—novel-eca-miR-42, b—eca-miR-29c, c—eca-miR-34b-3p, d—eca-miR-205) in mare endometrial fibroblasts (n = 4) cultured in vitro isolated from endometria category I (no fibrosis), IIA (mild fibrosis), and IIB (moderate fibrosis). Results are presented as arbitrary units of relative expression means. Asterisks denote statistical differences (*P < 0.05, nonparametric Mann–Whitney U-test).
TGF-β1 decreases novel-eca-miR-42 expression in mare endometrial fibroblasts
The in vitro experiment with the use of the most potent profibrotic cytokine—TGF-β1, revealed that after 24 h of treatment, the expression level of novel-eca-miR-42 was decreased in fibroblasts derived from category IIA endometria compared to non-treated fibroblasts (P < 0.05; Fig. 5b). Interestingly, TGF-β1 did not affect the expression of novel-eca-miR-42 in mare endometrial fibroblasts isolated from endometria without fibrosis (category I, P > 0.05; Fig. 5a). It indicates a possible influence of TGF-β1 on novel-eca-miR-42 expression levels at the early stage of endometrosis development. There was no statistically significant influence of TGF-β1 on the expression of eca-miR-29c (P > 0.05; Fig. 5c,d), eca-miR-34b-3p (P > 0.05; Fig. 5e,f), and eca-miR-205 (P > 0.05; Fig. 5g,h) in cultured fibroblasts derived from category I and IIB endometria (P > 0.05); however, we observed the tendency of TGF-β1 to increase their expression in fibroblasts isolated from endometria category IIB.

The effect of TGF-β1 treatment (10 ng/ml) on eca-miR-42 (a,b), eca-miR-29c (c,d), eca-miR-34b-3p (e,f), eca-miR-205 (g,h) expression in in vitro cultured fibroblast (n = 4 per group) derived from endometria with category I (no fibrosis; a,c,e,g), IIA (mild fibrosis; b), and IIB (moderate fibrosis; d,f,h). Results are presented as a fold change. Asterisks denotes statistical difference (*P < 0.05, nonparametric Mann–Whitney U-test).
Discussion
A growing body of evidence demonstrates disturbances in miRNA expression across multiple tissues fibrosis. However, to date, the miRNA expression profile—miRNAome of mare endometrium with endometrosis has not been described. Thus, we compared the endometrial miRNA expression profiles at every stage of endometrosis development. Further analyses showed most DEmiRs in category IIB compared to I endometria, indicating the possible importance of miRNAs in the development of mare endometrial fibrosis with the greatest importance in the advanced stage. Different DEmiRs were revealed along with the advancement of fibrosis, which may potentially regulate the expression of genes involved in such processes as focal adhesion, ECM-receptor interaction, and PI3-Akt signalling in mare endometrium.
The expression regulation of important in fibrotic disorders genes can be an essential mode of action of the identified DEmiRs in the development of endometrosis. Identified in the current study DEmiRs (inter alia: eca-miR-29b, eca-miR-29c, eca-miR-205, eca-miR-34c, novel-eca-miR-42, eca-miR-503) are associated with fibrosis via their target genes. These genes belong to ECM components: collagens (including COL1A1, COL5A1, COL6A3, COL8A2, COL9A1, and COL24A1), fibronectin (FN), elastin (ELN), and laminin (LAMA-1, -2, -3, -4, and -5). Moreover, also matrix metalloproteinases (MMP2, -13, -17, -19) and their tissue inhibitor (TIMP4) are targeted by the identified DEmiRs. Interestingly, these DEmiRs possibly regulate also immune response by affecting expression levels interferons (IFNA1, IFNA2, IFNK), interleukins (IL1A, IL10, IL17A, IL17B, IL18, IL27), interleukins receptors (IL1R1, IL6R, IL10RA, IL13RA2, IL17RA), and interleukin 4-induced gene-1 (IL4I1).
In mild fibrosis (category IIA), we found one, highly downregulated DEmiR—novel-eca-miR-42 compared to non-fibrous endometrium (category I). The target genes of the miRNA are engaged in pathways significant for fibrotic diseases, including focal adhesion, ECM-receptor interaction, PI3K-Akt signalling pathway, and regulation of actin cytoskeleton26,27,28,29,30,31. Moreover, we showed that TGF-β1 treatment decreased novel-eca-miR-42 expression in endometrial fibroblasts derived from category IIA endometria, in contrast to fibroblasts from category I endometria. miR-1388 (Danio rerio, Ornithorhynchus anatinus, and Taeniopygia guttata), which shares sequence with novel-eca-miR-42 with, was shown to inhibit NF-κB signalling pathway32, which in turn plays a role in liver fibrosis33. A recent study, concerning gene expression of NF-κB-dependent pathway in mare endometrium, indicated most changes in the follicular phase of the oestrous cycle34, confirming the possible important role of NF-κB-pathway in mare endometrosis progression. To the best of our knowledge, no research indicated the significance of miR-42 and its regulation by TGF-β1 in fibrotic disorders so far. Our findings suggest the contribution of novel-eca-miR-42 to signalling pathways associated with the development of fibrosis, and the regulation of its expression by TGF-β1. Thus, the downregulation of its expression in category IIA endometria as well as after TGF-β1 treatment of fibroblasts may denote miR-42 importance in molecular mechanisms at the early stage of fibrosis development. Since the foaling rate even decreases to 50% in IIA category of endometrosis mares35 it is important to find key drivers of initial and further development fibrosis in mare endometrium.
The highest number of DEmiRs (14 up- and 12 downregulated) were found in the endometrial tissue in category IIB compared to category I. It suggests that miRNAs are important regulators of processes taking part in the moderate stage of fibrosis. In the current study, eca-miR-58 was the most downregulated in category IIB vs. I endometria. To the best of our knowledge, there is no information on the action of this molecule in processes related to the development of fibrosis. However, the KEGG enrichment analysis revealed the potential role of novel-eca-miR-58 in the regulation of key processes associated with the development of fibrosis, such as Hippo, Wnt, MAPK, JAK-STAT, Hedgehog, and mTOR signalling pathways, as well as ECM-receptor interaction and focal adhesion. A recent study indicated the cross-species anti-inflammatory and pro-autophagia role of Salvia miltiorrhiza-derived sal-miR-5836. As the level of novel-eca-miR-58 was significantly reduced in our study, it can be hypothesized, that it may lead to the upregulation of proinflammatory and profibrotic genes expression. Restoring its level may be a potential treatment in fibrotic diseases16.
Among the identified in the present study DEmiRs, miR-29 (eca-miR-29b, eca-miR-29c) and miR-34 (eca-miR-34a, eca-miR-34b-3p, eca-miR-34b-5p, eca-miR-34c) families were previously described as drivers of various organ fibrosis37. The importance of the role of miR-29 and miR-34 in the development of fibrosis in many organs may be indicated by the attempts to target their action for the treatment of fibrotic diseases38,39. The miR-29 family was identified in this study, and seems to have multiple regulatory functions, controlling cell differentiation40, proliferation, migration41, and apoptosis42. Moreover, miR-29 family has even been called the master regulator of fibrosis due to their high impact on the development of fibrosis16. Our findings indicate the potential role of the members of miR-29 family in the development of fibrosis via regulation of multiple signalling pathways, including MAPK43,44,45, Hippo46,47, phosphatidylinositol, calcium, PI3K-Akt28,29,30,31,48, AMPK49, and Wnt47, as well as focal adhesion50, and ECM-receptor interaction. All these signalling pathways and processes were described as crucial in the progression of fibrosis. Previous studies indicated also a control role of miR-34 family over cell proliferation, migration51, and apoptosis52. The significant role of miR-34 family members in renal53, cardiac54, and hepatic fibrosis55 was also demonstrated.
In categories IIB and III (severe fibrosis), compared to category I endometria, we identified, out of others, the following DEmiRs: eca-miR-151-5p, eca-miR-370, novel-eca-miR-25, and eca-miR-205. Interestingly, our findings indicate the potential involvement of mentioned DEmiRs in the regulation of genes involved in oxytocin (OXT) signalling, such as OXT receptor, prostaglandin-endoperoxide synthase 2, phospholipase C beta 1 and nitric oxide synthase 3. Proper OXT responsiveness and signalling are crucial in the proper regulation of the oestrous cycle, and thus embryo implantation, and pregnancy in mares56. Additionally, previous research showed that miR-151-5p, miR-370, miR-25, miR-20557,58,59,60 are connected with processes accompanying fibrosis. Since, endometrosis is related to the decline of foaling rate1, altered both OXT signalling and pro-fibrotic action of DEmiRs may be important causes of reduced fertility in mares with endometrosis. However, these findings need further studies.
In category III vs. I endometria, we identified two downregulated miRNAs: eca-miR-146b-5p and eca-miR-49. These DEmiRs have already been described as related to fibrosis spread in both in vivo as well as in in vitro studies61,62,63. miR-146b-5p was shown to regulate IL-17-dependent activation of NF‐κB64, while miR-49 was found to regulate important in fibrotic disorders: focal adhesion kinase signalling65. Interestingly, a recent study found the possible role of miR-146b-5p in attenuating dermal fibrosis62. The authors of this study showed that miR-146b-5p may repress Platelet-derived growth factor receptor α-induced profibrotic activities, thus decreasing the development of skin fibrosis. Another study implicated the important role of miR-146b-5p in cardiac remodelling66.
The mechanisms involved in the development of fibrosis are tissue-specific; however, molecular processes between tissues share many similarities67. Finding molecules and biological pathways that are commonly differed in various organs undergoing fibrotic changes are essential to understand molecular background of these changes and design effective therapeutic strategies against fibrosis68. Mare endometrial fibrosis occurs naturally, in contrast to experimental animal models in which fibrosis is chemically induced69. What is more, endometrial biopsies can be taken routinely by veterinarians multiple times from each mare, what does not cause significant discomfort to the animals. Moreover, it is possible to culture in vitro cells and tissues taken from mares with various advancement of fibrosis to study molecular mechanisms. Thus, the use of the horse as a model of organ fibrosis would reduce the sacrifice of animals at the initial stages of research. Altogether, mare endometrosis could serve as a model to study the mechanism of the development of fibrotic diseases in humans.
Increasing number of studies concerning miRNAs as crucial players in the development of fibrosis indicate the need to study their mechanism of action in fibrotic diseases. However, the action of miRNAs is not unambiguous due to the characteristic feature of miRNAs16, as each miRNA can regulate the expression of many genes due to the imperfect nature of miRNA binding to its target mRNA. In turn, the expression of each gene can be regulated by multiple miRNAs. This nature of miRNAs therefore offers a plethora of opportunities for each miRNA to regulate the expression of target genes. This characteristic miRNAs regulatory action creates both many opportunities and causes difficulties both in analysing the action of miRNAs and in targeting their action70. These limitations, however, cannot overcome the plethora of possibilities of using miRNAs both in the search for markers of fibrosis and in the treatment that would target miRNAs action. To the date, despite limitations, miRNAs as biomarkers of various diseases are growing field of research71,72. Nevertheless, the studies of miRNAs as biomarkers of disease severity are still on its early stage due to numerous limitations. The results of these studies must be critically interpreted, and perhaps in the near future, miRNA-based markers and therapies will form a significant part of diagnosis and treatment73.
In conclusion, the current study showed changes in the miRNA expression signature at different stages of endometrosis, suggesting the potential role of miRNAs in the development and progression of mare endometrial fibrosis. In category IIA vs. I endometria, we identified one downregulated miRNA (novel-eca-miR-42), in category IIB vs. I—26 DEmiRs (14 up- and 12 downregulated), while in category III vs I—five DEmiRs (2 up- and 3 downregulated). The functional enrichment analysis revealed that identified DEmiRs may regulate multiple processes, such as focal adhesion, ECM-receptor interaction, Hippo and PI3K-Akt signalling pathways, which can contribute to endometrosis development and progression. Moreover, we demonstrated that the most potent profibrotic cytokine—TGF-β1 decreased the expression of novel-eca-miR-42 in fibroblasts derived from category IIA endometria. Likewise, we showed that along with the development of endometrosis, the miRNA expression becomes disturbed and some of DEmiRs can regulate the expression of genes associated with OXT signalling. These results may suggest impaired response to OXT in endometria with endometrosis, which can be regulated by miRNAs. Further in-depth understanding of the roles of miRNAs in processes associated with endometrosis initiation and progression may help guide the diagnosis and create targeted therapies in the future.
Materials and methods
Ethics approval
All experimental protocols were approved by the Local Ethics Committee for Experiments on Animals in Olsztyn, Poland (Approval No. 60/2014/DTN). All methods were carried out in accordance with relevant guidelines and regulations. All methods are reported in accordance with ARRIVE guidelines for the reporting of animal experiments.
Experimental design
Experiment 1: endometrial miRNA expression profiling at different stages of endometrial fibrosis in the mare
To perform miRNA expression profiling in mare endometrium at each stage of endometrosis severity, miRNA Next Generation Sequencing (NGS) was performed. For this purpose, total RNA was isolated from mare endometria at follicular phase of oestrous cycle (n = 9 per category I, IIA, IIB, and III, n = 36 in total). Then, the NGS analysis followed by bioinformatic analyses were performed.
Experiment 2: the effect of TGF-β1 on selected DEmiRs expression
To determine if expression of selected DEmiRs is regulated by most potent profibrotic cytokine—TGF-β1, in vitro experiment was performed. Thawed fibroblasts (n = 4 per category I, IIA and IIB, n = 12 in total) were seeded on T75 cm2 flasks and cultured in 38.0 °C in 5% CO2. After reaching 90% of confluence, fibroblasts were passaged and seeded on 6-well plates. After reaching 80% confluence, the culture medium was replaced with fresh Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham (DMEM/Ham’s F-12; D2906; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 0.01% of AA solution (A5955; Sigma-Aldrich, St. Louis, MO, USA), ascorbic acid (100 ng/ml; A4544; Sigma-Aldrich, St. Louis, MO, USA) and 0.1% (w/v) BSA (A1470, Sigma-Aldrich, St. Louis, MO, USA), and the cells were incubated at 38.0 °C in 5% CO2. After serum starvation, cells were treated with the vehicle (culture medium alone) or TGF-β1 (10 ng/ml; human recombinant; 100-21; PeproTech, Rocky Hill, NJ, USA) for 24 h. The dose of TGF-β1 was chosen based on previous experiments8. After treatment, the cells were washed with sterile PBS (P4417; Sigma-Aldrich, St. Louis, MO, USA), trypsinized, flash-frozen in liquid nitrogen, and stored in − 80 °C for subsequent RNA extraction and RT-qPCR analysis. The RNA isolation and RT-qPCR were performed as described below. The analysis of expression of eca-miR-42 was done in fibroblasts, cultured with and without TGF-β1, derived from categories I and IIA, while expression of eca-miR-29c, eca-miR-34b-3p, and eca-miR-205 was done in categories I and IIB. Moreover, we compared the expression level of selected DEmiRs in mare endometrial fibroblasts cultured in vitro between categories IIA (novel-eca-miR-42) or IIB (eca-miR-29c, eca-miR-34b-3p, and eca-miR-205) and category I.
Materials
Tissue collection for NGS analysis, fibroblast isolation and histological analysis
Procedures were reviewed and accepted by the Local Ethics Committee for Experiments on Animals in Olsztyn, Poland (Approval No. 60/2014/DTN). The study was conducted between April and June 2016 (Exp.1) and 2021 (Exp. 2). Sixty-four clinically healthy, normally cycling cold-blooded mares (weighing 500 ± 100 kg) at age 2–20 were used in this study. The animals were slaughtered to obtain meat as part of routine breeding as slaughter animals. Endometrial tissue samples were collected from mare endometrium in each category of endometrosis. The endometria were collected 5 min after the slaughter. The follicular phases of the estrous cycle were identified based on P4 analysis of blood plasma and the macroscopic observation of ovaries. The follicular phase was characterized by the absence of an active CL and the presence of follicles of various sizes, but always > 35 mm in diameter, with a concentration of P4 < 1 ng/ml as described previously74. Endometrial tissue was excised, rinsed with cold sterile RNAse-free saline solution, divided, and put in RNAlater (R0901; Sigma-Aldrich, St. Louis, MO, USA) and stored in − 80 °C for further RNA analyses and in 4% formaldehyde for histological examination. After haematoxylin–eosin staining, endometria were retrospectively assigned to Kenney and Doig classification35. This classification takes into account primarily the presence of inflammation and fibrosis, but also endometrial atrophy and dilatation of lymphatic vessels. Endometrium without pathological changes was classified as category I (Supplementary Data 10). Category IIA includes endometrium with mild to moderate inflammation, with few dilated lymphatic vessels. A small degree of fibrosis affects individual branches of the mammary glands, and there are no fibrotic gland sockets in 4 adjacent visual fields. Category IIB includes moderately inflamed endometrium with numerous dilated lymphatic vessels. Fibrosis is more severe than category IIA, with 2–4 fibrotic glandular pockets in 4 adjacent visual fields. Changes affect up to 60% of the uterine glands. Category III includes an endometrium with massive inflammation and greatly dilated lymphatic vessels. The fibrosis is massive, 5 or more fibrotic glands are found in 4 adjacent visual fields. Changes affect over 60% of the uterine glands in the most severe endometrosis stage75.
The isolation and culture of fibroblasts from mare endometrium
The fibroblasts were isolated from mare endometrium (n = 4 per category I, IIA and IIB, n = 12 in total), cultured, and passaged, as previously described with modifications8. The cells were counted using a haemocytometer. The viability of endometrial cells was higher than 95% as assessed by the trypan blue exclusion test.
The dispersed cells were seeded separately at a density of 5 × 105 viable cells/ml and cultured at 38.0°C in a humidified atmosphere of 5% CO2 in the air. To purify the fibroblast population, the medium was changed 18 h after plating, by which time selective attachment of fibroblasts had occurred. The fibroblast homogeneity was confirmed using immunofluorescent staining for vimentin based on the protocol described recently76. The purity of fibroblast after isolation was around 96%. After reaching 90% of confluency, the cells were cryopreserved as described previously77. After haematoxylin–eosin staining endometria were retrospectively assigned as a category I, IIA, IIB, or III according to the Kenney and Doig classification35.
Experimental procedures
RNA extraction
RNAextraction for NGS analysis
Total RNA was extracted using the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, California, USA) according to the manufacturer’s protocol. RNA concentration and quality were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and a 2200 TapeStation instrument (Agilent, Santa Clara, USA); only samples with ratio A260 nm/230 nm between 1.8 and 2.2 and RNA integrity number (RIN) values greater than 8.0 were used. The pooling of samples was performed to reduce the experimental costs and the variability among individual samples and at the same time have more individuals within an experiment.
RNA extraction and RT-qPCR analysis for NGS results validation and in vitro experiment
To validate the NGS results, four DEmiR were selected for qRT-PCR. The most changes novel-eca-miR-42 (Assay ID: 008434), and miRNAs whose roles in different tissues fibrosis were described previously: eca-miR-29c78,79 (Assay ID: 000587), eca-miR-34b-3p51 (Assay ID: 002618), and miR-20559 (Assay ID: 000509) were selected. Due to the prolonged period between the NGS analysis and RT-qPCR analysis, different RNA templates were used for NGS and RT-qPCR analyses. Additionally, RT-qPCR was used to determine the effect of TGF-β1 on the expression of the selected DEmiRs in mare endometrial fibroblasts in vitro.
Total RNA was extracted either from endometrial tissue (Experiment 1, n = 4 per category I, IIA, IIB, and III according to Kenney and Doig35, n = 16 in total) or cultured fibroblasts (Experiment 2, n = 12) using mirVana isolation kit (AM1560, Invitrogen, Carlsbad, CA, USA).
The concentration and quality of total RNA were determined spectrophotometrically. The ratio of absorbance at 260 and 280 nm (A260/280) was approximately 2. Total RNA (10 ng) was reverse transcribed using a TaqMan MicroRNA Reverse Transcription Kit (4,366,597; Invitrogen) and specific RT primers according to the manufacturer’s directions. The cDNA was stored at − 20 °C until qPCR. Further, qPCR was performed in a final volume of 20 µl, using 1.33 µl (1.9 ng) of cDNA, 1 µl of specific primers with probes, 10 µl TaqMan Universal PCR Master Mix II (4,440,049; Applied Biosystems; Thermo Fisher Scientific, Wilmington, NC, USA), and 7.67 µl nuclease-free water, on 96-well plates. All samples were run in duplicates. Amplification was performed with initial denaturation for 10 min at 95 °C, followed by 48 cycles of 15 s at 95 °C and 60 s at 60 °C with ViiA 7 Real-Time PCR System (Agilent Technologies, Waltham, MA, USA). As a negative control, nuclease-free water instead of template cDNA was used. The efficiency of miR-42, miR-29c, miR-34b-3p, and miR-205 were 80%, 80%, 83%, and 83%, respectively.
The qPCR data were analyzed by the method described previously80. It is a method for quantifying qPCR results using calculations based on the kinetics of individual PCR reactions without the need for the standard curve, independent of any assumptions or subjective judgments, which allows direct calculation of efficiency and CT. The relative concentration of mRNA (R0) for each target and reference miRNAs (selected based on our previous study: RNU6B and miR-19517) was calculated using the equation R0 = 1/(1 + E)Ct, where, E is the average gene efficiency and Ct is the cycle number at the threshold. The relative gene expression was calculated as R0target gene/R0reference gene and was expressed in arbitrary units. NormFinder81 was used to confirm the selection of the most stable reference genes for the normalization of the results.
RT-qPCR data analysis
All data were tested using Gaussian distribution using the Shapiro–Wilk test (GraphPad Software version 9; GraphPad, San Diego, CA). Whenever the assumptions of normal distribution were not met, nonparametric statistical analyses were done. In experiment 1 the significant differences were determined by the parametric T-test, while in experiment 2, using Mann–Whitney U test. The results were considered significantly different when P < 0.05. GraphPad Prism 9.0 was used both to perform statistical analysis and for generating bar graphs.
Small RNA library preparation and NGS sequencing
MicroRNA libraries were prepared with the use of the NEB Next Multiplex Small RNA library Prep Set for Illumina (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. Specifically, following 3′ adapter ligation, hybridization of the Reverse Transcription Primer and 5′ adapter ligation, Reverse Transcription and PCR amplification of obtained products were performed. The PCR was prepared with the use of 12 different indexed primers containing a 6 nt long unique sequence, which allows for barcoding the single library and multiplexing of the samples during sequencing. The next step included size selection (Novex 6% TBE PAGE gel; Invitrogen, Carlsbad, CA, USA) of libraries. Subsequently, the quantity of obtained libraries were measured with a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), while a 2200 TapeStation instrument (Agilent Technologies, Santa Clara, CA, USA) was used to assess their size. Afterward, the obtained libraries were sequenced on a HiScanSQ sequencing instrument (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol.
NGS data analysis
Identification of miRNAs
The quality of raw reads was evaluated by means of FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low quality reads and adapter sequences were removed by cutadapt software (2.8)82 excluding reads: without a 3’-adapter, containing non-determined nucleotides (nt) “N”, shorter than 18 nt or larger than 30 nt. Next, clean reads were mapped to the horse non-coding RNA (ncRNAs; EquCab3.0, Ensembl release 106) with the use of bowtie2 software (2.4.2)83. Read mapped to the horse ncRNA were removed from further analysis. The remaining clean reads were then mapped to the mature miRNAs sequences from mirBbase database (22.1)84. The miRDeep2 software (0.1.3)85 was used to predict new miRNAs from unidentified miRNA reads. After described analysis, due to low number of reads and identified miRNAs, comparing to the remaining samples (median of 3,368,155 reads), one sample from category III (with pooled samples from mares 28, 29 and 30; no of reads: 18 800) was excluded from further analysis. The differentially expressed miRNAs (DEmiRs) and corresponding P-adjusted values were determined by means of R statistical software (4.1.3) using DESeq2 package (1.34.0)86. The threshold for the significantly different expression was set at P-adjusted ≤ 0.05 and log2 fold change (log2FC) ≥ 1.0 or log2FC ≤ − 1.087,88. The visual presentation of the results was performed by R software using the ggplot2 package (version 3.3.6).
Prediction for targets of miRNAs
The prediction of target genes of the DEmiRs was performed by means of Tools4mirs server89 using miRanda90, PITA91, rna2292 and RNAhybrid93 tools. The 5′ UTR, CDS and 3′ UTR sequences of horse protein coding genes were used as potential targets. Only miRNA-mRNA pairs predicted in at least three out of four tools were used. Additionally, miRNA-mRNA pairs with the interaction binding free energy above − 8.0 kcal mol−1, predicted by PITA and miRanda, were excluded from further analysis.
Functional enrichment analysis
Functional analysis of the identified targets genes for differentially expressed miRNAs was performed based on the Gene Ontology (GO) database using clusterProfiler94, DOSE95, biomaRt96 and AnnotationHub97 packages of R software, with the established criterion P-adjusted ≤ 0.05. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to ascribe identified miRNA targets to particular biological mechanisms and cellular pathways (the established criteria: P-adjusted ≤ 0.05). The KEGG enrichment analysis was performed by clusterProfiler, DOSE and AnnotationHub packages of R software. The visual presentation of the results was performed by R software using ggplot2.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request. The miRNA profiling results were submitted to the NCBI BioProject under the Accession Number PRJNA880660.
References
Schoniger, S. & Schoon, H. A. The healthy and diseased equine endometrium: A review of morphological features and molecular analyses. Animals 10, 40625. https://doi.org/10.3390/ani10040625 (2020).
Bracher, V., Mathias, S. & Allen, W. R. Influence of chronic degenerative endometritis (endometrosis) on placental development in the mare. Equine Vet. J. 28, 180–188. https://doi.org/10.1111/j.2042-3306.1996.tb03771.x (1996).
Kenney, R. M. Cyclic and pathologic changes of the mare endometrium as detected by biopsy, with a note on early embryonic death. J. Am. Vet. Med. Assoc. 172, 241–262 (1978).
Hoffmann, C. et al. The equine endometrosis: New insights into the pathogenesis. Anim. Reprod. Sci. 111, 261–278. https://doi.org/10.1016/j.anireprosci.2008.03.019 (2009).
Lamceva, J., Uljanovs, R. & Strumfa, I. The main theories on the pathogenesis of endometriosis. Int. J. Mol. Sci. 24, 254. https://doi.org/10.3390/ijms24054254 (2023).
Liu, I. K. & Troedsson, M. H. The diagnosis and treatment of endometritis in the mare: Yesterday and today. Theriogenology 70, 415–420. https://doi.org/10.1016/j.theriogenology.2008.05.040 (2008).
Ricketts, S. W. & Alonso, S. The effect of age and parity on the development of equine chronic endometrial disease. Equine Vet. J. 23, 189–192. https://doi.org/10.1111/j.2042-3306.1991.tb02752.x (1991).
Szostek-Mioduchowska, A. Z., Lukasik, K., Skarzynski, D. J. & Okuda, K. Effect of transforming growth factor-beta1 on alpha-smooth muscle actin and collagen expression in equine endometrial fibroblasts. Theriogenology 124, 9–17. https://doi.org/10.1016/j.theriogenology.2018.10.005 (2019).
Szostek-Mioduchowska, A. Z., Baclawska, A., Rebordao, M. R., Ferreira-Dias, G. & Skarzynski, D. J. Prostaglandins effect on matrix metallopeptidases and collagen in mare endometrial fibroblasts. Theriogenology 153, 74–84. https://doi.org/10.1016/j.theriogenology.2020.04.040 (2020).
Szostek-Mioduchowska, A. Z., Baclawska, A., Okuda, K. & Skarzynski, D. J. Effect of proinflammatory cytokines on endometrial collagen and metallopeptidase expression during the course of equine endometrosis. Cytokine 123, 154767. https://doi.org/10.1016/j.cyto.2019.154767 (2019).
Szostek-Mioduchowska, A., Slowinska, M., Pacewicz, J., Skarzynski, D. J. & Okuda, K. Matrix metallopeptidase expression and modulation by transforming growth factor-beta1 in equine endometrosis. Sci. Rep. 10, 1119. https://doi.org/10.1038/s41598-020-58109-0 (2020).
Rebordao, M. R. et al. Neutrophil extracellular traps formation by bacteria causing endometritis in the mare. J. Reprod. Immunol. 106, 41–49. https://doi.org/10.1016/j.jri.2014.08.003 (2014).
Alpoim-Moreira, J. et al. Metallopeptidades 2 and 9 genes epigenetically modulate equine endometrial fibrosis. Front. Vet. Sci. 9, 970003. https://doi.org/10.3389/fvets.2022.970003 (2022).
Szóstek-Mioduchowska, A. et al. 5th International Conference on Uterine Disorders in Farm Animals: Endometritis as a Cause of Infertility in Domestic Animals 22.
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338. https://doi.org/10.1038/nrneph.2016.48 (2016).
O’Reilly, S. MicroRNAs in fibrosis: Opportunities and challenges. Arthritis Res. Ther. 18, 11. https://doi.org/10.1186/s13075-016-0929-x (2016).
Ibrahim, S., Szostek-Mioduchowska, A. & Skarzynski, D. Expression profiling of selected miRNAs in equine endometrium in response to LPS challenge in vitro: A new understanding of the inflammatory immune response. Vet. Immunol. Immunopathol. 209, 37–44. https://doi.org/10.1016/j.vetimm.2019.02.006 (2019).
Adwent, I. et al. The influence of adalimumab and cyclosporine A on the expression profile of the genes related to TGFbeta signaling pathways in keratinocyte cells treated with lipopolysaccharide A. Mediat. Inflamm. 2020, 3821279. https://doi.org/10.1155/2020/3821279 (2020).
O’Brien, J., Hayder, H., Zayed, Y. & Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 9, 402. https://doi.org/10.3389/fendo.2018.00402 (2018).
Ma, Y. et al. MiR-130b increases fibrosis of HMC cells by regulating the TGF-beta1 pathway in diabetic nephropathy. J. Cell Biochem. 120, 4044–4056. https://doi.org/10.1002/jcb.27688 (2019).
Ye, M., Wang, S., Sun, P. & Qie, J. Integrated microRNA expression profile reveals dysregulated miR-20a-5p and miR-200a-3p in liver fibrosis. Biomed. Res. Int. 2021, 9583932. https://doi.org/10.1155/2021/9583932 (2021).
Zhang, W. et al. MicroRNA-26a protects the heart against hypertension-induced myocardial fibrosis. J. Am. Heart Assoc. 9, e017970. https://doi.org/10.1161/JAHA.120.017970 (2020).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Hinz, B. Masters and servants of the force: The role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 85, 175–181. https://doi.org/10.1016/j.ejcb.2005.09.004 (2006).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801. https://doi.org/10.1038/nrm3904 (2014).
Yang, L. et al. Periostin facilitates skin sclerosis via PI3K/Akt dependent mechanism in a mouse model of scleroderma. PLoS ONE 7, e41994. https://doi.org/10.1371/journal.pone.0041994 (2012).
Wei, L. et al. Asiatic acid attenuates CCl4-induced liver fibrosis in rats by regulating the PI3K/AKT/mTOR and Bcl-2/Bax signaling pathways. Int. Immunopharmacol. 60, 1–8. https://doi.org/10.1016/j.intimp.2018.04.016 (2018).
Qin, W., Cao, L. & Massey, I. Y. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell Biochem. 476, 4045–4059. https://doi.org/10.1007/s11010-021-04219-w (2021).
Wang, J. et al. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm. Sin. B 12, 18–32. https://doi.org/10.1016/j.apsb.2021.07.023 (2022).
Chang, R., Zheng, W., Sun, Y. & Xu, T. microRNA-1388-5p inhibits NF-kappaB signaling pathway in miiuy croaker through targeting IRAK1. Dev. Comp. Immunol. 119, 104025. https://doi.org/10.1016/j.dci.2021.104025 (2021).
Luedde, T. & Schwabe, R. F. NF-kappaB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 8, 108–118. https://doi.org/10.1038/nrgastro.2010.213 (2011).
Domino, M. et al. Expression of genes involved in the NF-kappaB-dependent pathway of the fibrosis in the mare endometrium. Theriogenology 147, 18–24. https://doi.org/10.1016/j.theriogenology.2020.01.055 (2020).
Kenney, R. M. & Doig, P. A. Equine endometrial biopsy. Curr. Therapy Theriogenol. 723–729, 723–729 (1986).
Qin, Y. et al. Salvia miltiorrhiza-derived Sal-miR-58 induces autophagy and attenuates inflammation in vascular smooth muscle cells. Mol. Ther. Nucleic Acids 21, 492–511. https://doi.org/10.1016/j.omtn.2020.06.015 (2020).
Jiang, X. P., Ai, W. B., Wan, L. Y., Zhang, Y. Q. & Wu, J. F. The roles of microRNA families in hepatic fibrosis. Cell Biosci. 7, 34. https://doi.org/10.1186/s13578-017-0161-7 (2017).
Zhao, M. et al. MicroRNA-34a: A novel therapeutic target in fibrosis. Front. Physiol. 13, 895242. https://doi.org/10.3389/fphys.2022.895242 (2022).
He, Y., Huang, C., Lin, X. & Li, J. MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie 95, 1355–1359. https://doi.org/10.1016/j.biochi.2013.03.010 (2013).
Roderburg, C. et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218. https://doi.org/10.1002/hep.23922 (2011).
Huang, D. et al. Inhibiting effect of miR-29 on proliferation and migration of uterine leiomyoma via the STAT3 signaling pathway. Aging (Albany NY) 14, 1307–1320. https://doi.org/10.18632/aging.203873 (2022).
Mott, J. L., Kobayashi, S., Bronk, S. F. & Gores, G. J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 26, 6133–6140. https://doi.org/10.1038/sj.onc.1210436 (2007).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83. https://doi.org/10.1128/MMBR.00031-10 (2011).
Ihn, H. Pathogenesis of fibrosis: Role of TGF-beta and CTGF. Curr. Opin. Rheumatol. 14, 681–685. https://doi.org/10.1097/00002281-200211000-00009 (2002).
Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. https://doi.org/10.1038/nature02006 (2003).
Mia, M. M. & Singh, M. K. New insights into Hippo/YAP signaling in fibrotic diseases. Cells 11, 2065. https://doi.org/10.3390/cells11132065 (2022).
Jorgenson, A. J. et al. TAZ activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ECM gene expression. Am. J. Physiol. Cell Physiol. 312, C277–C285. https://doi.org/10.1152/ajpcell.00205.2016 (2017).
Engelman, J. A., Luo, J. & Cantley, L. C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619. https://doi.org/10.1038/nrg1879 (2006).
Li, M. et al. Icaritin inhibits skin fibrosis through regulating AMPK and Wnt/beta-catenin Signaling. Cell Biochem. Biophys. 79, 231–238. https://doi.org/10.1007/s12013-020-00952-z (2021).
Goffin, J. M. et al. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol. 172, 259–268. https://doi.org/10.1083/jcb.200506179 (2006).
Cordova-Rivas, S. et al. 5p and 3p strands of miR-34 family members have differential effects in cell proliferation, migration, and invasion in cervical cancer cells. Int. J. Mol. Sci. 20, 545. https://doi.org/10.3390/ijms20030545 (2019).
Tarasov, V. et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 6, 1586–1593. https://doi.org/10.4161/cc.6.13.4436 (2007).
Zhang, L. & Yang, F. Tanshinone IIA improves diabetes-induced renal fibrosis by regulating the miR-34-5p/Notch1 axis. Food Sci. Nutr. 10, 4019–4040. https://doi.org/10.1002/fsn3.2998 (2022).
Huang, Y., Qi, Y., Du, J. Q. & Zhang, D. F. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 18, 1355–1365. https://doi.org/10.1517/14728222.2014.961424 (2014).
Li, W. Q. et al. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J. 278, 1522–1532. https://doi.org/10.1111/j.1742-4658.2011.08075.x (2011).
Sharp, D. C., Thatcher, M. J., Salute, M. E. & Fuchs, A. R. Relationship between endometrial oxytocin receptors and oxytocin-induced prostaglandin F2 alpha release during the oestrous cycle and early pregnancy in pony mares. J. Reprod. Fertil. 109, 137–144. https://doi.org/10.1530/jrf.0.1090137 (1997).
Chen, C. et al. Mesenchymal stem cell transplantation in tight-skin mice identifies miR-151-5p as a therapeutic target for systemic sclerosis. Cell Res. 27, 559–577. https://doi.org/10.1038/cr.2017.11 (2017).
Jeong, D. et al. miR-25 tough decoy enhances cardiac function in heart failure. Mol. Ther. 26, 718–729. https://doi.org/10.1016/j.ymthe.2017.11.014 (2018).
Sun, B. et al. miR-205 suppresses pulmonary fibrosis by targeting GATA3 through inhibition of endoplasmic reticulum stress. Curr. Pharm. Biotechnol. 21, 720–726. https://doi.org/10.2174/1389201021666191210115614 (2020).
Yuan, H. & Gao, J. The role of miR370 in fibrosis after myocardial infarction. Mol. Med. Rep. 15, 3041–3047. https://doi.org/10.3892/mmr.2017.6397 (2017).
Mullenbrock, S. et al. Systems analysis of transcriptomic and proteomic profiles identifies novel regulation of fibrotic programs by miRNAs in pulmonary fibrosis fibroblasts. Genes 9, 120588. https://doi.org/10.3390/genes9120588 (2018).
Fujisawa, C. et al. The role for miR-146b-5p in the attenuation of dermal fibrosis and angiogenesis by targeting PDGFRalpha in skin wounds. J. Investig. Dermatol. 142, 1990–2002. https://doi.org/10.1016/j.jid.2021.11.037 (2022).
Gong, L. et al. S1PR3 deficiency alleviates radiation-induced pulmonary fibrosis through the regulation of epithelial–mesenchymal transition by targeting miR-495-3p. J. Cell Physiol. 235, 2310–2324. https://doi.org/10.1002/jcp.29138 (2020).
Tu, Z. et al. Loss of miR-146b-5p promotes T cell acute lymphoblastic leukemia migration and invasion via the IL-17A pathway. J. Cell Biochem. 120, 5936–5948. https://doi.org/10.1002/jcb.27882 (2019).
Guo, B., Hui, Q., Xu, Z., Chang, P. & Tao, K. miR-495 inhibits the growth of fibroblasts in hypertrophic scars. Aging (Albany NY) 11, 2898–2910. https://doi.org/10.18632/aging.101965 (2019).
Liao, Y. et al. Therapeutic silencing miR-146b-5p improves cardiac remodeling in a porcine model of myocardial infarction by modulating the wound reparative phenotype. Protein Cell 12, 194–212. https://doi.org/10.1007/s13238-020-00750-6 (2021).
Friedman, S. L., Sheppard, D., Duffield, J. S. & Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 5, 167. https://doi.org/10.1126/scitranslmed.3004700 (2013).
Mehal, W. Z., Iredale, J. & Friedman, S. L. Scraping fibrosis: Expressway to the core of fibrosis. Nat. Med. 17, 552–553. https://doi.org/10.1038/nm0511-552 (2011).
Yanguas, S. C. et al. Experimental models of liver fibrosis. Arch. Toxicol. 90, 1025–1048. https://doi.org/10.1007/s00204-015-1543-4 (2016).
Witwer, K. W. Circulating microRNA biomarker studies: Pitfalls and potential solutions. Clin. Chem. 61, 56–63. https://doi.org/10.1373/clinchem.2014.221341 (2015).
Christopher, A. F. et al. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 7, 68–74. https://doi.org/10.4103/2229-3485.179431 (2016).
Condrat, C. E. et al. miRNAs as biomarkers in disease: Latest findings regarding their role in diagnosis and prognosis. Cells 9, 20276. https://doi.org/10.3390/cells9020276 (2020).
Hanna, J., Hossain, G. S. & Kocerha, J. The potential for microRNA therapeutics and clinical research. Front. Genet. 10, 478. https://doi.org/10.3389/fgene.2019.00478 (2019).
Szostek, A. Z., Lukasik, K., Galvao, A. M., Ferreira-Dias, G. M. & Skarzynski, D. J. Impairment of the interleukin system in equine endometrium during the course of endometrosis. Biol. Reprod. 89, 79. https://doi.org/10.1095/biolreprod.113.109447 (2013).
Snider, T. A., Sepoy, C. & Holyoak, G. R. Equine endometrial biopsy reviewed: Observation, interpretation, and application of histopathologic data. Theriogenology 75, 1567–1581. https://doi.org/10.1016/j.theriogenology.2010.12.013 (2011).
Szostek-Mioduchowska, A. Z. et al. Effects of cortisol on prostaglandin F2alpha secretion and expression of genes involved in the arachidonic acid metabolic pathway in equine endometrium—In vitro study. Theriogenology 173, 221–229. https://doi.org/10.1016/j.theriogenology.2021.08.009 (2021).
Szostek, A. Z. et al. Effects of cell storage and passage on basal and oxytocin-regulated prostaglandin secretion by equine endometrial epithelial and stromal cells. Theriogenology 77, 1698–1708. https://doi.org/10.1016/j.theriogenology.2011.12.015 (2012).
Huang, H. et al. The microRNA MiR-29c alleviates renal fibrosis via TPM1-mediated suppression of the Wnt/beta-catenin pathway. Front. Physiol. 11, 331. https://doi.org/10.3389/fphys.2020.00331 (2020).
Wang, X. et al. Upregulated miR-29c suppresses silica-induced lung fibrosis through the Wnt/beta-catenin pathway in mice. Hum. Exp. Toxicol. 37, 944–952. https://doi.org/10.1177/0960327117741750 (2018).
Zhao, S. & Fernald, R. D. Comprehensive algorithm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 12, 1047–1064. https://doi.org/10.1089/cmb.2005.12.1047 (2005).
Andersen, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. https://doi.org/10.1158/0008-5472.CAN-04-0496 (2004).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 3. https://doi.org/10.14806/ej.17.1.200 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. https://doi.org/10.1093/nar/gky1141 (2019).
Friedlander, M. R., Mackowiak, S. D., Li, N., Chen, W. & Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. https://doi.org/10.1093/nar/gkr688 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Du, K. et al. Integrated analysis of microRNAs, circular RNAs, long non-coding RNAs, and mRNAs revealed competing endogenous RNA networks involved in brown adipose tissue whitening in rabbits. BMC Genom. 23, 779. https://doi.org/10.1186/s12864-022-09025-2 (2022).
He, J. et al. Integrated analysis of miRNAs and mRNA profiling reveals the potential roles of miRNAs in sheep hair follicle development. BMC Genom. 23, 722. https://doi.org/10.1186/s12864-022-08954-2 (2022).
Lukasik, A., Wojcikowski, M. & Zielenkiewicz, P. Tools4miRs—One place to gather all the tools for miRNA analysis. Bioinformatics 32, 2722–2724. https://doi.org/10.1093/bioinformatics/btw189 (2016).
John, B. et al. Human microRNA targets. PLoS Biol. 2, e363. https://doi.org/10.1371/journal.pbio.0020363 (2004).
Kertesz, M., Iovino, N., Unnerstall, U., Gaul, U. & Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 39, 1278–1284. https://doi.org/10.1038/ng2135 (2007).
Miranda, K. C. et al. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell 126, 1203–1217. https://doi.org/10.1016/j.cell.2006.07.031 (2006).
Kruger, J. & Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454. https://doi.org/10.1093/nar/gkl243 (2006).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Yu, G., Wang, L. G., Yan, G. R. & He, Q. Y. DOSE: An R/bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31, 608–609. https://doi.org/10.1093/bioinformatics/btu684 (2015).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191. https://doi.org/10.1038/nprot.2009.97 (2009).
Munz, M. et al. Qtlizer: Comprehensive QTL annotation of GWAS results. Sci. Rep. 10, 20417. https://doi.org/10.1038/s41598-020-75770-7 (2020).
Acknowledgements
The authors would like to thank Agnieszka Bacławska and Witold Krzywiec from the Institute of Animal Reproduction and Food Research, PAS, Olsztyn for their technical support.
Funding
This study was supported by National Science Centre, Poland (Miniatura, 2017/01/X/NZ5/01528) and UID/CVT/00276/2020; LA/0059/2020/AL4AnimalS; 2022.09161. PTDC funded by Fundação para a Ciência e Tecnologia (FCT; Portugal). The cost of Open Access publication was covered by the Society for Biology of Reproduction in Poland.
Author information
Authors and Affiliations
Contributions
A.W.—writing-original draft, methodology, formal analysis, visualization, investigation, statistical analysis; T.M.—bioinformatics analysis of NGS results, visualization; K.L., E.Ż.—investigation; K.P.-T., A.G., T.Sz., M.B.-P.—formal analysis;. G.F.-D., D.S.—editing original draft, A.Sz.-M.—conception and design, funding acquisition, methodology, investigation, formal analysis, editing original draft. All authors edited, revised and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wójtowicz, A., Molcan, T., Lukasik, K. et al. The potential role of miRNAs and regulation of their expression in the development of mare endometrial fibrosis. Sci Rep 13, 15938 (2023). https://doi.org/10.1038/s41598-023-42149-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42149-3
This article is cited by
-
Bioinformatic analysis of endometrial miRNA expression profile at day 26–28 of pregnancy in the mare
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}