
Abstract
Anaerobic digestion is widely used to process and recover value from food waste. Commercial food waste anaerobic digestion facilities seek improvements in process efficiency to enable higher throughput. There is limited information on the composition of microbial communities in food waste prior to digestion, limiting rational exploitation of the catalytic potential of microorganisms in pretreatment processes. To address this knowledge gap, bacterial and fungal communities in food waste samples from a commercial anaerobic digestion facility were characterised over 3 months. The abundance of 16S rRNA bacterial genes was approximately five orders of magnitude higher than the abundance of the fungal intergenic spacer (ITS) sequence, suggesting the numerical dominance of bacteria over fungi in food waste before anaerobic digestion. Evidence for the mass proliferation of bacteria in food waste during storage prior to anaerobic digestion is presented. The composition of the bacterial community shows variation over time, but lineages within the Lactobacillaceae family are consistently dominant. Nitrogen content and pH are correlated to community variation. These findings form a foundation for understanding the microbial ecology of food waste and provide opportunities to further improve the throughput of anaerobic digestion.
Similar content being viewed by others


In 2017, 2 billion tonnes of municipal solid waste was generated globally. In which 84% were collected, and only 15% was recycled1. Approximately 60% of this waste stream is organic2 and can be anaerobically digested for energy recovery. In the 2018 Australian Government Waste Generation Report, 87% of food waste was landfilled, creating landfill gas and leachate problems. Only 1% of food waste went to energy recovery facilities3. The mismanagement of the organic fraction of municipal waste can cause generation of greenhouse gases, landfill leachate, and other harmful products from the uncontrolled decomposition of organic waste4, 5. Landfill gas and leachate are harmful to the environment and raise safety concerns6, 7. Engineered anaerobic digestion (AD) of organic waste can relieve pressure from landfills by harvesting biogas and nutrients from organic waste. This study focuses on the food waste fraction of the organic waste stream.
Anaerobic digestion relies on microorganisms that decompose organic substances in the absence of oxygen8. The digestion process involves four stages (hydrolysis, acidogenesis, acetogenesis and methanogenesis) each carried out by different groups of microorganisms. Complex food matrices are hydrolysed extracellularly into simpler compounds, then acidified and acetified, which then ultimately fermented to acetate, carbon dioxide and dihydrogen by bacteria9. These products then serve as substrates for the production of methane by methanogenic archaea10. Commercial anaerobic digestion systems have been optimised over decades, focusing on higher biogas yields, higher methane:carbon dioxide ratios, and lower residual solids yields11. Rarely has attention been paid to increased digestor throughput, despite the economic trade-off an increased loading capacity can offer12. This is surprising given that most of the revenue for AD facilities comes from payments for food waste disposal. Therefore, increasing the loading rate of the digestor improves the financial viability of such facilities and diverts more organic waste from landfills.
Food has an associated microbial community and is highly susceptible to abiotic decomposition and biodegradation. Perish starts as soon as food is harvested, processed, or produced. Anaerobic digestion facilities receive food waste at an early stage of decay from an increasingly active indigenous microbial community13. Despite the potential of the microbial community indigenous to food waste to play a role in downstream anaerobic digestion, there is limited data available on the microbial community of food waste feedstock (food waste prior to anaerobic digestion) for AD facilities. For example, the diversity and evenness of bacterial and fungal communities in food waste and how the composition of the microbial community is impacted by environmental parameters such as pH, water content, and element content are unknown. Given that the composition of food waste can vary, it is reasonable to expect that the composition of the microbial community also varies, although this has never been investigated.
This study investigated food waste samples received and pulped by a commercial Australian food waste anaerobic digestion facility. Samples from two stages of basic pretreatment were analysed for microbial community variations and physico-chemical characteristics including pH and element content. Sequencing and quantification of 16S rRNA and ITS showed the structure of and shifts in food waste microbial communites prior to anaerobic digestion. Changes in microbial community composition over the 3 month sampling period were linked to physico-chemical parameters, specifically pH and nitrogen content.
Materials and methods
Feedstock for the food waste digestor
EarthPower, Sydney, NSW, Australia, provided food waste feedstock. The facility receives solid and liquid food waste from food production facilities, supermarkets, and restaurants. Food waste is sorted and pulped upon receipt. Liquid food waste is mainly oil trap waste and spadable sludge with inconsistent receival rate. The liquid stream is mixed with the pulped solid waste before anaerobic digestion. Two types of feedstock were sampled, hydropulper shredder and digestor feed. Hydropulper samples are pulped fresh food waste with water added in a 3:1 ratio (waste/water), excluding plastic packaging and other undesirable materials. Digestor feeding tank samples include both food waste pulp and liquid food waste. Figure 1a shows a basic process flow diagram of the facility. Samples were collected on 38 dates between 22 June 2020 and 17 September 2020, then stored at − 20 °C individually before processing. EarthPower provided on site water content and pH data for the samples. Non-consistant sampling frequency was decided by EarthPower at their operational convenience. There are approximately 16–20 h of detention time between food waste pulp and digestor feeding tank. Food waste delivered to the facility is processed in the hydropulper shredder on a daily basis. Processed food waste is then stored for 2–3 days in the digester feed tank. Both facilities are operated at ambient temperature (8.7–24 °C, average 12.9 °C for the sampling period).

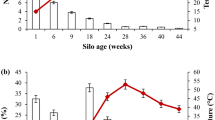
(a) Basic process flow chart of the food waste digestion facility. The sampling points are shaded in grey at the hydropulper and digestor feed tank. Liquid food waste includes mainly grease traps and spadable liquid waste. The abundance of (b) bacteria and (c) fungi per gram of sample (wet weight) taken from the hydropulper (triangles) and digestor feed (squares) was determined by quantitative PCR targeting 16S rRNA gene copies for bacteria and ITS copies for fungi. Gene copy counts represent cell concentration measurements, thus are relative to the wet weight of the sample, and are the average number of technical triplicates.
DNA extraction and PCR
Genomic DNA was extracted from 0.2 g of food waste samples (wet weight) using the QIAamp PowerFecal Pro DNA extraction kit (Qiagen). Manufacturer protocols were followed, except DNA-free PCR grade water (Sigma) was used instead of the C6 solution for final preservation. Genomic DNA concentration was measured using a Qubit 2.0 fluorometer. Polymerase chain reactions (PCR) were performed on the extracted genomic DNA. The primers used were 1048F (5′-GTGSTGCAYGGYTGTCGTCA-3′) and 1294R (5′-GCCTACGATCTGAACTGAGC 3′) for bacteria, and ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) & ITS4 (5′-TCCTCCGC TTATTGATA TGC-3′) for fungi. The thermal cycle setup of bacteria PCR is initial denaturation at 95 °C for 5 min, then followed by 30 cycles of 30 s of denaturation at 94 °C, 30 s of annealing at 58 °C, and 40 s of extension at 72 °C and the final extension period is 10 min at 72 °C. The fungus PCR setting begins with an initial denaturation at 94 °C for 5 min, then followed by 30 cycles of 45 s of denaturation at 94 °C, 30 s of annealing at 60 °C, and 45 s of extension at 72 °C and the final extension period is 10 min at 72 °C. Gel electrophoresis was used to identify the integrity of the PCR product with 1% agarose gel run at 90 V for 30 min.
Quantitative PCR (qPCR) was used to quantify bacteria and fungi in food waste samples with a Bio-Rad CFX qPCR machine. The primers used for qPCR are 1048F (5′-GTGSTGCA YGGYTGTCGTCA-3′) and 1194R (5′-ACGTCATCCCCACCTTCC-3′) for the total copy count of the 16S rRNA gene representing the bacteria community14 and ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and 4 (5′-TCCTCCGCTTATTGATATGC-3′) for the total copy count representing the fungi community. The working solution mix contained 5 µL of SsoFast EvaGreen Supermix (BIO-RAD), 0.1 µL of each primer, 0.1 µL of BSA (20 mg/mL) and 2.7 µL of molecular water. When plating, 96 well plates with 8 µL of working solution and 2 µL of sample were used in each well. The total bacterial qPCR protocol was 3 min of denaturation at 98 °C, forty cycles of DNA segment replication at 90 °C for 20 s, and 62 °C for 50 s. The temperature decreased to 60 °C and was maintained for 10 s for fluorescent reads, following a temperature increase of 60 to 90 °C in 0.5 °C intervals for melt curve analysis. Fungi quantification qPCR protocol was 2 min denaturation at 95 °C, forty cycles of DNA segment replication at 90 °C for 20 s, 55 °C for 30 s, and 72 °C for 60 s. The temperature stays at 72 °C for 10 min15, 16. The average efficiency of the qPCR of the 16S rRNA gene and ITS gene copies was 95% and 96%, with an average slope of (− 3.42) and (− 3.46) and an average R2 equal to 0.993 and 0.995, respectively.
DNA sequencing
Illumina sequencing was used to identify species of bacteria and fungi. The sequencing platform was MiSeq v2 2 × 250 bp. The Ramaciotti Center at UNSW provided the sequencing service, including library preparation and sequencing runs. The bacteria assays were prepared with the 16S rRNA gene amplicon library and the fungi assays with the ITS2 amplicon library (UNSW, Sydney, Australia). Paired-end sequencing results were analysed using QIIME 2 2019.7 pipeline17. The sampling depth was set to 1600 for quality control. The Dada2 plugin18 was used to denoise the data with trimming length determined using FastQC (https://www.bioinformatics.babraham.Ac.uk/projects/fastqc) for quality assurance. Taxonomy was assigned using the q2-feature classifier19 classify-sklearn naïve Bayes taxonomy classifier on Silva 138 release20 for bacteria and UNITE 8.021 for fungi taxonomy classification. The reference sequence was modified to increase accuracy to exclude primer overhang. The reference sequence was not cut with the advice of the QIIME2 team22. The genomic sequence entries were then BLASTed (https://blast.ncbi.nlm.nih.gov) to acquire species information. Sequence reads extracted from the Illumina results were aligned using MUSCLE. A maximum likelihood phylogenetic tree was generated using MEGA-723.
Elemental analysis for C, N, and S content
The carbon, nitrogen, and sulphur content of freeze-dried food waste (every other sample) were analysed with a varioMARCO cube at the Solid State & Elemental Analysis Unit XRF at the UNSW Mark Wainwright Analytical Centre (UNSW Sydney, Australia). The manufacturer’s suggested operational protocol was followed24.
Data analysis
Within-sample diversity is represented by Margalef richness25 and Simpson evenness26 categorized based on sample type, pH, and N%. The data were calculated with Qiime 2 pipeline. The diversity of bacteria and fungi samples were presented in weighted Unifrac PcoA graphs27 ategorized by sample type, pH, and nitrogen element content. The bacteriological taxonomy data were filtered to exclude chloroplast sequences. The relative abundance of OTUs is considered the reference abundance of microorganisms in the samples. Due to countless substrains that are low in abundance (< 1%) or only present in a few samples, the top four bacteria species (making up > 40% of community) were kept for further correlation calculations. Similarly, the analysis of the community of fungi included the four most abundant species. GraphPad Prism 9 was used to analyse the correlation between environmental parameters and variances in community structure. Correlations are examined using nonparametric Spearman correlations to cover more than linear relations and to generate heat maps. The P values were calculated with the student t-test in Prism 9.
Bacterial growth modelling
Bacterial growth in the digestor feed tank was modelled using the Monod equation and key reaction kinetic parameters (Figure S1). The equations reflect the specific growth rate of bacteria (\(\mu\)max), which depends on the temperature, pH, and nutrient level of the medium. The chosen (\(\mu\)max) used in the model was conservative since the aim of the model was to determine if there was enough residence time (steady state) for bacterial growth to occur between the two sampling points. Other kinetic parameters used include the biomass yield (Yx/s) and the saturation constant (Ks). Growth kinetics were based on Lactobacillus, which represents more than 70% of the bacterial community.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Results
The abundance of bacteria and fungi in food waste feedstock
To investigate the variation in the microbial communities of food waste, samples were taken from the hydropulper shredder and digestor feeding tank (Fig. 1a) approximately weekly for 3 months from winter to spring. DNA was extracted and the total abundance of 16S rRNA (bacteria) and ITS gene sequences (fungi) was determined using quantitative PCR (Fig. 1b,c). Archaeal relative abundance was below 1%. The 16S rRNA gene copy abundance was 4–6 orders of magnitude higher than the abundance of ITS sequences, indicating that bacteria are numerically dominant. The abundance of fungi was similar in the hydropulper and digestor feed samples (average 4.3 ± 2.0 × 104 and 4.4 ± 2.0 × 104 copies/g, respectively). Bacterial abundance in the digestor feed (average 1.6 ± 2.4 × 1010 copies/g) was 26-fold higher than in the hydropulper (average 6.2 ± 6.4 × 108 copies/g) indicating proliferation.
To estimate the growth rate and threshold of bacteria in food waste, a bacterial growth model was developed based on growth rates of Lactobacillus (> 70% relative abundance of bacteria community, Fig. 3a,b) under comparable conditions and estimates of available growth substrates (Supplement information 1). The model was used to determine whether the residence time between the hydropulper and the digestor feed tank (~ 16 h) was sufficient to account for the observed 26-fold increase in bacterial abundance. From a starting point of 6.2 × 108 copies/g, the model reached a 16S rRNA gene copy density of 1 × 1010 copies/g after 16 h and plateaued at 1.3 × 1010 copies/g after 18 h. Growth model data and qPCR data are consistent with the proliferation of bacteria in food waste between the hydropulper and the anaerobic digestor feeding tank.
Composition of the fungal community in food waste
Figure 2 shows the structure variation of the fungal community in the hydropulper and digestor feed samples. Sequencing was done on the Illumina ITS platform, and QIIME 2 pipelines were used to process reads. The most abundant fungal lineages belong to the genera Saccharomyces and Kazachstania. The digestor feeding tank samples held more Saccharomyces and the < 1% abundance fungi strains decreased in relative abundance. The composition of the fungal community in the feedstock of food waste was relatively consistent over the 3 month sampling period. The consistent community structure with the lack of proliferation of fungi between the hydropulper and digestor feed samples indicated limited fungal activity in the food waste.

Composition of the fungal community in (a) hydropulper and (b) digestor feed samples.
Composition of the bacterial community in food waste
Figure 3a,b shows the variation of the bacterial communities in the hydropulper and digestor feed samples. Illumina 16S rRNA sequencing was used to generate raw reads, and QIIME 2 pipelines were used to process the reads. Lactobacillaceae lineages (including Lactobacillus and Lactiplantibacillus) dominated throughout the sampling period, with an average total relative abundance in the hydropulper of 64% and 78% in the digestor feed. The relative abundance of Lactobacillaceae was more variable in the hydropulper than in the digestor feed, ranging from 32 to 93%. Sequences within the Lactobacillaceae family and closest relatives from the NCBI database were used to generate a phylogenetic tree to allow species assignation (Fig. 3c). Leuconostoc and Klebsiella strains were added as outgroups. Three of the most abundant Lactobacillaceae lineages belonged to Lactobacillus amylovorus (Lin1), Lactobacillus sanfranciscensis (Lin2), and Lactiplantibacillus plantarum (Lin3). Lactobacillus amylovorus (Lin1) was the most abundant species in the hydropulper, while Lactobacillus sanfranciscensis (Lin2) and Lactiplantibacillus plantarum (Lin3) were the most abundant in digestor feed samples. Other bacterial lineages above 1% relative abundance include Klebsiella, Leuconostoc, Acetobacter, Pseudomonas, Weissella, Brachymonas, Cloacimonadaceae W5 and Unclassified Enterobacteriaceae. They were more abundant in hydropulper samples than digestor feed samples. Bacterial lineages with less than 1% relative abundance in the hydropulper had further decreased in relative abundance in the digestor feed.

Composition of the bacterial community in food waste samples from (a) the hydropulper and (b) digestor feed determined with Illumina sequencing. Genus-level identities are presented where possible. Relative abundance excludes chloroplast entries (< 25%). The variation in the abundance of Lactobacillaceae in the bacterial community includes only Lactobacillaceae lineages with > 1% relative abundance (Lin1-7). Minor Lactobacillaceae include all Lactobacillaceae lineages with less than 1% relative abundance. Lin 1–3 on the graphs correspond to Lactobacillus/Lactiplantibacillus Lin1-3 in the legend. (c) Maximum likelihood phylogenetic tree of Family Lactobacillaceae extracted from the Illumina 16S rRNA Illumina sequence entries (Lin1-7 indicated by solid circle). Number and scale in figure showing the phylogenetic relationship between bacterial lineages observed in food waste and their closest cultured relatives. Numbers represent bootstrap (branch point confidence) values from 500 replicates.
Correlations between environmental parameters and community variation
To understand the relationship between food waste physico-chemical characteristics and the composition variations of the microbial community, three characteristics of food waste were measured (Supplementary information). Figure S2a shows the water content and pH variation of the samples over 3 months. Figure S2b shows the proportions of carbon, nitrogen and sulfur in the samples. The digestor feed and hydropulper samples have similar carbon (average of 43.38% and 42.47%, respectively), nitrogen (average of 2.40% and 2.46%, respectively) and sulphur content (average of 0.18% and 0.32%, respectively). Although the sample characteristics between the two sampling points are similar in average, on a daily basis, the sample can be vastly different. Although average elemental contents were similar, the carbon content variation pattern in hydropulper were different from the digestor feed sample from the same day. Similarly, nitrogen content had disagreed abundance variation between same day samples from the two sampling locations.
Figure 4 shows the bacterial community diversity among samples. The fungal community (not shown) did not return any prominent grouping for all metadata categories. For the bacterial community, two metadata categories, pH and nitrogen element content, were identified with nestings associated with specific ranges. The bacterial community experienced structural shifts from the hydropulper to digestor feeding tank samples. The lower the pH or higher the nitrogen content, the more closely related the bacterial communities were. The pH range of 3.5–4 and the nitrogen element content of 3–3.5% showed a strong influence on the bacteria community.

Weighted PCoA graphs show the relationship between environmental parameters and bacterial community composition. Hydropulper (squares) and digestor feed (stars) communities are distinct (a). pH (b) and nitrogen (c) content showed specific grouping at pH 3.5–4 and nitrogen content 3–3.5% (indicated by black color).
The diversity within each sample regarding the three metadata criteria, type of sample, pH, and N% of feedstock, are summarised in Table 1. Hydropulper samples had higher bacterial diversity (richness) than digestor feed samples (P = 4.72E−07). When pH was higher than 5, the richness of the bacterial community increased (P = 0.039), and a higher percentage of nitrogen was associated with an increase in bacterial community evenness (P = 0.040).
Figure 5 illustrates the correlations between environmental parameters, physico-chemical characteristics, abundance species variation, and community variance for both bacterial and fungal communities. Factor scales from − 1 to 1 indicate a total negative or positive nonparametric Spearman correlation. Underlined correlation scores had significant differences (P < 0.05).

The bacterial heat map indicates the relationships among the environmental parameters (pH, nitrogen content, the variance in relative abundance, and the number of bacterial 16S rRNA gene copies in the sample separated into hydropulper (a) and digestor feed (b) samples. The negative (dark green) heat map cell indicates a negative nonparametric Spearman correlation and vice versa. The fungal heat map indicates the relationships among environmental parameters, the variance in strain relative abundance, and the number of fungal ITS gene copies in the sample separated into hydropulper (c) and digestor feed (d) samples. The negative (dark green) heat map cell indicates a negative nonparametric Spearman correlation and vice versa. The underlined entries have a P value less than 0.05 from sigificance test.
Between the hydropulper and digestor feed tank, nitrogen content was positively correlated with abundant bacterial genera (Lactobacilus, Leunocostoc, and Klebessila) and negatively correlated to pH. Furthermore, the BLAST search of Illumina entries allowed for analysing the abundance variations of Lactobacillaceae species under various conditions. The highly abundant L. plantarum (Lin3), L. sanfranciscensis (Lin2), and L. amylovorus (Lin1) were correlated with pH, nitrogen and carbon content with varied dependency.
In the fungal community (Fig. 5c,d), the sulphur and nitrogen element content impacted the hydropulper community, where fungal strains are negatively connected. The pH was not clearly correlated with the relative abundance of fungal strains or the number of ITS gene copies.
Discussion
Methane yield during anaerobic digestion correlates with the relative abundance of specific bacterial and archaeal lineages in the incumbent microbial community28. The composition of microbial communities is determined by the growth and death rates of community members and immigration from outside the system harbouring the community29. In the context of anaerobic digestion of food waste, immigration is logically driven by the indigenous community of the foodwaste feedstock. The microbiota associated with feedstock is additionally relevant with respect to chemical transformations it catalyses prior to feedstock entering a digester (biological pretreatment).
Despite its relevance to anaerobic digestion of food waste, relatively little is known about the composition of microbial communities in food waste. One study described the microbial community in food from a canteen from the moment it was discarded and over the ensuing 72 h13. Another described changes in community composition of food waste in response to aeration30. In addition, a study has explained the variation of food waste composition in response to storage environment31. None have described how the community composition of food waste feedstock received by a digestion facility varies over time or within different units prior to entering digesters. The food waste that undergoes anaerobic digestion is mainly composed of bacteria, especially Lactobacillaceae. However, the bacterial community is not stable and varies every day along with the pH and nitrogen levels of the waste. Ammonia, as a form of nitrogen, can inhibit the growth of various species of bacteria and pose selective pressure and shift the microbial community in food waste32. This knowledge underpins future attempts to rationally engineer the foodwaste community for improved downstream resource recovery.
In the current study of an industrial anaerobic digestion facility handling food waste, bacteria and fungi were quantified in food waste samples from the hydropulper unit and from the downstream storage unit from which food waste is transferred into the digester. pH ranged from 3 to 5.5 trending downwards throughout the sampling campaign. Water content sat between 80 and 90%. The pH shift of the food waste could result either from changes in the food waste composition as lower pH is associated with carbohydrate-rich food waste and higher pH associated with protein-rich food waste or the storage temperature that affects the production of acidic fermentation products31.
Quantification of fungal and bacterial sequences revealed that bacteria are numerically dominant in food waste by several orders of magnitude. This suggests that fungi play a limited role in foodwaste decay or depolymerisation prior to anaerobic digestion. The closely related yeasts Saccharomyces and Khazachstania were the dominant fungal lineages. Khazachstania has been observed previously in food waste and is common in food fermentation communities33 Saccharomyces are commonly found in food fermentation waste such as the washdown of brewery and bakery factories34, 35. The low abundance of moulds may be due to the homogenisation of food waste in handling, transport and ultimately pulping, and the limited availability of oxygen36, but the low abundance of fungi generally was suprising. Fungi can be tolerant of low pH and oxygen limitation, so it is possible that the fungi in foodwaste are suppressed through production of antifungal agents by bacteria. The bacteria Lactobacillus sanfranciscensis (Lin2) and Lactiplantibacillus plantarum (Lin3), observed to dominate the bacterial community in this study, are known to produce antifungal substances37. Regardless of the driver of low fungal relative abundance, it appears that the biodegradative abilities of fungi are not being exploited in foodwaste.
The bacterial community was dominated by lineages within the Lactobacillaceae Family (principally Lactobacillus species). Lactobacillus species are well known for their ability to ferment sugars under oxygen limitation and for the associated release of volatile fatty acids that results in a drop of pH38. This has long been exploited in the preservation of foods because lowering the pH below 5 creates conditions unfavourable to most microbes39, 40. In essence, Lactobacillus species exclude competitors in food waste through pH manipulation. This limits the biodegradation ability of the microbial community in foodwaste to degradative enzymatic activity generated by Lactobacillus. Regarding extracellular enzyme activity, abundant L. amylovorus, L. sanfranciscensis, and L. plantarum lineages, are known to produce amylase41,41,42,44. L. amylovorus and L. plantarum are also known to produce lipase and protease45,45,47 though proteolytic activity is moderate48, 49. L. plantarum also produces extracellular feruloyl esterase50, while L. amylovorus has only been reported to produce intracellular esterase51. Therefore, the three most abundant bacteria observed can produce amylase, lipase, protease, and feruloyl esterase, but they are not known to produce cellulase. They probably play a significant role in starch degradation, but have a limited ability to hydrolyse lignocellulosic biomass. There may be potential in bioaumenting cellulase producing bacteria to increase downstream digestion efficiency.
Proteobacteria were notable in their absence in foodwaste13, 40, 52. Proteobacteria are capable of degrading various complex substances and are prevalent in anaerobic digestion systems53, 54. This supports the potential for bioaugmenting foodwaste with Proteobacteria or altering conditions for the growth of Proteobacteria to exploit their degradation capabilities including cellulase activity55.
Aerobic pretreatment of organic substrates can improve anaerobic digestion56,56,58. This is believed to be a consequence of reducing the concentration of easily digestible substrates that can result in rapid decreases in pH due to VFA production under anaerobic conditions, accelerated oxidation of VFAs and the depolymerisation of relatively recalcitrant biopolymers resulting in more extensive digestion and reduced biosolids59, 60. Aerobic pre-treatment can increase digestion stability and efficiency61,61,63. These benefits derived from aeration logically hinge on the composition of microbial communities indigenous to food waste and the catalytic abilities encoded therein30.
This study showed the variability of microbial community structure in food waste. The samples showed dominance and growth of the bacterial community but not the fungal community. Lactobacillaceae were dominant, and their activity was mainly influenced by pH and nitrogen content. The results inform the potential for adjusting the community via bioaugmentation or supplying air to achieve higher downstream digestion efficiency. These observations form a foundation from which rational engineering of food waste pretreatment conditions can be developed to increase anaerobic digestion throughput.
Data availability
The sequencing results can be found in the NCBI Sequence read archive PRJNA805020. Other datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Kaza, S., Yao, L. C., Bhada-Tata, P. & Van Woerden, F. What a Waste 20: A Global Snapshot of Solid Waste Management to 2050 (World Bank, 2018). https://doi.org/10.1596/978-1-4648-1329-0.
Lim, S. L., Lee, L. H. & Wu, T. Y. Sustainability of using composting and vermicomposting technologies for organic solid waste biotransformation: Recent overview, greenhouse gases emissions and economic analysis. J. Clean. Prod. 111, 262–278. https://doi.org/10.1016/j.jclepro.2015.08.083 (2016).
Pickin, J. et al. 9 NOVEMBER 2018 PREPARED FOR Department of the Environment and Energy PREPARED IN ASSOCIAT ION WITH Disclaimer Report title National Waste Report 2018 Client Department of the Environment and Energy Status Final (2018).
Metz, B., Davidson, O., Bosch, P., Dave, R. & Meyer, L. Climate Change 2007—Mitigation of Climate Change (Cambridge University Press, 2007).
Wang, K. S., Chiang, K. Y., Lin, S. M., Tsai, C. C. & Sun, C. J. Effects of chlorides on emissions of toxic compounds in waste incineration: Study on partitioning characteristics of heavy metal. Chemosphere 38, 25 (1999).
Hickey, M. E. & Ozbay, G. Food waste in the United States: A contributing factor toward environmental instability. Front. Environ. Sci. Eng. China 2, 25 (2014).
Tonini, D., Albizzati, P. F. & Astrup, T. F. Environmental impacts of food waste: Learnings and challenges from a case study on UK. Waste Manage. 76, 744–766 (2018).
Pain, B. F. & Hepherd, R. Q. Anaerobic Digestion of Farm Waste (1985).
Syaichurrozi, I. & Rusdi, R. Kinetics studies impact of initial pH and addition of yeast Saccharomyces cerevisiae on biogas production from tofu wastewater in Indonesia. Int. J. Eng. 29, 25 (2016).
Wang, P., Wang, H., Qiu, Y., Ren, L. & Jiang, B. Microbial characteristics in anaerobic digestion process of food waste for methane production—a review. Bioresour. Technol. 248, 29–36 (2018).
Zhang, R. et al. Characterization of food waste as feedstock for anaerobic digestion. Bioresour. Technol. 98, 929–935 (2007).
Usack, J. G. et al. An evaluation of anaerobic co-digestion implementation on New York State dairy farms using an environmental and economic life-cycle framework. Appl. Energy 211, 28–40 (2018).
Wu, S. et al. Bacterial communities changes during food waste spoilage. Sci. Rep. 8, 25 (2018).
Maeda, H. et al. Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol. Med. Microbiol. 39, 81–86 (2003).
Bokulich, N. A. & Mills, D. A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 79, 2519–2526 (2013).
De Beeck, M., Lievens, B., Busschaert, P., Declerck, S. & Vangronsveld, J. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS One 9, 97629 (2014).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Unite, C. UNITE QIIME release for Fungi. UNITE https://doi.org/10.15156/BIO/786334 (2019).
QIIME 2 community. Training feature classifiers with q2-feature-classifier—QIIME 2 2021.2.0 documentation. https://docs.qiime2.org/2021.2/tutorials/feature-classifier/ (2021).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Ea. Operating instructions vario MACRO cube CHNOS Elemental Analyzer, 47–50 (2017).
Margalef, R. Information theory in Ecology. J. Gener. Syst. http://www.sciepub.com/reference/72223 (1958).
Allaby, M. A Dictionary of Ecology (Oxford University Press, 2010). https://doi.org/10.1093/acref/9780199567669.001.0001.
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. https://doi.org/10.1128/AEM.01996-06 (2007).
Ma, G., Chen, Y. & Ndegwa, P. Association between methane yield and microbiota abundance in the anaerobic digestion process: A meta-regression. Renew. Sustain. Energy Rev. 135, 110212 (2021).
Dottorini, G. et al. Mass-immigration determines the assembly of activated sludge microbial communities. Proc. Natl. Acad. Sci. USA 118, 25 (2021).
Fisgativa, H., Tremier, A., Saoudi, M., Le Roux, S. & Dabert, P. Biochemical and microbial changes reveal how aerobic pre-treatment impacts anaerobic biodegradability of food waste. Waste Manage. 80, 119–129 (2018).
Daly, S. E. et al. Systematic analysis of factors that affect food-waste storage: Toward maximizing lactate accumulation for resource recovery. ACS Sustain. Chem. Eng. 8, 13934–13944 (2020).
Regueiro, L. et al. Comparing the inhibitory thresholds of dairy manure co-digesters after prolonged acclimation periods: Part 2-correlations between microbiomes and environment. Water Res. 87, 458–466 (2015).
Fisgativa, H., Tremier, A. & Dabert, P. Characterizing the variability of food waste quality: A need for efficient valorisation through anaerobic digestion. Waste Manage. 50, 264–274 (2016).
Krogerus, K., Preiss, R. & Gibson, B. A unique Saccharomyces cerevisiae × Saccharomyces uvarum hybrid isolated from norwegian farmhouse beer: Characterization and reconstruction. Front. Microbiol. 10, 2253 (2018).
Zhang, G., Sun, Y., Sadiq, F. A., Sakandar, H. A. & He, G. Evaluation of the effect of Saccharomyces cerevisiae on fermentation characteristics and volatile compounds of sourdough. J. Food Sci. Technol. 55, 2079 (2018).
Criado, M. V., Fernández Pinto, V. E., Badessari, A. & Cabral, D. Conditions that regulate the growth of moulds inoculated into bottled mineral water. Int. J. Food Microbiol. 99, 343–349 (2005).
Black, B. A., Zannini, E., Curtis, J. M. & Gänzle, M. G. Antifungal hydroxy fatty acids produced during sourdough fermentation: Microbial and enzymatic pathways, and antifungal activity in bread. Appl. Environ. Microbiol. 79, 1866 (2013).
Kleerebezem, M. et al. Lifestyle, metabolism and environmental adaptation in Lactococcus lactis. FEMS Microbiol. Rev. 44, 804–820. https://doi.org/10.1093/femsre/fuaa033 (2020).
Laughton, J. M., Devillard, E., Heinrichs, D. E., Reid, G. & McCormick, J. K. Inhibition of expression of a staphylococcal superantigen-like protein by a soluble factor from Lactobacillus reuteri. Microbiology 152, 1155–1167 (2006).
Maldonado-Barragán, A. & West, S. A. The cost and benefit of quorum sensing-controlled bacteriocin production in Lactobacillus plantarum. J. Evol. Biol. 33, 101–111 (2020).
Amapu, T., Ameh, J., Ado, S., Abdullahi, I. & Dapiya, H. Amylolytic Potential of lactic acid bacteria isolated from wet milled cereals, cassava flour and fruits. Brit. Microbiol. Res. J. 13, 25 (2016).
Burgess-Cassler, A. & Imam, S. Partial purification and comparative characterization of α-amylase secreted by Lactobacillus amylovorus. Curr. Microbiol. 23, 25 (1991).
Corsetti, A. et al. Combined effect of sourdough lactic acid bacteria and additives bread firmness and staling. J. Agric. Food Chem. 48, 25 (2000).
Nakamura, L. K. Lactobacillus amylovorus, a new starch-hydrolyzing species from cattle waste-corn fermentations. Int. J. Syst. Bacteriol. 31, 25 (1981).
De Angelis, M., Gobbetti, M. & Corsetti, A. Esterase and lipase activities of Lactobacillus sanfranciscensis strains used in sourdough fermentation. Ital. J. Food Sci. 11, 25 (1999).
Maroju, P. A., Tata, P., Balapure, A., Ray Dutta, J. & Ganesan, R. Lactobacillus amylovorus derived lipase-mediated silver derivatization over poly(ε-caprolactone) towards antimicrobial coatings. Enzyme Microb. Technol. 150, 25 (2021).
Uppada, S. R., Akula, M., Bhattacharya, A. & Dutta, J. R. Immobilized lipase from Lactobacillus plantarum in meat degradation and synthesis of flavor esters. J. Genet. Eng. Biotechnol. 15, 331–334 (2017).
Zeng, X. et al. Technological properties of Lactobacillus plantarum strains isolated from chinese traditional low salt fermented whole fish. Food Control 40, 25 (2014).
Johnson, B. R. et al. The S-layer associated serine protease homolog prtX impacts cell surface-mediated microbe-host interactions of Lactobacillus acidophilus NCFM. Front. Microbiol. 8, 25 (2017).
Esteban-Torres, M., Reverón, I., Mancheño, J. M., De las Rivas, B. & Muñoz, R. Characterization of a feruloyl esterase from Lactobacillus plantarum. Appl. Environ. Microbiol. 79, 25 (2013).
Xu, Z., He, H., Zhang, S., Guo, T. & Kong, J. Characterization of feruloyl esterases produced by the four lactobacillus species: L. amylovorus, L. acidophilus, L. farciminis and L. fermentum, isolated from ensiled corn stover. Front. Microbiol. 8, 15 (2017).
Almeida, O. G. G. et al. Does Quorum Sensing play a role in microbial shifts along spontaneous fermentation of cocoa beans? An in silico perspective. Food Res. Int. 131, 109034 (2020).
Wang, Y., Tang, Y., Li, M. & Yuan, Z. Aeration rate improves the compost quality of food waste and promotes the decomposition of toxic materials in leachate by changing the bacterial community. Bioresour. Technol. 340, 125716 (2021).
Nzeteu, C. et al. Resource recovery from the anaerobic digestion of food waste is underpinned by cross-kingdom microbial activities. Bioresou. Technol. Rep. 16, 100847 (2021).
Sethi, S., Datta, A., Gupta, B. L. & Gupta, S. Optimization of cellulase production from bacteria isolated from soil. ISRN Biotechnol. 2013, 985685 (2013).
Lim, J. W., Chiam, J. A. & Wang, J.-Y. Microbial community structure reveals how microaeration improves fermentation during anaerobic co-digestion of brown water and food waste. Bioresour. Technol. 171, 132–138 (2014).
Ruan, D. et al. Enhancing methane production of anaerobic sludge digestion by microaeration: Enzyme activity stimulation, semi-continuous reactor validation and microbial community analysis. Bioresour. Technol. 289, 121643 (2019).
Fu, S.-F., Wang, F., Shi, X.-S. & Guo, R.-B. Impacts of microaeration on the anaerobic digestion of corn straw and the microbial community structure. Chem. Eng. J. 287, 523–528 (2016).
Lim, J. W. & Wang, J.-Y. Enhanced hydrolysis and methane yield by applying microaeration pretreatment to the anaerobic co-digestion of brown water and food waste. Waste Manage. 33, 813–819 (2013).
Nguyen, D. & Khanal, S. K. A little breath of fresh air into an anaerobic system: How microaeration facilitates anaerobic digestion process. Biotechnol. Adv. 36, 1971–1983 (2018).
Rafieenia, R. et al. Effect of aerobic pre-treatment on hydrogen and methane production in a two-stage anaerobic digestion process using food waste with different compositions. Waste Manage. 59, 194–199 (2017).
Xu, S., Selvam, A. & Wong, J. W. C. Optimization of micro-aeration intensity in acidogenic reactor of a two-phase anaerobic digester treating food waste. Waste Manage. 34, 363–369 (2014).
Charles, W., Walker, L. & Cord-Ruwisch, R. Effect of pre-aeration and inoculum on the start-up of batch thermophilic anaerobic digestion of municipal solid waste. Bioresour. Technol. 100, 2329–2335 (2009).
Acknowledgements
The authors thank EarthPower for providing samples, the Ramaciotti Centre (UNSW) for sequencing, and the Mark Wainwright Analytical Facility for elemental analysis. In addition, the author thank NSW DPI for the financial support.
Funding
This work was funded through an NSW Department of Planning, Industry, and Environment grant collaborating with UNSW Sydney.
Author information
Authors and Affiliations
Contributions
L.T. was responsible for the manuscript writing, research design, and experiment execution, J.O. contributed to part of data generation and manuscript writing, O.K provided technical support on experiments and data interpretation, M.M. guided the design of research and provided feedback on research results. O.K. and M.M. reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, L., O’Dwyer, J., Kimyon, Ö. et al. Microbial community composition of food waste before anaerobic digestion. Sci Rep 13, 12703 (2023). https://doi.org/10.1038/s41598-023-39991-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-39991-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
