
Abstract
Ultrasmall silver clusters in reduced state are difficult to synthesize since silver atoms tend to rapidly aggregate into bigger entities. Here, we show that dimers of reduced silver (Ag2) are formed within the framework of a metal–organic framework provided with thioether arms in their walls (methioMOF), after reduction with NaBH4 of the corresponding Ag+-methioMOF precursor. The resulting Ag2-methioMOF catalyzes the methanation reaction of carbon dioxide (CO2 to CH4 hydrogenation reaction) under mild reaction conditions (1 atm CO2, 4 atm H2, 140 °C), with production rates much higher than Ag on alumina and even comparable to the state-of-the-art Ru on alumina catalyst (Ru–Al2O3) under these reaction conditions, according to literature results.
Similar content being viewed by others
Introduction
Ligand-free sub-nanometric metal clusters are metastable chemical species where most of the atoms are sited at the surface of the particle, available to interact with external molecules1,2. This topology, combined with electronic and cooperative effects, makes metal clusters particularly useful in catalysis, since all metal atoms can participate during the catalytic chemical events3,4. From an economy and efficiency point of view, the smaller the cluster is the better the catalytic production per metal atom can be, the limit being on single metal atoms (single atom catalysts or SACs)5. However, the latter (SACs) lack potential metal-to-metal cooperation processes, which can be of undoubtable importance during the catalytic reaction. Following this rationale, metal dimers (two atoms) bring the best of both worlds for catalysis: optimized metal atom efficiency and metal cooperativity.
Coinage metal atoms are particularly suitable to form ultrasmall metal clusters, including dimers, since their electron-rich, loosely bound delocalized valence shells allow to bind few metal atoms in stable entities, without extensive aggregation under selected reaction conditions6. Not in vain, gold7, palladium8 and platinum9 clusters are commonly found in catalysis. However, silver is an exception here, since the extraordinary tendency of silver atoms to reduce (even with just light) and aggregate in nano- and micro-particles hampers the formation of ultrasmall metal clusters10,11. In particular, catalytic ligand-free silver dimers in reduced state (Ag2) have been obtained with electrochemistry techniques12, under size-selected synchrotron conditions13,14,15 or by supporting them in porous solids16.
During the last two decades, a type of porous materials, so-called metal–organic frameworks (MOFs), have been intensely studied given their excellent performances in many important fields 17,18, which mainly arise from their unique host–guest chemistry19,20. For instance, MOFs have been shown to be very effective for the encapsulation of different ultrasmall metal clusters21 and even for their in-situ step-by-step preparation within the functional channels of the MOF4,22, which acts as an efficient chemical nanoreactor. In this context, we have recently reported that the use of a highly robust and crystalline MOF, as a chemical nanoreactor, allowed to synthesise and stabilize Ag1 and Ag2 within the microporous networks, in gram amounts23. Our synthetic approach required the use of extensive solid-to-solid post-synthetic steps, including at least three metal cation exchanges before the final reduction of the Ag+–MOF precursor, which could be achieved given the high robustness and crystallinity of the selected MOF23.
In the present work, a previously reported MOF, with formula {CaIICuII6[(S,S)-methox]3(OH)2(H2O)}·16H2O (1)24 [where methox is bis[(S)-methionine]oxalyl diamide], whose channels are densely decorated with thioether arms, is efficiently used as chemical reactor to prepare Ag2 nanoclusters within its functional channels. Thus, the functional pore environment of 1, considering the high affinity of sulfur towards silver atoms, enables the incorporation and chemical reduction of Ag+ in the MOF’s structure without additional steps, to generate the targeted Ag0@1 with precise atomicity and in gram-scale25. This new solid material catalyzes very efficiently the hydrogenation of carbon dioxide (CO2) to methane (CH4) under mild reaction conditions (1 atm CO2, 4 atm H2, 140 °C), with productivity rates comparable to an industrial catalyst under these reaction conditions, according to the literature26,27. Ag0@1 is relatively stable under reaction conditions and can be recovered and reused at least three times without severe depletion of the catalytic activity. These results bring ultrasmall Ag0 species as a new catalytic tool for hydrogenation reactions, moreover considering the reluctance of the CO2 molecule towards the hydrogenation reaction. Besides, the synthesis of this material, which consists in the formation of the MOF from highly available amino acids and calcium and copper salts24, and the incorporation of Ag and its subsequent reduction, proceeds with a very high atomic economy, since the only reagent that does not incorporates directly in the material is the final reducing agent, and all steps occur at room temperature. Therefore, the MOF synthesis is not neither energetically intensive nor material costly, so with a low carbon footprint, amply compensated in terms of global warming effects during the catalytic carbon dioxide hydrogenation reaction.
Results and discussion
Synthesis and characterization of 1, Ag+@1 and Ag0@1
MOF 1 was prepared as previously reported. The MOF-driven preparation of Ag01 single atoms and Ag2 clusters takes place in two consecutive steps (Fig. 1). Firstly, the highly robust water-stable MOF 1, featuring channels decorated with sulfur-containing chains capable to retain silver(I) cations, was soaked in a saturated AgNO3 aqueous solution to give an intermediate material with formula (AgNO3)3@{CaIICuII6[(S,S)–methox]3(OH)2(H2O)} . 13H2O (Ag+@1) (Fig. 2). Then, Ag01 single atoms, and the Ag2 dimers could be obtained by reducing Ag+@1 with NaBH4, under visible light, yielding the final material (Ag01)(Ag02)@{CaIICuII6[(S,S)-methox]3(OH)2(H2O)}·14H2O (Ag0@1) (Fig. 3).

The schematic (based on the three real crystal structures) showing the two-steps protocol starting from 1 (c), soaked in AgNO3 aqueous solution to give Ag+@1 (b) before preparation of Ag0@1 (c). Color scheme: sulfur, yellow spheres; calcium, pastel orange polyhedra; copper, dark cyan polyhedra, silver(I), blue spheres, silver(0) sky blue spheres, oxygen and nitrogen from nitrate anions, red and violet spheres, atoms from the ligand, gray sticks.

Crystal structure of Ag+@1: views of a fragment (a) and one single channel (b) of Ag+@1 along the c axis; details of Ag+–S binding sites (c). Yellow and blue spheres represent S and Ag atoms whereas all the porous network is depicted as gray sticks. Sky blue and red spheres represent N and O atoms of nitrate anions. Blues dashed lines represent the Ag···S interactions. In (c) copper and calcium atoms from the network are represented by cyan and pastel orange spheres, respectively.

Crystal Structure of Ag0@1. Views of a fragment (a) and one single channel (b) along the c and [111] direction (c); Details of crystal structure and binding sites for Ag01 and Ag02 (d). Yellow and light blue spheres represent S and Ag0 atoms, respecively whereas all the porous network is depicted as gray sticks except for methionine arms represented as yellow sticks. Light blue dashed lines represent the Ag···S interactions.
Indeed, functional thio–alkyl chains decorating the pores of MOF 1 played a dual key role by retaining silver(I) cations within the MOF channels and also by limiting the number of silver(I) and allowing their homogeneous distribution along the channels (see structural section) cations inserted and allowing the ulterior formation of ultrasmall silver single atoms and clusters after reduction.
Regarding the synthetic protocol, single crystals of the MOF 1, suitable for single crystal X-ray diffraction (SCXRD), could be obtained by slow diffusion techniques (see Experimental Section for complete description), as previously reported24,28. Then, given the high crystallinity and robustness of 1, crystals of the MOF were used to obtain crystals of Ag+@1 and Ag0@1 after a soaking and reduction process, respectively, whose structures could be solved using synchrotron radiation. The obtention of the crystal structures of Ag+@1 and Ag0@1 allowed unique insights about the clusters nature, as described in the crystal structure section. Alternatively, a multi-gram protocol was also developed to obtain large quantities of polycrystalline samples of 1, Ag+@1 and Ag0@1 (see Experimental Section) which were used for catalytic experiments.
The crystal structures of both Ag+@1 and Ag0@1 are isomorphic to 1. They both crystallise in the P63 chiral space group of the hexagonal system confirming the robustness of the 3D porous network of 1, after AgNO3 capture and even after Ag+ reduction. In the crystal structure of Ag+@1, Ag+ metal ions together with NO3- anions are trapped in the hexagonal nanopores of 1, being recognized by the thioether arms of the methionine residues. Thanks to the robustness of the network, detailed analysis of synchrotron X-ray data (at T = 45 K) gave Ag0@1 crystal structure allowing also to give new insights on the structural parameters and binding motifs of Ag2 clusters in MOF structure.
Crystal stucture of Ag+@1 unambigueusly shows silver(I) cations captured and hosted in hydrophilic hexagonal pores [virtual diameter of 0.8 nm] open-framework structure of 1 (Figs. 1a,b, 2) and smaller voids of the neutral CaIICuII6 porous network. In Ag+@1 and Ag0@1, the thioether chains from methiomox ligand show as detected conformation one of the two crystallographically distinct moieties in a distended conformation towards the pores, and the other one pretty bent with the terminal methyl groups pointing in smaller interstitial voids developing along a axis (Fig. 3c,d) and (Figs. S1–S3). Both conformations allow sulfur to target efficiently Ag+ ions by S binding sites as confirmed by crystal structure of Ag+@1 [Ag+···S distances of 2.52(2) and 2.54(4) Å]. Nitrate anions, residing within pores, are well stabilized either being coordinated to Ag+ metal ions in a chelating motif (Fig. 2c) [Ag+···O distances of 2.14(4) and 2.24(9) Å] or by static and non-covalent interactions. Both the Ag+ ions residing in the most accessible pores and smaller voids could be reduced to Ag0, as confirmed by XPS spectrum of Ag0@1 (vide infra and Fig. S4). This feature is likely due to visible light-assisted photoreduction process involving Ag+ ions.
After the reduction process, the crystal structure of Ag0@1 unambiguously reveals Ag(0) confined into the pores with metal sites pretty reminiscent of that found in Ag+@1. Ag+ ions atoms were reduced to Ag10 single atoms and Ag2 dimers, to give Ag0@1 (Fig. 3). Ag(0) single atoms are only found in smaller voids (Fig. 3c,d) whereas Ag20 dimers are generated in situ from straight Ag+ ions reduction and controlled aggregation (being well fixed in pores). Such dimers are retained by sulfur atoms and stabilized through a double binding mode [Ag0···S distances of 2.471(9) Å for Ag10single atoms and 2.32(3) and 2.732(11) Å for Ag20 dimers, featuring an Ag0···Ag0 separation of 2.59(3) Å] (Fig. 3). The coordinated nitrate anions might play a role in the mechanism of formation of the Ag20 dimers in Ag0@1, by supporting interactions and by a synergistic effect with the flexible dimethyl thioether chains from the methionine residues, facilitating the approach of silver in bigger hexagonal pores. On the contrary, the reduction process on the silver cations located in the less accessible pores, grew Ag0 single atoms still clutched and stabilized by Ag–S interactions.
Apart from the crystal structures, the nature of Ag+@1 and Ag0@1 was further established by combining inductively coupled plasm–mass spectrometry (ICP–MS, Table S1, Supporting Information), powder X–ray diffraction (PXRD), Fourier transform infrared spectroscopy (FT-IR) experiments and elemental C, H, S, N and thermo–gravimetric (TGA) analyses, in combination with XPS, which was used to establish the structure and oxidation state of Ag in both Ag+@1 and Ag0@1 (see Supporting Information, Figs. S4–S8 and Table S1).
TGA analyses for Ag+@1 and Ag0@1 (Fig. S5) allowed to ascertain the water contents for both hybrid MOFs, thus determining their chemical formulas (see experimental section). PXRD patterns of Ag+@1 and Ag0@1 (Fig. S6) confirm that bulk (polycrystalline) samples are pure and isostructural to crystals selected for SCXRC in both cases. Moreover they also suggest that open-framework structures remain unaltered after cation insertion (Ag+@1) and the reduction process (Ag0@1), which is also confirmed by the corresponding FT-IR spectra (Fig. S7). In addition, any peaks related to silver metal nanoparticles or oxide crystal structures were not observed in the PXRD pattern of Ag0@1, which confirms that only small nanoclusters should be present.
XPS spectra for Ag+@1 and Ag0@1 can be observed in Fig. S4. First, XPS spectrum of Ag+@1 (Fig. S4a) shows the expected two bands at 367.4 and 373.4 eV, which can be unambiguosly attributted to Ag 3d5/2 and Ag 3d3/2 binding energies, typical of Ag+23,29. On the other side, XPS spectrum for Ag0@1 (Fig. S4b) shows the same two bands shifted to 368.2 and 374.2 eV, which suggest that all Ag+ cations have been reduced to Ag0 atoms. The N2 and CO2 adsorption isotherms for Ag+@1 and Ag0@1, and also MOF 1 for the sake of comparison, were also studied (Fig. S8). N2 adsorption isotherms (Fig. S8a) suggest a certain permanent porosity for 1, Ag+@1 and Ag0@1, respectively. Ag+@1 exhibits lower N2 adsorption which can be attributed to the presence of Ag+ cations and NO3− anions occupying the pores. In turn, Ag0@1 not only exhibits larger N2 adsorption than Ag+@1, which could be expected as NO3− anions are not longer present, but also larger than that of MOF 1. This behaviour is reproduced in CO2 adsorption isotherm (Fig. S8b), which shows a ca. 25% uptake increase for Ag0@1. Overall, gas adsorption measurements confirm both that Ag0@1 possess permanent porosity and also that it is capable to adsorb CO2, which are prerequisites for the selected catalytic reaction.
Catalytic methanation of CO2
The hydrogenation of CO2 to methane (Sabatier reaction) has gained attention in the last years as a means to recycle anthropogenic CO2 and combat climate change, besides generating renewable methane26. Despite the industrial production costs of H2 precluded this approach in a first approximation, the raise of a cheap green H2 industry could boost this reaction back, with grounded environmental and economic basis30. Figure 4 shows the catalytic results for the hydrogenation of CO2 with Ag0@1. For the sake of comparison, the catalytic behaviour of Ag+@1, our previously reported Ag2-MOF catalyst23, and a house-made sample of Ag–Al2O3, was also studied. The results show that Ag0@1 catalyzes the methanation reaction much more efficiently than the other solid catalysts, and with a productivity rate per metal atom comparable to the state-of-the-art Ru–Al2O3 catalyst under these reaction conditions27. Notice here that the reaction temperature (140 °C) was chosen to achieve the maximum reaction rates possible, since Ru–Al2O3 shows the highest productivity at > 120 °C reaction temperature27 and lower temperatures considerable decrease the initial rate of the industrial catalyst31. The productivity of the solid catalyst Ag0@1 was also compared to that of the industrially used Ru on alumina catalyst by calculating the corresponding µmolCO2 g−1catalyst h−1 values (Table S3), under similar reaction conditions27. It can be seen that the productivity for Ag0@1 is not far from the currently used industrial catalyst (68 vs. 93 µmolCO2 g−1catalyst h−1, respectively), which illustrates the remarkable catalytic activity of Ag0@1.

Kinetic results for the methanation of CO2 with different solid catalysts. Reaction conditions: 5 bars of the gas mixture N2 (internal standard), CO2 and H2 (1:1:4), solid catalyst (0.008 mmol of metal, 5 mol% respect to CO2), 140 °C. Error bars account for a 5% uncertainty.
Complete hydrogenation of CO2 to CH4 was found for all catalysts, and neither carbon monoxide, formaldehyde or methanol were not observed in the gas phase, despite conversions are typically < 3%. A kinetic isotopic experiment with D2 shows the incorporation of all deuterium atoms in the final methane product (no scrambling with adsorbed water on the solid) and that the breaking of the H–H bond is participating in the limiting step of the hydrogenation reaction, since the kinetic isotopic effect (KIE) = 2.7 (Fig. S9).
Ag0@1 is easily recovered from the reaction mixture after simple evacuation of the gas reagents, since liquid products are not formed, and can be reused at least three times without severe depletion in the final yield of CH4 (Fig. S10). However, an abrupt decrease in catalytic activity occurred after the third use. Characterization of the five-times used solid catalyst by XRD (Fig. S11), XPS (Fig. S12) and high-angle annular dark-field scanning transmission electron microscopy (HAADF–STEM, Fig. S13) confirmed the stability of the Ag2 dimer inside the MOF. However, the FT-IR spectrum of this reused catalyst showed a new signal at 1770 cm−1, in the typical area for carbon monoxides (CO) species, characteristic of Agx(CO)x species (Fig. S14)32. Thus, we can ascribe the poisoning of the catalyst to the formation of CO during reaction, which does not completely evolve to the gas phase but may stay stuck to the Ag0 species. The fact that the hydrogenation reaction stops after formation of these Agx(CO)x species, together with the robustness of the Ag0@1 during reaction, the inactivity of the Ag nanoparticles (Ag–Al2O3) and the lack of any induction time in the kinetic profile, strongly supports that the Ag species in Ag0@1 are the truly catalytic active species for the methanation reaction. We placed the recovered catalyst, having the adsorbed CO molecules, under an atmosphere of H2, to check if the CO molecules could react, and the analysis of the reaction by GC–MS showed the formation of methane, confirming that the reverse water gas-shift reaction (RWGS) also occurs.
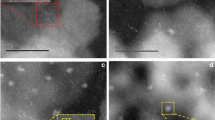
Our previously reported Ag2-MOF catalyst23, much harder to prepare and without thioether moieties to further stabilize Ag2, does not show any catalytic activity for the methanation reaction (see Fig. 4 above), which supports the role of thioether MOF 1 not only for an easy preparation but also for a better catalytic activity and stability of the metal dimer. To check this hypothesis, the analysis of the inactive Ag2-MOF material after reaction was accomplished, by PXRD (Fig. S15) and also by HAADF–STEM (Fig. S16). The PXRD analysis (Fig. S15) shows the appearance of peaks at 38° and 44° in the used MOF, which correspond to the (111) and (200) crystallographic planes of Ag NPs. The HAADF–STEM images clearly show the formation of Ag NPs, some of them > 10 nm, confirmed by the corresponding mapping. Notice that Ag NPs are catalytically inactive under our reaction conditions according to the lack of catalytic activity of Ag NPs on alumina (see Fig. 4). These results, together, strongly support the instability of the Ag dimers in the absence of Ag–S interactions within the MOF (compare the new results with the stable Ag0@1 MOF after reaction, Figs. S11–S14), in other words, the Ag–S bond makes the Ag dimers to be more stable during the methanation reaction.
To further corroborate these conclusions, we performed periodic DFT calculations about the stability of the different Ag species on both Ag+@1 and Ag0@1 materials. The results (Figs. S17–S18 and Table S4) show that the isolated Ag1 atoms, both in the channels and in the interstitial regions, interact with two S at ⁓ 2.4–2.5 Å, in agreement with the experimental characterization, and that this interaction leaves a net positive charge of < 0.5e on the Ag1 atoms. Ag2 species in the interstitial region are not stable and break to interact with available S atoms in the surroundings, to have again each Ag atom interacting with two S atoms at ⁓ 2.4–2.5 Å and with a net positive charge of ⁓ 0.5e. In contrast, Ag2 dimers are stable in the large channel, with an optimized Ag–Ag distance of ⁓ 2.89 Å and net positive charges of only 0.18 and 0.38e. One of the Ag atoms is interacting with two S atoms at 2.5 and 2.6 Å, and the second Ag atom is only bonded to one S at 2.6 Å, in agreement with the experimental characterization. All these results indicate that the Ag dimers in the channel are energetically stable and that the interstitial Ag atoms prefer to be in single cationic form. The new computational results in combination with the experimental results confirm that the Ag cations located in the less accessible pores (i.e. interstitial) are not catalytically active, since these interstitial Ag atoms are similarly present and stable in both the active Ag@1 and inactive Ag2-MOF, but only the former catalyzes the methanation reaction. Thus, by substration, the interstitial Ag atoms should not be active for the reaction. Overall, these results reflect the high sensitiveness of the catalysed reaction to the metal atomicity, in this case to just a metal dimer, in line with classical postulates for this type of metal catalysis33,34.
Conclusions
Ag2(0) dimers are prepared and stabilized in methioMOF 1, and act as a solid catalyst for the methanation reaction of CO2. Selectivity towards CH4 is complete under mild reaction conditions, and the new solid catalyst can be easily recovered and reused three times without severe catalytic depletion. These results open the way to employ these ultrasmall Ag clusters as catalysts in hydrogenation reactions. The findings here shown constitute a new example of how MOFs can host metal species otherwise unstable and difficult to synthesize, to be efficiently used in catalysis.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Wang, N., Sun, Q. & Yu, J. Ultrasmall metal nanoparticles confined within crystalline nanoporous materials: A fascinating class of nanocatalysts. Adv. Mater. 31, 1803966 (2019).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Mitchell, S. et al. Nanoscale engineering of catalytic materials for sustainable technologies. Nat. Nanotechnol. 16, 129–139 (2021).
Viciano-Chumillas, M. et al. Metal-organic frameworks as chemical nanoreactors: Synthesis and stabilization of catalytically active metal species in confined spaces. Acc. Chem. Res. 53, 520–531 (2020).
Ye, C. et al. Single atomic site catalysts: Synthesis, characterization, and applications. Chem. Commun. 56, 7687–7697 (2020).
Leyva-Pérez, A. Sub-nanometre metal clusters for catalytic carbon–carbon and carbon-heteroatom cross-coupling reactions. Dalton Trans. 46, 15987–15990 (2017).
Oliver-Meseguer, J., Cabrero-Antonino, J. R., Dominguez, I., Leyva-Perez, A. & Corma, A. Small gold clusters formed in solution give reaction turnover numbers of 10(7) at room temperature. Science 338, 1452–1455 (2012).
Fortea-Perez, F. R. et al. The MOF-driven synthesis of supported palladium clusters with catalytic activity for carbine-mediated chemistry. Nat. Mater. 16, 760–766 (2017).
Rivero-Crespo, M. et al. Cyclic metal(oid) clusters control platinum catalysed hydrosilylation reactions: From soluble to zeolite and MOF catalysts. Chem. Sci. 11, 8113–8124 (2020).
Stamplecoskie, K. G. & Scaiano, J. C. Kinetics of the formation of silver dimers: Early stages in the formation of silver nanoparticles. J. Am. Chem. Soc. 133, 3913–3920 (2011).
Schulze, W., Rabin, I. & Ertl, G. Formation of light-emitting Ag and Ag species in the course of condensation of Ag atoms with Ar. ChemPhysChem 5, 403–407 (2004).
Buceta, D. et al. Ag and Ag clusters: Synthesis, characterization, and interaction with DNA. Angew. Chem. Int. Ed. 54, 7612–7616 (2015).
Vajda, S. & White, M. G. Catalysis applications of size-selected cluster deposition. ACS Catal. 5, 7152–7176 (2015).
Socaciu-Siebert, L. D. et al. Ultrafast nuclear dynamics induced by photodetachment of Ag2 and Ag2O2: Oxygen desorption from a molecular silver surface. Phys. Chem. Chem. Phys. 7, 2706–2709 (2005).
Fedrigo, S., Harbich, W. & Buttet, J. Optical response of silver dimer, silver trimer, gold dimer, and gold trimer in argon matrixes. J. Chem. Phys. 99, 5712–5717 (1993).
Gomez, R. S. et al. Zeolite-supported silver and silver–iron nanoclusters and their activities as photodecomposition catalysts. Res. Chem. Intermed. 37, 729–745 (2011).
Freund, R. et al. The current status of MOF and COF applications. Angew. Chem. Int. Ed. 60, 23975–24001 (2021).
Zhang, X. et al. Design and applications of water-stable metal–organic frameworks: Status and challenges. Coord. Chem. Rev. 423, 213507 (2020).
Kondo, M. et al. Three-dimensional framework with channeling cavities for small molecules: {[M2(4, 4′-bpy)3(NO3)4]·xH2O}n (M = Co, Ni, Zn). Angew. Chem. Int. Ed. 36, 1725–1727 (1997).
Mon, M. et al. Crystallographic snapshots of host–guest interactions in drugs@metal–organic frameworks: Towards mimicking molecular recognition processes. Mater. Horiz. 5, 683–690 (2018).
Young, R. J. et al. Isolating reactive metal-based species in metal–organic frameworks—viable strategies and opportunities. Chem. Sci. 11, 4031–4050 (2020).
Escamilla, P. et al. Metal–organic frameworks as chemical nanoreactors for the preparation of catalytically active metal compounds. Chem. Commun. 59, 836–851 (2023).
Tiburcio, E. et al. Highly efficient MOF-driven silver subnanometer clusters for the catalytic Buchner ring expansion reaction. Inorg. Chem. 61, 11796–11802 (2022).
Mon, M. et al. Selective gold recovery and catalysis in a highly flexible methionine-decorated metal–organic framework. J. Am. Chem. Soc. 138, 7864–7867 (2016).
Zhuang, P. et al. Silver nanoclusters encapsulated into metal–organic frameworks for rapid removal of heavy metal ions from water. Molecules 24, 2442 (2019).
Ghaib, K., Nitz, K. & Ben-Fares, F. Z. Chemical methanation of CO2: A review. ChemBioEng Rev. 3, 266–275 (2016).
Mon, M. et al. Synthesis of densely packaged, ultrasmall Pt(0)2 clusters within a thioether-functionalized mof: Catalytic activity in industrial reactions at low temperature. Angew. Chem. Int. Ed. 57, 6186–6191 (2018).
Mon, M. et al. Selective and efficient removal of mercury from aqueous media with the highly flexible arms of a bioMOF. Angew. Chem. Int. Ed. 55, 11167–11172 (2016).
Firet, N. J. et al. Operando EXAFS study reveals presence of oxygen in oxide-derived silver catalysts for electrochemical CO2 reduction. J. Mater. Chem. A 7, 2597–2607 (2019).
Steinlechner, C. & Junge, H. Renewable methane generation from carbon dioxide and sunlight. Angew. Chem. Int. Ed. 57, 44–45 (2018).
Moon, W. K. et al. Guidelines to prepare active, selective, and stable supported metal catalysts for CO2 methanation with hydrogen. J. Catal. 413, 221–238 (2022).
Jiang, L. & Xu, Q. Infrared spectra of the (AgCO)2 and AgnCO (n = 2–4) molecules in rare-gas matrices. J. Phys. Chem. A 110, 11488–11493 (2006).
Boudart, M. Catalysis by supported metals. Adv. Catal. 89, 153–166 (1969).
Somorjai, G. A. & Carrazza, J. Structure sensitivity of catalytic reactions. Ind. Eng. Chem. Fundam. 25, 63–69 (1986).
Acknowledgements
Financial support by the Spanish Ministry of Science and Innovation (CEX2021-001230-S grant funded by MCIN/AEI/10.13039/501100011033) is gratefully acknowledged. This work was also supported by the MICIIN (Spain) through projects PID2020-115100GB-I00, PID2019-104778GB-I00 and “Maria de Maeztu” CEX2019-000919-M. The Ministero dell’Università e della Ricerca (Italy) and the Generalitat Valenciana (Project PROMETEO/2021/054) are also acknowledged. This study forms part of the Advanced Materials programme (MFA/2022/048) and was supported by MCIN with funding from European Union NextGenerationEU (PRTR–C17.I1) and by Generalitat Valenciana. Thanks are also extended to the Ramon y Cajal Program (RYC2019-027940-I, J. F.-S). M.M. thanks MICIIN from a contract under the Juan de la Cierva program (FJC2019-040523-I). E.P. acknowledges the financial support of the European Research Council under the European Union's Horizon 2020 research and innovation programme/ERC Grant Agreement No 814804, MOF-reactors. Y.Z. thanks to the China Scholarship Council (CSC No: 202009350009) for a Ph.D. fellowship.
Author information
Authors and Affiliations
Contributions
Y.Z. carried out the catalytic experiments, N.M. prepared the MOFs, M.B. performed and interpreted the computational calculations, J.F.-S. and M.M. supervised the experimental work and interpreted the results, D.A. carried out the XRD analyses and interpreted the results, E.P. and A.L.-P. conceived the idea, supervised the whole work and wrote the manuscript. All authors have revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, Y., Martín, N., Boronat, M. et al. Ag2(0) dimers within a thioether-functionalized MOF catalyze the CO2 to CH4 hydrogenation reaction. Sci Rep 13, 10376 (2023). https://doi.org/10.1038/s41598-023-37600-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37600-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
