
Abstract
Understanding the main steps involved in the activation of passive metals is an extremely important subject in the mechanical and energy industry and generally in surface science. The titanium-H2SO4 system is particularly useful for this purpose, as the metal can either passivate or corrode depending on potential. Although several studies tried to hypothesise the surface state of the electrode, there is no general consensus about the surface state of Ti in the active–passive transition region. Here by combining in-situ atomic force microscopy (AFM) and Raman spectroscopy, operating in an electrochemical cell, we show that the cathodic electrification of Ti electrodes causes the dissolution of the upper TiO2 portion of the passive film leaving the electrode covered by only a thin layer of titanium monoxide. Fast anodic reactions involved the acidification of the solution and accumulation of sulphur containing anions. This produces a local increase of the solution turbidity, allowing to distinguish favourable regions for the precipitation of TiOSO4·2H2O. These results give a clear answer to the long-stated question of the physical origin behind the formation of negative polarization resistances, sometimes occurring in corroding systems, and a rationale about the proton-induced degradation of passive surfaces in presence of sulphur containing species.
Similar content being viewed by others


Introduction
A physical–chemical system constituted by an activating electrode, like titanium Gr. 2 immersed in sulphuric acid, offers the opportunity of studying an electrochemical interface characterised by spatial inhomogeneities and time dependent reactions responsible of multiple steady states. The importance of studying the dissolution process of Ti in sulphuric acid was highlighted in the previous works1,2,3,4,5,6, regarding the complex electrochemical interface developing over the metal, as a result of the multitude possible valence states assumed by the metal cation and the formation of complexes from the coupling of Ti cations and S bearing anions. Titanium continues to deserve attention7 as it is a strategic metal particularly employed, for example, in the aerospace industry where it comes in contact with sulphuric acid when airplanes flight in the troposphere and stratosphere8. This causes de-passivation to occur upon the reduction of its oxide layer, a process still far to be completely understood9,10. Here the attack can be very dangerous when the metal is employed, for example, in the construction of fan blades for military engines. This can severely affect the creep resistance and the components capability to sustain the high load condition. Therefore, understanding the main steps leading to titanium activation can be of interest to put the basis for future mitigation strategies involving alloying or surface coatings11,12. This could be done exploiting specific in-situ analysis combining topographic and surface spectroscopic acquisitions while controlling the electrochemical reactions by a potentiostat. For example, reduced titanium oxide phases, developed upon cathodization, are quite sensitive to atmospheric oxygen so that their investigation should involve the protecting effect of a strong reducing environment like concentrated sulphuric acid, to preserve their stoichiometry. Following this idea, the reader is advised to compare the Raman spectrum, collected in deionised water, (Supplementary Fig. 1) of the air exposed concentration cell with the in-situ analysis of Fig. 3f–k.
A Ti electrode, immersed inside a reducing acidic solution, allows the corrosion potential (Ecorr) to fall in correspondence of values around − 300 mV/SCE (Saturated Calomel Electrode), where the oxide is considered to be almost proton transparent4,13. From a phenomenological point of view, three tentative models have been developed to account for the surface state of the material during the active to passive transition, hypothesising (i) the formation of a monolayer of adsorbed species, (ii) the presence of a reduced oxide or (iii) an interface characterised by a mix of both species4. Here, we definitively prove that, in the transition region, the Ti surface is always characterised by a thin oxide mainly composed by TiO and located between passivity and the critical electrochemical potential (Ecrit.), confirming the validity of the second hypothesis previously advanced. In these regions, fast anodic kinetics followed by cations hydrolysis implies the local acidification of the electrode surface, resulting at least in the decrease of one unit in solution pH. According to electroneutrality, those regions are affected by high accumulation of sulphates and bisulphates forming a supersaturated concentration cell, having diameters of tens of micrometres that evolves with the process. Here, the involved chemical-electrochemical reactions lead to the precipitation of a film of TiOSO4·2H2O.
Methods
All the experiments are performed over 1 cm2 circular titanium Gr. 2 (UNS R50400) samples cut by metal shearing whose chemical composition is as follows: Fe (0.30%) + O (0.25%) + C (0.08%) + N (0.03%) + H (0.015%) + Ti (balance) supplied by RL3 S.r.l.. Samples are mechanically polished with silicon carbide papers and alumina particles to obtain a mirror like surface. Before the electrochemical experiments, samples are cleaned in ultrasound with acetone and then washed in deionised water. Titanium is always allowed to passivate for 24 h air exposure in order to saturate the oxide thickness9. According to the literature, a layer ~ 5 nm thick composed by a mix of 2 +, 3 + and 4 + oxidation states is expected9. All the electrochemical tests are performed using a Metrohm Autolab PGSTAT equipped with a FRA32M module for EIS using a 4 ml PTFE 3 electrode cell (ASTM G514) with a platinum wire used as a pseudo-reference and counter electrode. A pseudo-reference is preferred to avoid any solution contamination. Values are then converted with respect to a saturated silver-silver chloride (+ 0.197 V/SHE) reference electrode (SSCsat.). The experiment is repeated for a total of 10 times in an electrolytic solution of 40 %v/v H2SO4 (7.46 M, Merck 99.999%) at room temperature (21 °C). The impedances are evaluated at fixed potential values: at the corrosion potential in passive condition Ecorr (~ + 0.15 V/SSCsat.), at − 0.2 V/ SSCsat., at − 0.3 V/ SSCsat., below the conduction band (Ecb) edge − 0.4 V/ SSCsat., near the critical potential − 0.5 V/SSCsat. and in the active region − 0.6 V/SSCsat.. All the EIS acquisitions are performed after 30 min of stabilisation both in free corrosion and after the application of a potential, in a frequency window between 10−2 to 105 Hz, collecting 10 points per decade with a voltage amplitude, of the sinusoidal perturbation, of 10 mVrms considering no repetition for each data point acquired by the system. The procedure can be better visualised in Fig. 1 where a cyclic voltametric experiment, carried out with a scan rate of 0.001 V/s, presents the potential window and the kinetics of interest. In-situ electrochemical atomic force microscopy (EC-AFM) and in-situ Raman spectroscopy acquisitions are performed in correspondence of previously mentioned potential values. EC-AFM is performed using a commercial NTEGRA Spectra set-up (NT-MDT). Images are collected in-situ (acquisition time ~ 1000 s), in non-contact mode (υ0 ~ 130 kHz) by employing VIT P/IR tips (TipsNano). Raman spectroscopy is performed by employing an excitation laser source at 532 nm, having a power of 5 mW. Quantification of sulphate molarities of the uncontaminated solution is afforded considering pure solutions of sodium sulphate (see Supplementary Fig. 2 and Table 1 for details). Crystal structure is characterized by x-ray diffraction using a Philips PW3020 goniometer with Cu Kα1 radiation (1.54058 Å) in Bragg–Brentano geometry. X-ray photoelectron spectroscopic (XPS) experiments is performed using a non-monochromatized Mg-Kα x-ray source (hν = 1253.6 eV) and the photoelectron kinetic energy is measured by a 150 mm hemispherical analyser (PHOIBOS150) from SPECS15. Core level regions are acquired with pass energy of 20 eV, with energy resolution of 1 eV. The base pressure of the vacuum chamber during XPS experiments was \(3\cdot {10}^{-10} \mathrm{Torr}\) and the sample is transferred from the EC cell to the vacuum system in Ar atmosphere to prevent surface contamination and reoxidation by air exposure. Considering that a reduction of reflectivity (surface roughness) decreases the signal-to-noise ratio and precludes the acquisition of significant data, all the in-situ analysis are carried out only along the descendent potential sweep before the activation of the electrode.

Topographic and electrochemical investigation. (a) image acquired at Ecorr. (b) image acquired at − 0.4 V/SSCsat.. (c) image acquired at − 0.5 V/SSCsat.. (d) Nyquist representation of impedance data evaluated at: Ecorr, − 0.2 V/SSCsat. and − 0.3 V/SSCsat.. (e) Nyquist representation of impedance data evaluated at: − 0.4 V/SSCsat., − 0.5 V/SSCsat. and − 0.6 V/SSCsat. (f) and (g) cyclic voltametric experiment performed on Ti Gr. 2 immersed in 40 %v/v H2SO4.
Results and discussion
Proton-induced degradation of the upper TiO2 layer
Once a cathodic potential is applied to the electrode, the Fermi level of the oxide, covering the metal, is raised favouring the reduction of protons, the flow of a cathodic current and degradation of the upper layers. Figure 1a–c, which explore an area of (10 × 10) µm2, reports the topographical evolution of the electrode surface as a consequence of the modification induced by the cathodic sweep applied to the polycrystalline Ti substrate. While AFM images at Ecorr (Fig. 1a) were acquired immediately, ~ 2.5 h are required to induce the surface modifications seen in Fig. 1b and ~ 4 h for the ones of Fig. 1c. The reader is referred to Supplementary Fig. 3 for comparing the nanometric modification seen in Fig. 1a–c and Supplementary Fig. 4, induced by the corrosion process, with the topography of the pristine sample. In Fig. 1a, it is possible to see that, once the sample is in free corrosion and up to potentials above Ecb, the pristine morphology is preserved: longitudinal ripples are consequence of the surface mechanical preparation. This result is in agreement with another study16, which demonstrates that dissolution initially occurs between grains and evolves in the formation of multitude local peaks as highlighted in Fig. 1b. The cathodic wave observed in the insert of Fig. 1g near − 0.18 V/SSCsat.17 is assigned to the redox couple Ti4+/Ti3+ while the one at − 0.3 V/SSCsat.18 to the flat band potential of TiO2. Those waves prove that proton insertion occurred also above Ecb (~ − 0.34 V/SSCsat.19,20,21,22), resulting in the substantial degradation of the upper TiO2 layer. An additional confirmation comes from in-situ Raman spectroscopy analysis: (1) the rutile 2nd order feature at ~ 230 cm−1 of Fig. 2a progressively redshifts and decreases in the integrated signal intensity; (2) a new peak at 218 cm−1 (Fig. 2b) appears and it is attributed to the H3O+ insertion inside the octahedral arrangement of the Ti and O ions23,24; (3) the Eg(symmetric stretching)-rutile (444 cm−1) and A1g(anti-symmetric bending)-rutile (614 cm−1) modes disappears (see Supplementary Fig. 5 and Table 2) and (4) a peak rises at 271 cm−1 (Fig. 2b). The latter could be related to the Eg of Ti2O3 or to the vibrational mode, as found by Tao et al.25,26, occurring upon proton insertion in titanate structures.

Low frequency Raman spectroscopy analysis. (a) Raman spectrum acquired at Ecorr. (b) Raman spectrum acquired at − 0.4 V/SSCsat.. (c) Raman spectrum acquired at − 0.5 V/SSCsat.. (d) Raman spectrum acquired at − 0.6 V/SSCsat..
Apart from oxide related lines, the spectra highlighted in Fig. 2 are characterised by the presence of peaks belonging to vibrational modes related to H bonds present among H2O molecules, for further details the reader should refer to Table 3. Knowledge about those features is gained by acquiring Raman spectra during cathodization over a pure Au substrate in the tested solution (see Supplementary Figs. 6, 7 and Table 4). Oxide reduction is confirmed by the sudden decrease of the arc diameter of Fig. 1d (see also Supplementary Fig. 8), indicative of the charge transfer resistance (Rct) that passes from 1.09·106 Ω·cm2 at Ecorr to 8.46·104 Ω·cm2 at − 0.2 V/SSCsat..
Consequently, the capacitance of the system27 increases from 13.9 µF/cm2 to 16.8 µF/cm2, denoting charge accumulation in the space charge region at the semiconductor-electrolyte interface. The above process description can be summarised in Eq. (1), which describes the process of proton insertion coupled by electron transfer.
With the increase of the charge transfer reaction kinetics, the system becomes partially controlled by mass transport28. For fitting purpose, of the impedance spectra, semi-infinite diffusion is considered (Supplementary Fig. 9 and Table 5 collecting fitting result). Given the high acidity it is reasonable to assume mass transport to be mainly related to solid state diffusion of protons inside the TiO2 lattice21 obtaining diffusivities29,30 (Eq. (1) in Supplementary material) equal to \({D}_{{H}_{3}{O}^{+}}^{-0.2 V/SSCsat.}=3.10\cdot {10}^{-18} \frac{{cm}^{2}}{s}\) and \({D}_{{H}_{3}{O}^{+}}^{-0.3 V/SSCsat.}=6.67\cdot {10}^{-18} \frac{{cm}^{2}}{s}\), entailing a proton penetration (Eq. (2) in Supplementary material) of 0.97 nm at − 0.2 V/SSCsat. and 2.01 nm at − 0.3 V/SSCsat. according to present polarization periods. This is purely speculative but we denote a certain similarity between diffusion length extracted by EIS and the Ra parameter obtained by EC-AFM, an hypothesis deserving further investigation in future researches. Having obtained titanium oxide hydrogenation, via an electrochemical route, this induces the formation of shallow donor defects, lying immediately below Ecb31. Under those circumstances the specie moves interstitially forming almost covalent bond with lattice O without interacting with O vacancies31.The oxide protectiveness vanishes below Ecb, where the current changes from cathodic to anodic, inducing substantial topological modifications (Fig. 1b).
This fact highlights that the kinetic of the process is dictated by the oxidation of the metal cation as \(Ti\to {Ti}^{3+}+3{e}^{-}\). In the literature, this is generally attributed to the fact that, below Ecb, the phase oxide ceases to exist leading to the formation of an adsorbed monolayer4. Conversely, our study demonstrates this assumption to be partially wrong, because a thin reduced oxide layer always covers Ti in the transition region.
In-situ surface analysis with an optical microscope (Fig. 3a–c) reveals the presence of fluorescent spots randomly distributed on the sample surface when the latter is polarised below − 0.4 V/SSCsat.. Colour centres over titanium oxide are generally the result of the local oxide reduction with formation of electronic defects. This local dissolution may arise from regions of high electronic conductivity32. Keeping this in mind and focusing on EIS (Fig. 3d and e), the Nyquist representation shows the occurrence of a low frequency capacitive loop with negative real impedances (negative curl). This apparently strange behaviour, also observed during anodic dissolution of iron in sulphuric acid33,34, does not violate the principle of linearity, stability and causality giving consistency to the EIS results35.

Investigation of the corrosion triggering mechanism. (a) Optical image of uncorroded sample. (b) optical image showing fluorescence due to oxide reduction. (c) Optical image showing formation of a concentration cell. (d) Nyquist plot collected in correspondence of image b. (e) Nyquist plot collected in correspondence of concentration cell formation. (f) low frequency Raman spectrum extracted in correspondence of the fluorescent region seen in image b. (g) 300–800 cm−1 Raman spectral portion collected inside the fluorescent region. (h) 800–1200 cm−1 Raman spectral portion collected inside the fluorescent region. (i) low frequency Raman spectrum extracted in correspondence of the concentration cell seen in image c. (j) 300–800 cm−1 Raman spectral portion collected inside the concentration cell seen in image c. (k) 800–1200 cm−1 Raman spectral portion collected inside the concentration cell seen in image c.
Here, we point out that the negative polarization resistance, in the Nyquist representation and in polarization plots is consequence of the enlargement of the fluorescent regions (FR), affected by the dissolution of the protective upper TiO2 layer (see below), causing a progressively increasing anodic current in opposition to the decreasing anodic potential. The phenomenon develops in a time scale compatible with the low frequency spectrum mainly affecting the zero frequency value of the impedance, i.e., the polarisation resistance. Raman analysis localised on FR (Fig. 3f–k) shows the presence of clear components at 187 (Fig. 3f), 215 (Fig. 3f) and 603 cm−1 (Fig. 3g), whose positions and reciprocal intensities perfectly agree with the TiO reference spectrum36. The TiO2 upper layer is thus locally degraded, leaving the lower unprotective TiO structure exposed to the solution.
Formation of the concentration cell
Previously highlighted steps result, at the end, in macroscopic surface modifications of the electrode. However, it will be shown that the opportunity offered by the present experimental apparatus is to capture the very first instant of formation of local phenomena responsible for the metal activation. Fast anodic currents are expected to result in feeding those unprotected regions that are affected by the dissolution of the upper TiO2 layer. Since Ti3+ is a strong reducing agent, it is reasonable to assume this ion to be readily reactive with one coordinated water molecule to form Ti4+. As a result, Ti4+ ions can readily hydrolyse leading to local acidification37. This can be demonstrated, electrochemically, according to the cyclic voltammetry reported in Fig. 1f. Here the shift (~ 70 mV) of the anodic peak in the positive direction is consistent with the pH decrement of one unit4. This positive surface charge is compensated by an anion accumulation (SO42− and HSO4−) as seen by the enhancement of the Raman lines intensity in the spectral region above 800 cm−1 (see Fig. 4). The main features are assigned to the stretching mode of the bisulphate (ν1-HSO4− at ~ 1034 cm−1)38,39,40 and sulphate (at ~ 980 cm−1)41,42,43.

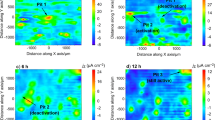
Analysis of the concentration cell by Raman spectroscopy. (a) Raman spectrum of the 800–1200 cm−1 region collected outside the concentration cell. (b) Raman spectrum of the 800–1200 cm−1 region collected at the border of the concentration cell. (c) Raman spectrum of the 800–1200 cm−1 region collected at the centre of the concentration cell. (d) Optical image collected in correspondence of a concentration cell delimited by the white dotted profile. (e) enlarged view of image d. (f) Raman map of the intensity of the 991 cm−1 component of the ML2 complex. (g) Raman map of the intensity of the 1029 cm−1 component of the HSO4−·H3O+.
The Raman shift found for the former line is indicative of protons pairing. This chemical state almost completely substitutes the free bisulphate ion inside the concentration cell, as confirmed by the disappearance of the ν4-HSO4− line found generally around 890 cm−1. Here, there is a particular increment in the solution turbidity, denoting the formation of a circular concentration cell as in Fig. 3c. In this regions, Raman features (Fig. 3i,j) resemble the spectra acquired over the electrode surface when the potential is swept below − 0.5 V/SSCsat. (Fig. 2c and d). At those cathodic potentials, the surface appears black coloured by the presence of a corrosion product. The latter can be characterised by AFM (Fig. 1c), which detects an increase of the Ra (14.2 nm). The increase of surface coverage by the corrosion product contributes to the pinch-off of the anodic current, resulting in positive Rp.
The deposit are characterised by photoemission experiments and x-ray diffraction (Supplementary Fig. 10) indicating TiOSO4·2H2O as the main constituent. The Raman lines at 192 cm−1 and 391 cm−1, collected inside the concentration cell and in regions covered by the black corrosion product, coincide with the vibrations of the TiO6 octahedra composing an oxy-sulphate crystal structure44. Moreover, depending on the hydration level of the sulphonated film doublet and triplet may arise from the ν2-SO42− and ν4-SO42− vibrational mode according to the symmetry lowering occurring when passing from the liquid to the solid state41.
Precipitation of the corrosion product
A detailed Raman spectroscopic investigation of the concentration cell and its neighbouring regions can favour the understating of the chemical-electrochemical steps leading to the precipitation of the corrosion product. Some authors45,46 found no precipitation of hydrated TiOSO4 until the concentration of H2SO4 reached 65 wt.%, quite far from the nominal concentration employed in the present work, confirming the suitability of the model involving the formation of the concentration cells. There is further evidence of a lower local pH, triggering concentration cell formation, according to the redshifts (1029 cm−1, Supplementary Table 6) of the HSO4−·H3O+ stretching line, resulting from paring with protons47,48. Raman quantification process indicates a bulk solution characterised by 1.23 M of sulphates and 6.23 M of bisulphates according to literature49. The speciation of previous anions with Ti4+ considerably modified previous numbers, creating favourable conditions for corrosion product deposition. In particular, inside the concentration cell there is evidence of formation of two Ti IV-sulphates complexes, found in literature to be bidentate (ML2—991 cm−1) and unidentate systems (ML1—1010 cm−1)38,39,50. This is the result of their considerably higher stability constant found for sulphate complexes with respect to bisulphate systems37,38,51,52. The larger integrated intensity found for the ML2 complex, inside the concentration cell, allows to hypothesise the establishment of a larger sulphate to Ti IV concentration ratio (Fig. 4d). We have found substantial changes in the integrated intensity of peaks related to the main ions checking different regions outside and inside the concentration cell. In particular, at the periphery of the concentration cell, sulphate species start to complex with Ti4+ with a progressively increasing trend as the centre of the concentration cell is approached. Analysis of the stretching vibration of bisulphates allows to see that the overall integrated intensity is consistently reduced according to the strong decrease of peak width at the full width at half maximum (FWHM) (from ~ 46 cm−1 outside the concentration cell to ~ 15 cm−1 inside the latter). In these regions, the strong modifications seen at the previous bisulphate ion line are assumed to be related to the interaction with the electrode surface, according to the electron transfer reaction described by Eq. (2). This is capable to explain the bisulphate concentration decrement inside the concentration cell. This reaction is energetically favoured for potentials more cathodic than 0.16 V/SHE in acidic solutions.
This contributes to the accumulation of SO2 (see Raman peak at ~ 1180 cm−1 inside the concentration cell). According to Eq. (3), SO2 may react with water and give SO42−, thus providing the excess of sulphate species necessary to complex with Ti4+.
This mechanism gives a rationale of the corrosion products precipitation and the formation of a deposit according to a polymerisation reaction46. In conclusion, it is demonstrated how the activation of a Ti Gr. 2 electrode starts in local weak spots where the dissolution of the upper TiO2 layer results in the formation of fluorescent regions characterised by a greenish hue. Here, the fast anodic kinetic is responsible of the local acidification resulting from cations hydrolysis. Consequent charge compensation involves the migration of S containing anions, entailing the formation of growing concentration cells visually distinguished from the background according to an enhanced solution turbidity. Reaction between Ti4+ ions and SO42− implies the formation of unidentate and bidentate complexes which, according to a polymerisation reaction, promote the precipitation of a corrosion product. This in-situ investigation confirms those regions to be the preferential zones affected by the precipitation of TiOSO4·2H2O, demonstrating Ti to be always covered by a titanium monoxide layer surmounted by an oxy-sulphate corrosion product before complete activation of the electrode is reached.
Data availability
The datasets generated and/or analysed during the current study are available in the Supplementary material.
References
Armstrong, R. D., Harrison, J. A., Thirsk, H. R. & Whitfield, R. The anodic dissolution of titanium in sulfuric acid. J. Electrochem. Soc. 117, 1003 (1970).
Caprani, A. & Frayret, J. P. Steady state anodic behaviour of titanium in concentrated hydrochloric acid. J. Less-Common Met. 69, 29–36 (1980).
Thomas, N. T. & Nobe, K. Kinetics of the hydrogen evolution reaction on titanium. J. Electrochem. Soc. 117, 622–626 (1970).
Kelly, E. J. Electrochemical behaviour of titanium. Mod. Asp. Electrochem. 14, 557–639 (1982).
Liu, J., Alfantazi, A. & Asselin, E. The anodic passivity of titanium in mixed sulfate-chloride solutions. J. Electrochem. Soc. https://doi.org/10.1149/2.0451512jes (2015).
Vaughan, J. & Alfantazi, A. Corrosion of titanium and its alloys in sulfuric acid in the presence of chlorides. J. Electrochem. Soc. 153, B6 (2005).
Carrozza, A. et al. Materials characterization an investigation on the effect of different multi-step heat treatments on the microstructure, texture and mechanical properties of the DED-produced Ti-6Al-4V alloy. Mater. Charact. 189, 111958 (2022).
Yacovitch, T. I. et al. Infrared spectroscopy of hydrated bisulfate anion clusters: HSO 4-(H2O)1–16. J. Phys. Chem. Lett. 2, 2135–2140 (2011).
Zhang, Y. et al. Mitigation of the surface oxidation of titanium by hydrogen. J. Phys. Chem. C 122, 20691–20700 (2018).
Lu, G., Bernasek, S. L. & Schwartz, J. Oxidation of a polycrystalline titanium surface by oxygen and water. Surf. Sci. 458, 80–90 (2000).
Casanova, L. et al. A nanoscale investigation on the influence of anodization parameters during plasma electrolytic oxidation of titanium by high-resolution electron energy loss spectroscopy. Appl. Surf. Sci. 570, 151133 (2021).
Casanova, L., Belotti, N., Pedeferri, M. P. & Ormellese, M. Sealing of porous titanium oxides produced by plasma electrolytic oxidation. Mater. Corros. https://doi.org/10.1002/maco.202112612 (2021).
Vezvaie, M., Noël, J. J., Tun, Z. & Shoesmith, D. W. Hydrogen absorption into titanium under cathodic polarization: An in-situ neutron reflectometry and EIS study. J. Electrochem. Soc. 160, C414–C422 (2013).
ASTM G5 Standard Reference Test Method for Making Potentiodynamic Anodic Polarization Measurements. Annu. B. ASTM Stand. (2014). doi:https://doi.org/10.1520/G0005-13E02.2
Berti, G. et al. Direct observation of spin-resolved full and empty electron states in ferromagnetic surfaces. Rev. Sci. Instrum. 85, 5–9 (2014).
Brown, G. M., Thundat, T., Allison, D. P. & Warmack, R. J. Electrochemical and in situ atomic force microscopy and scanning tunneling microscopy investigations of titanium in oxalic acid solution. J. Vac. Sci. Technol. A 10, 3001–3006 (1992).
Pourbaix, M. Atlas of electrochemical equilibria in aqueous solutions. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry (1974).
Latimer, W. M. The oxidation states of the elements and their potentials in aqueous solutions. Soil Sci. https://doi.org/10.1097/00010694-193910000-00009 (1939).
Casanova, L. et al. Influence of stoichiometry on the corrosion response of titanium oxide coatings produced by plasma electrolytic oxidation. Corros. Sci. 203, 110361 (2022).
Makivić, N. et al. Evidence of bulk proton insertion in nanostructured anatase and amorphous TiO2 electrodes. Chem. Mater. 33, 3436–3448 (2021).
Kim, Y. S. et al. Evidencing fast, massive, and reversible H+ insertion in nanostructured TiO2 electrodes at neutral pH. Where do protons come from?. J. Phys. Chem. C 121, 10325–10335 (2017).
Lyon, L. A. & Hupp, J. T. Energetics of the nanocrystalline titanium dioxide/aqueous solution interface: Approximate conduction band edge variations between H0 = -10 and H- = +26. J. Phys. Chem. B 103, 4623–4628 (1999).
Denisova, T. A. et al. Metatitanic acid: Synthesis and properties. Russ. J. Inorg. Chem. 51, 691–699 (2006).
Beuvier, T., Richard-Plouet, M. & Brohan, L. Ternary morphological diagram for nano(tube-ribbon-sphere) sodium titanate deduced from Raman spectra analysis. J. Phys. Chem. C 114, 7660–7665 (2010).
Tao, G., Fjellvåg, H. & Norby, P. Raman scattering properties of a protonic titanate HxTi 2–x/4□x/4O4·H2O (□, vacancy; x = 0.7) with lepidocrocite-type layered structure. J. Phys. Chem. B 112, 9400–9405 (2008).
Kukovecz, Á., Hodos, M., Kónya, Z. & Kiricsi, I. Complex-assisted one-step synthesis of ion-exchangeable titanate nanotubes decorated with CdS nanoparticles. Chem. Phys. Lett. 411, 445–449 (2005).
Brug, G. J., Van den Eeden, A. L. G., Sluyters-Rehbach, M. & Sluyters, J. H. The analysis of electrode impedances complicated by the presence of a constant phase element. J. Electroanal. Chem. 176, 275–295 (1984).
Lasia, A. Electrochemical impedance spectroscopy and its application. Impedance Spectrosc. Appl. https://doi.org/10.1007/978-1-4614-8933-7 (2014).
Pound, B. G. Hydrogen ingress in titanium. Corrosion 47, 99–104 (1991).
Qin, Z., Zeng, Y. & Shoesmith, D. W. Modeling hydrogen permeation through a thin titanium oxide film and palladium. Thin Solid Films https://doi.org/10.1016/j.tsf.2013.02.030 (2013).
Mo, L. B. et al. Hydrogen impurity defects in rutile TiO2. Sci. Rep. 5, 0–7 (2015).
Boxley, C. J., White, H. S., Gardner, C. E. & Macpherson, J. V. Nanoscale imaging of the electronic conductivity of the native oxide film on titanium using conducting atomic force microscopy. J. Phys. Chem. B 107, 9677–9680 (2003).
Keddam, M., Mottos, O. R. & Takenouti, H. Reaction model for iron dissolution studied by electrode impedance: I. Experimental results and reaction model. J. Electrochem. Soc. 128, 257–266 (1981).
Keddam, M., Mattos, O. R. & Takenouti, H. Reaction model for iron dissolution studied by electrode impedance: II. Determination of the reaction model. J. Electrochem. Soc. 128, 266–274 (1981).
Vivier, V. & Orazem, M. E. Impedance analysis of electrochemical systems. Chem. Rev. 122, 11131–11168 (2022).
Hasegawa, G. et al. Hierarchically porous monoliths based on N-doped reduced titanium oxides and their electric and electrochemical properties. Chem. Mater. 25, 3504–3512 (2013).
Liberti, A., Chiantella, V. & Corigliano, F. Mononuclear hydrolysis of titanium (IV) from partition equilibria. J. Inorg. Nucl. Chem. https://doi.org/10.1016/0022-1902(63)80192-X (1963).
Baillon, F., Provost, E. & Fürst, W. Study of titanium(IV) speciation in sulphuric acid solutions by FT-Raman spectrometry. J. Mol. Liq. 143, 8–12 (2008).
Szilágyi, I., Königsberger, E. & May, P. M. Characterization of chemical speciation of titanyl sulfate solutions for production of titanium dioxide precipitates. Inorg. Chem. 48, 2200–2204 (2009).
Turner, D. J. Raman spectral study of bisulphate ion hydration. J. Chem. Soc. Faraday Trans. 2 68, 643–648 (1972).
Ben Mabrouk, K., Kauffmann, T. H., Aroui, H. & Fontana, M. D. Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions. J. Raman Spectrosc. 44, 1603–1608 (2013).
Chen, H. & Irish, D. E. A raman spectral study of bisulfate-sulfate systems. III. Salt effects. J. Phys. Chem. 75, 2681–2684 (1971).
Sharma, S. K. et al. Standoff Raman spectroscopy for future Europa Lander missions. J. Raman Spectrosc. 51, 1782–1793 (2020).
Kiyomi, L., Ronil, F. & Sanches, N. Titanyl sulphate, an inorganic polymer: Structural studies and vibrational assignment. Quim. Nov. 42, 1112–1115 (2019).
Roche, E. G., Prasad, J., Philip E., G. & Stuart, A. D. Patents A sulfate process EP2064356A1. 1–33 (2007).
Ahmed, M. A. K., Fjellvag, H. & Kjejshus, A. Syntheses and crystal structures of titanium oxide sulfates. Acta Chem. Scand. 50, 275–283 (1996).
Lund Myhre, C. E., Christensen, D. H., Nicolaisen, F. M. & Nielsen, C. J. Spectroscopic study of aqueous H2SO4 at different temperatures and compositions: Variations in dissociation and optical properties. J. Phys. Chem. A 107, 1979–1991 (2003).
Clegg, S. L. et al. Comment on the ‘thermodynamic dissociation constant of the bisulfate ion from Raman and ion interaction modeling studies of aqueous sulfuric acid at low temperatures’. J. Phys. Chem. A 109, 2703–2709 (2005).
Casas, J. M., Alvarez, F. & Cifuentes, L. Aqueous speciation of sulfuric acid-cupric sulfate solutions. Chem. Eng. Sci. 55, 6223–6234 (2000).
Hefter, G. When spectroscopy fails: The measurement of ion pairing. Pure Appl. Chem. 78, 1571–1586 (2006).
Pichavant, A., Provost, E., Fürst, W. & Hochepied, J. F. Determination of the temperature dependence of Titanium(IV) hydrolysis and complexation constants in aqueous sulfuric or chlorhydric solutions. J. Chem. Thermodyn. 131, 184–191 (2019).
Baes, C. F. & Mesmer, R. E. The Hydrolysis of Cations by Charles Frederick Baes, Robert Eugene Messmer. 489 (1976).
Acknowledgements
This in-situ study was carried out at Solinano-Sigma, an interdepartmental facility of Politecnico di Milano. The authors thank M. Tommasini and A. Luccotti for fruitful discussions and advice on Raman spectroscopy.
Author information
Authors and Affiliations
Contributions
L.C.: conceptualisation, experimental design, data collection, editing, writing. M.M.: conceptualisation, experimental design, data collection, editing, writing. F.G.: conceptualisation, experimental design, data collection, editing, writing. M.P.: editing, writing. L.D.: editing, writing. M.O.: editing, writing. G.B.: conceptualisation, experimental design, data collection, editing, writing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Casanova, L., Menegazzo, M., Goto, F. et al. Investigating the activation of passive metals by a combined in-situ AFM and Raman spectroscopy system: a focus on titanium. Sci Rep 13, 6117 (2023). https://doi.org/10.1038/s41598-023-33273-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33273-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
