
Abstract
Dracaena (Asparagaceae family) tree is famous for producing "dragon blood"—a bioactive red-colored resin. Despite its long history of use in traditional medicine, little knowledge exists on the genomic architecture, phylogenetic position, or evolution. Hence, in this study, we sequenced the whole chloroplast (cp) genomes of D. serrulata and D. cinnabari and performed comparative genomics of nine genomes of the genus Dracaena. The results showed that the genome sizes range from 155,055 (D. elliptica) to 155,449 (D. cochinchinensis). The cp genomes of D. serrulata and D. cinnabari encode 131 genes, each including 85 and 84 protein-coding genes, respectively. However, the D. hokouensis had the highest number of genes (133), with 85 protein coding genes. Similarly, about 80 and 82 repeats were identified in the cp genomes of D. serrulata and D. cinnabari, respectively, while the highest repeats (103) were detected in the cp genome of D. terniflora. The number of simple sequence repeats (SSRs) was 176 and 159 in D. serrulata and D. cinnabari cp genomes, respectively. Furthermore, the comparative analysis of complete cp genomes revealed high sequence similarity. However, some sequence divergences were observed in accD, matK, rpl16, rpoC2, and ycf1 genes and some intergenic spacers. The phylogenomic analysis revealed that D. serrulata and D. cinnabari form a monophyletic clade, sister to the remaining Dracaena species sampled in this study, with high bootstrap values. In conclusion, this study provides valuable genetic information for studying the evolutionary relationships and population genetics of Dracaena, which is threatened in its conservation status.
Similar content being viewed by others

Introduction
Dracaena is an important genus from the family Asparagaceae that includes wild and indoor exquisite plants1. The genus comprises 190 species2 and is also known as Dragon trees. These are distributed across the drylands in Africa, Arabia, and the Americas3. In response to incisions, these plants produce a red resin called “Dragon Blood” that is medicinally important and has an ancient history in traditional herbal medicine4. The resin has been known to act as an anti-cancer, hemostatic, anti-ulcer, anti-viral, anti-microbial, anti-inflammatory, and anti-oxidant5. Dracaena resin is also used for giving colors to certain materials like toothpaste, varnishes, and plasters6. The highest levels of species diversity occur in tropical Africa and Southeast Asia. These species grow in various habitats, including tropical monsoon, semi-evergreen, and evergreen rain forests. Some species grow in specialized habitats such as escarpments, littoral forest edges, and riverbeds with strongly fluctuating water levels, where they become facultative rheophytes7.
Among Dracaena species, D. serrulata and D. cinnabari (Fig. 1) are regional, endemic species found in southern Oman, Saudi Arabia, and Yemen (Socotra Island). These endangered species are currently threatened by mining operations, agriculture, drought, and possibly climate change. The known populations are threatened by grazing (camels, goats, and sheep) during the dry season8,9,10. Dracaena, along with other globally important genera Sansevieria Thunb and Pleomele Salisb (family Asparagaceae and Nolinoideae subfamily) are collectively referred to as ‘dracaenoids’. These have had various taxonomic and evolutionary unsolved problems since the eighteenth century11,12. The classification of these three genera was always unclear, and that's why these were shifted from one family to another like Agavaceae, Liliaceae13,14, Dracaenaceae7,15,16, Ruscaceae11, and lately in Asparagaceae17 over a period of times. Due to similar floral characters, Sansevieria and Dracaena were believed to be synonymous. However, their stature, leaf morphology, and plant habitats have distinct variations. Similarly, the dracaenoid genera have ambiguous systematic relationships, and extensive evolutionary history and biogeographic studies are needed18.

The dragon tree plants and their habitat. D. serrulata (A) and D. cinnabari (B).
The ambiguity is associated with Dracaena species identification and intra-generic relationships, including sub-species of D. serrulata2. According to Marrero et al.3 assessment based on morphological and ecological characteristics, the D. draco (Macaronesian species) would show closer affinities with the D. cinnabari (Socotran species) and D. tamaranae, which is located in the Horn of Africa, is more closely related to the D. serrulata (Arabian species). However, Durán et al.19 reported that based on barcoding genes (rbcL and matK) and intergenic spacers (trnQ-rps16 and rpl32-trnL), D. cinnabari (Socoteran) is more closely related to D. serrrulata (Omani species) than D. draco (Macaronesian). Genomic studies can resolve such species-relatedness, which is minimal for the genus Dracaena20. Next-generation sequencing combined with bioinformatic analysis can also help solve key taxonomic and genetic diversity issues21.
In this case, the chloroplast is one of the most important organelles and has its own independently replicating genome called chloroplast genome or plastome22. Chloroplast genome possesses a typical quadripartite structure having a large single-copy region (LSC), small single-copy region (SSC) and a pair of inverted repeats (IRa and IRb), which are mirror images of each other23. The chloroplast genome is highly conserved among typical land plants compared to other genomes present in the plant cell like mitochondria and nuclear genome24. Despite the conservative nature of the chloroplast genome, it still has variations like insertion and deletion and single nucleotide polymorphism, which provides sufficient information in plant identification25,26. Many chloroplast derived markers are used in the plant phylogenetic, population genetics, and phylogeographic analyses due to their low recombination, low nucleotide substitutions rate, and uniparental inheritance27. Many chloroplast genomes such as trnH-psbA, rbcL, and matK were commonly used as DNA barcodes for plant identification and discrimination of sub-species17.
In some cases, these barcodes cannot differentiate especially between the closely related species within Dracaenoid genera28, due to the little variation in the loci27,29. Complete chloroplast genome sequencing coupled with comparative analysis allows advanced phylogenetic reconstruction and can be used as super-barcodes to resolve identification at lower taxonomic levels27,30. Looking at these challenges, in the current study, we aim to sequence the D. serrulata and D. cinnabari and perform comparative chloroplast genome analysis to explain the basis of genome architecture and divergences across Dracaena species. Hence, we report the complete chloroplast genomes sequences of D. serrulata and D. cinnabari. Both the species and other species in the genus possess the least genomic information. Hence, current datasets will help understand the genome architecture, comparative genomics with related species, and in-depth phylogeny of Dracaena species.
Methodology
Sample collection
Fresh young leaves were collected from the D. serrulata and D. cinnabari plants growing in the Dhofar region (wild) of Oman. The habitat climate is arid with a low precipitation rate and temperate (25–46 °C). All plant specimens used for this study were collected from the wild to the best of our knowledge in compliance with local, institutional, national, or international regulations at the time of collection. A permission letter was retrieved from the Director-General of Nature Conservation, Ministry of Environment and Climate Affairs, Sultanate of Oman. The fresh specimens of D. cinnabari were donated by the Environmental Protection Authority Socotra, Yemen. The voucher specimen numbered UoN-DS1 (D. serrulata) and UoN-DC1 (D. cinnabari) were deposited in the University of Nizwa herbarium center. The identification of plants was carried out by Saif Al-Hathmi, an expert taxonomist at the Oman Botanical Garden in Muscat, Oman. The collected materials were transported in liquid nitrogen or dry ice and stored at − 80 °C for further processing.
DNA extraction and sequencing
With brief modifications, the cp DNA was isolated from collected samples as described in Shi et al.31. The construction of genomic libraries was carried out as per provided instructions (Life Technologies USA, Eugene, OR, USA). To arrange the cp DNA into 400 bp fragments (enzymatically) for libraries, the Ion Shear™ Plus Reagents kit and Ion Xpress™ Plus gDNA Fragment Library kit were used. Qubit 3.0 fluorometer and bioanalyzer (Agilent 2100 Bioanalyzer system, Life Technologies USA) were used to quantify the prepared libraries. The amplification of the template was performed using Ion OneTouch™ 2. The Ion OneTouchTM ES enrichment system enriched the amplified templates using Ion 530 and 520 OT2 reagents. Ion S5 protocol of sequencing was followed for loading the sample on S5 530 chip.
Genome assembly and annotation
The number of raw reads obtained for D. serrulata and D. cinnabari were 14,654,144 and 16,888,126, respectively. The reads were first screened for a Phred score < 30 to remove low-quality sequences. To ensure the accuracy of cp genome assembly, we employed two methods to assemble the cp genome. In the first method, obtained reads of cp genomes D. serrulata and D. cinnabari were mapped to the reference genome of D. cochinchinensis (MF943127) and D. combodiana (MN20094), respectively, by Geneious Pro (v.10.2.3) software using Bowtie2 (v.2.2.3)32,33. Assembly means coverage of D. serrulata was 876X, and D. cinnabari was 768X. In the second method, the cp genome of D. serrulata and D. cinnabari were de novo assembled using the GetOrganelle pipeline34, with SPAdes 3.10.1 assembler35. The cp genomes D. serrulata and D. cinnabari were annotated using CpGAVAS and DOGMA (http://dogma.ccbb.utexas.edu/, China)36. The tRNAs can-SE detected the tRNA genes (v.1.21)37. Intron boundaries, manual alteration and start and stop codon adjustments of genomes were carried out using Geneious Pro (v.10.2.3)33 and tRNAs can-SE37 by comparing the cp genomes to reference genomes. OGDRAW38 was utilized to illustrate the structural features in cp genomes.
Repeat identification
The determination of palindromic, forward and reverse repeats was performed using the online tool REPuter39 with 8 bp minimum repeat size and 50 maximum computed repeats. Furthermore, MISA software40 with conditions of ≥ 10 repeat units for 1 bp repeats; ≥ 8 repeat units for 2 bp repeats; ≥ 4 repeat units for 3 and 4 bp repeats and ≥ 3 repeat units for 5 and 6 bp repeats was used to calculate Simple sequence repeats (SSRs) and tandem repeats were calculated by Tandem Repeats Finder v.4.0941.
Genome divergence
The sequence divergence in shared genes and complete cp genomes of D. serrulata and D. cinnabari, and other closely related species were determined. Multiple sequence alignment was performed via comparative analysis, and the gene order was compared to clarify the missing and ambiguous gene annotation. The cp genomes were aligned with default parameters using MAFFT version 7.22242 with default parameters. Kimura’s two parameter model (K2P)43 was utilized to find the pairwise sequence divergence. The relative synonymous codon usage (RSCU) value analysis and variable sites (Pi) were calculated through sliding window analysis using DnaSP software version 6.13.0344. The mVISTA45 in shuffle-LAGAN mode was used to determine the genomic divergence while using cp genome of D. serrulata as a reference.
Phylogenetic analysis
To resolve the phylogenetic position of D. serrulata and D. cinnabari within the subfamily of Nolinoideae a total of 44 cp genomes were retrieved from NCBI database. Four Asparagus species, A. schoberioides, A. officinalis, A. racemosus and A. setaceus were selected as outgroups. The first tier alignment of complete cp genomes was performed according to the cp genome structure and conserved gene order46. The phylogenetic trees were constructed using four methods by employing the setting described previously by Asaf et al.48. Neighbour-joining (NJ) and maximum likelihood (ML) were implemented in MEGA 649; Bayesian inference (BI) was employed in MrBayes 3.1.250; and maximum parsimony (MP) by using PAUP version 4.051. For the ML run, the parameters were optimized by BIONJ tree52 as the starting tree with 1000 bootstrap replicates by employing the Kimura 2-parameter model with invariant sites gamma-distributed rate heterogeneity. For Bayesian inference, the best substitution model GTR + G was tested by jModelTest version v2.1.02100 according to the Akaike information criterion (AIC) for Bayesian posterior probabilities (PP) in BI analyses. The Markov Chain Monte Carlo (MCMC) method was run using four incrementally heated chains across 1,000,000 generations, starting from random trees and sampling 1 out of every 100 generations. To estimate the posterior probabilities, the values of first 30% of trees were discarded as burn-in. Similarly, the maximum parsimony run was based on a heuristic search with 1000 random addition of sequence replicates with the tree-bisection-reconnection (TBR) branch-swapping tree search criterion. In the second tier, 66 shared protein-coding genes from 46 cp genomes from subfamily Nolinoideae were aligned using MAFFT version 7.22294 under default parameters and making various manual adjustments to preserve and improve reading frames in the second tiers of phylogenetic analysis. The above four aforementioned phylogenetic inference models (ML, NJ, BI and MP) were employed to construct trees using 66 concatenated genes as mentioned above and suggested by Asaf et al.53.
Results and discussion
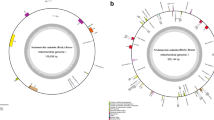
The results showed that the cp genomes of D. serrulata (MT408026) and D. cinnabari (OK235335) have the typical quadripartite structures like other related plants54,55,56 with a genome size of 155,398 bp and 155,351 bp respectively. Both the cp genomes comprised of 4 distinctive parts in which the LSC (83,871 bp, 83,818 bp) and SSC (19,247 bp, 18,579 bp) are separated by two IRs (26,140 bp, 26,477 bp) (Fig. 2, Table 1). The cp genomes of D. serrulata and D. cinnabari were analyzed and compared with D. angustifolia, D. cambodiana, D. cochinchinensis, D. cochinchinensis2, D. draco,D. elliptica, D. fragrans, D. hokouensis and D. terniflora (Table 1), which are closely related and belongs to the same genus. The sizes of these cp genomes range from 155,055 bp (D. elliptica) to 155,449 bp (D. cochinchinensis), as shown in Table 1. The cp genomes of D. serrulata and D. cinnabari encodes a total of 131 genes like all compared cp genomes except D. cambodiana, D. cochinchinensis and D. elliptica, which encode 130 genes while D. hokouensis encodes 133 genes. Similarly, among the total genes encoded by cp genomes of D. serrulata and D. cinnabari, 85 and 84 are protein-coding genes, respectively (Table 1). Furthermore, D. serrulata and D. cinnabari's cp genomes encode eight rRNA and 38 tRNA genes, respectively (Fig. 2). Similar results were reported previously in other angiosperms57,58,59. Among the protein-coding genes 12 genes (rps11,12, 14, 15, 16, 18, 2, 3, 4, 7, 7, 8) code for small ribosomal subunits, 9 genes (rpl14, 16, 2, 0, 22, 23, 32, 33, 36) for large ribosomal subunits, 44 genes (Table 2) photosynthesis related proteins, 4 (rpoA, rpoB, rpoC1, rpoC2) DNA dependent RNA polymerase, and 8 genes (accD, ccsA, cemA, matK, ycf1, ycf2, ycf3, ycf4) code for other proteins (Table 2). Furthermore, 20 genes containing introns were identified in 18 genes containing a single intron whereas two genes (ycf3, clpP) had two introns and three exons (Table 3). The trnK-UUU gene was identified with the largest intron (2,568 bp) and rpl2 gene with the smallest intron (652 bp). The rps12 gene was trans-spliced; the 5′ end exon was detected in the LSC region and the 3′ exon was identified in the IR region, as in most other angiosperms. These results are consistent with previous reports60,61,62. The overall GC content of D. serrulata and D. cinnabari cp genomes was 37.6% and 37.5%, respectively, similarly found in other cp genomes (Table 1) as reported previously28.

Genome Map of the D. serrulata and D. cinnabari cp genomes. Thick lines represent inverted repeat regions (IRs). IRs split the cp genome into large single copies (LSC) and single small copies (SSC) regions. The counter-clockwise transcribing genes are drawn outside while the clockwise are drawn inside the circle. Genes related to different functional groups are color coded. The GC and AC content is represented by the circle's dark and light green shades.
Repeats and simple sequence repeats SSR analysis in Cp genomes
A total of 80 and 82 repeats were identified in D. serrulata and D. cinnabari, respectively. In contrast, the cp genome of D. terniflora had the highest number of total repeats (103) and D. elliptica had the minimum (79). In D. serrulata and D. cinnabari, the palindromic repeats were 29 and 26, respectively (Fig. 3A). Similarly, both sequenced cp genomes had the forward repeats of 20 each (Fig. 3B) whereas the reverse repeats identified were zero in D. serrulata and 3 in D. cinnabari (Fig. 3C). Furthermore, the tandem repeats were also identified for both sequenced genomes, 31 and 33, respectively (Fig. 3D). Although, the highest and lowest number of forward repeats were detected in cp genome of D. terniflora (36) and D. hokouensis (19), while the reverse repeats were highest in D. cochinchinensis, D. draco and D. elliptica (4) and zero in D. serrulata. Most palindromic repeats were detected in D. serrulata and D. hokouensis i.e. 29. Similarly, the tandem repeats were most in the cp genome of D. cochinchinensis (38) and least in D. elliptica (30). The total number of repeats was highest (87) in D. cochinchinensis (Fig. 3E).

Repetitive sequences in D. serrulata, D. cinnabari and related Dracaena species cp genomes. (A) A total number of repetitive sequences in cp genomes, (B) Lengthwise frequency of palindromic repeats (C) Lengthwise frequency of forward repeats (D) Lengthwise frequency of reverse repeats (E) Lengthwise frequency of tandem repeats.
Simple sequence repeats (SSR) are used as genetic markers in evolutionary and population genetics studies63. These repeats also known as microsatellites are usually comprised of 1–6 bp repeat units64. Furthermore, SSRs are important because their relative lack of recombination, maternal inheritance, and haploid nature make them potential candidates for phylogenetic studies. SSRs play a role in estimating genetic variation, gene flow analysis, and studying the population history in plants and animals65,66. In this study, we analyzed SSRs in the cp genomes of D. serrulata and D. cinnabari and nine other Dracaena species Fig. 4A and B). Interestingly, the highest number of SSRs were identified in D. serrulata (176) followed by D. draco (163). In D. cinnabari, D. angustifolia and D. hokouensis, the identified SSRs were 159. The least number of SSRs were identified in D. cambodiana and D. cochinchinensis, which were 152 (Fig. 4A). Mononucleotide repeats were the most detected SSRs (Fig. 4C). The highest number of mono-nucleotide SSRs were detected in D. Serrulata (164), followed by D. hokouensis (151). The highest number of di-nucleotide SSRs were detected in the sequenced cp genome of D. cinnabari (5), followed D. serrulata (4) (Fig. 4D), while the tri-nucleotide SSRs were 3 in cp genomes of D. serrulata and D. cinnabari along with other compared cp genomes except in D. fragrance which were two and D. cochinchinensis2 with no tri-nucleotides (Fig. 4E). A total of 2 tetra-nucleotide SSRs were detected only in the cp genome of D. cochinchinensis. In contrast, in this study, the remaining cp genomes had no tetra-nucleotide SSRs, including the sequenced cp genomes of D. serrulata and D. cinnabari (Fig. 4F). The penta-nucleotide SSRs detected in D. serrulata were 5, while the D. cinnabari had zero (Fig. 4G). Contrastingly the hexanucleotide SSRs was found in only the D. cinnabari cp genome, as shown in Fig. 4H. Likewise, patterns in Dracaena and other angiosperms cp genomes were also reported previously67,68. Our results agree with the recent studies reporting that identified SSRs in cp genomes are made of polyadenine or polythymine repeats. The contrary is with guanine (G) and cytosine (C). Therefore, the cp genomes of D. serrulata and D. cinnabari are rich in ‘AT’ content, as reported previously69,70,71. As per earlier reports, the SSRs are randomly distributed across the cp genomes, revealing important information for selecting molecular markers for polymorphism (inter and intra-specific)72,73. The current results are in synergy with previous reports of angiosperms indicating the dominating abundance of ‘A’ or ‘T’ mono-nucleotides SSRs in cp genomes and resulting in ‘AT’ rich cp genomes74,75.

Simple sequence repeats (SSRs) in D. serrulata, D. cinnabari, and related Dracaena species cp genomes. (A) Total number of SSRs in cp genomes, (B) SSR motif frequency in cp genomes, (C) Mono-nucleotides SSRs (D) Di-nucleotides SSRs, (E) Tri-nucleotides SSRs, (F) Tetra-nucleotides SSRs, (G) Penta-nucleotides SSRs and (H) Hexa-nucleotides SSRs.
Comparative analysis and sequence divergence analyses
Comparative analysis of the cp genome plays a pivotal role in understanding plant species' genetic diversity and evolutionary relationships22,76 The cp genomes D. serrulata and D. cinnabari were compared to the closely related species for sequence divergence. The cp genome of D. serrulata was selected as a reference genome. The cp genomes of D. serrulata and D. cinnabari along with all the compared cp genomes, were highly conserved. All aligned sequences exhibit high similarities with only a few regions sequence variations in non-coding regions (Fig. 5). Interestingly, a higher degree of divergence was observed in non-coding regions in all cp genomes compared to the coding areas reported previously77,78. The current results revealed the high similarity of cp genome sequences of all species included in the study, suggesting that the cp genomes of Dracaena genus are highly conserved as reported for Dracaena28 and Camellia genus79. The petD, and clpP genes in the LSC region, and the ycf1 gene in the SSC region showed sequence divergence in the coding areas across all compared species, and these results agree with21,28,71,80.

Visual alignment of D. serrulata, D. cinnabari, and related Dracaena species cp genomes. VISTA-based identity plot showing sequence identities among eleven Dracaena species, using D. serrulata as a reference. Genome regions are color-coded as protein-coding, rRNA coding, tRNA coding, or conserved non-coding sequences (CNS). The x-axis represents the coordinate in the chloroplast genome. Annotated genes are displayed along the top. The sequences similarity of the aligned regions is shown as horizontal bars indicating the average percent identity between 50 and 100%.
Moreover, in IR regions, the rps19 gene showed sequence divergence in the cp genomes of D. cinnabari and D. cochinchinensis. In contrast, the ycf2 gene showed variation in the cp genome of D. cambodiana. In the LSC region, accD atpF, ycf3, and rps15 genes showed sequence divergence in some cp genomes compared to the D. serrulata cp genome (Fig. 5). Furthermore, in the non-coding areas such as rsp16-trnT, rps4-trnL, and petE-trnG in LSC while rps7-trnV in SSC showed sequence divergence across all the compared cp genomes, likewise pattern of divergence was also reported previously78,79,81.
Moreover, the average pairwise sequence divergence among the complete cp genomes (Table S2) and shared genes (Table S3) was calculated. D. cinnabari cp genome showed an average pairwise sequence divergence of 0.003. The cp genome of D. cinnabari showed the highest average pairwise sequence divergence with D. cochinchinensis and D. fragrance (0.0077). Other cp genomes included in the study and previous reports also showed similar results48,71. The most divergent genes were accD, matK, rpl16, rpoC2, and ycf1. The highest pairwise sequence divergence was identified for rpl16 (0.03) (Table S3). Similar results are also reported by Zhang, et al.28. Similarly, the relative synonymous codon usage (RSCU) value analysis was performed using coding regions of 10 Dracaena cp genomes. The most abundantly used codons were A/U-ending codons. These results exhibited a higher codon usage toward A/U- endings than G/C-ended codons in all cp genomes of Dracaena species28,82,83. Codons like CAA, GCU, GCA, and GUA UAC (yellow colored) have less than one RSCU value in one or more cp genomes (Fig. 6). Whereas the highest RSCU value was recorded for AGA (2) across all cp genomes, similar results were reported for Punica granatum84 and D. draco28. The codon characteristic pattern and frequency of usage are given in Table S1. The most frequently used codon was AAA (n = 2,036, 51.5%) in these genomes, which encodes lysine amino acid. In contrast, the least used codon was GCG (n = 257, 5.2%), coding the arginine amino acid (Table S1); these results agree with earlier reports28,85.

Heatmap plot of codon distribution of all shared protein-coding genes in 11 Dracaena species. Color key: yellow indicates lower, green indicates moderate, while purple indicates higher RSCU values.
Similarly, the nucleotide diversity (Pi) values were calculated in these cp genomes (Fig. 7). The Pi values ranged from 0 to 0.024 (LSC), 0 to 0.027 (LSC), and 0 to 0.049 (IRs) with a mean of 0.0030, which indicates that the variation is slight among these cp genomes and are highly conserved, similar variation patterns were previously reported in angiosperm cp genomes86. Furthermore, the IR region showed higher Pi values than LSC and SSC reported87. However, some genes like accD, psbL (LSC), and ycf1 (SSC) showed higher Pi values of 0.02, 0.02, and 0.026 than other protein-coding genes. Similarly, the trnV-rps7 (IR region) showed the highest Pi value of 0.05. these results also agree with mVISTA divergence analysis and previous reports21,88,89.

Sliding window analysis of nucleotide variability among the Dracaena species cp genomes (window length: 600 bp; step size: 200 bp).
Contraction and expansion of IRs and single copy regions
Inverted repeat regions are considered the most conserved regions. The size variations in cp genomes occur due to expansion/contraction of IRs and single copy regions76,90,91. The four junctions (JLA, JLB, JSA, and JSB) between the single copy regions (LSC, SSC) and IRs (IRA, IRB) in cp genomes of D. serrulata, D. cinnabari, and others were comprehensively assessed. The IRs regions are remarkably conserved across all the cp genomes in the current study. The IRs regions' lengths correlate across all the compared cp genomes with only slight expansion and contraction (Fig. 8). The cp genomes of D. serrulata and D. cinnabari have the shortest IRs regions of 26,140 bp and 26,477 bp, respectively. In comparison, D. angustifolia and D. terniflora possess the most extended IRs regions of 26,530 bp. The positions of rpl22 and rps19 genes at JLB are similar in all the cp genomes with only one base (D. elliptica, D. cochinchinensis) and three bases (D. draco) differences. Interestingly, the ndhF gene was found 40 and 22 base pairs away from the JSB in SSC in cp genome of D. serrulata and D. cinnabari (Fig. 8). In contrast, in other compared cp genomes it is found extended into IRb regions and overlaps with ycf1 (28–80 bp) as found previously92. Similarly, the ycf1 and rpl22 genes at JSA and JSB are slightly variable across some cp genomes. Previous reports support the results92,93,94.

Distances between adjacent genes and junctions of the small single-copy (SSC), large single-copy (LSC), and two inverted repeats (IR) regions among D. serrulata, D. cinnabari, and related Dracaena species cp genomes. Boxes above and below the primary line indicate the adjacent border genes. The figure is not scaled regarding sequence length and only shows relative changes at or near the IR/SC borders.
Phylogenetic analysis
Since the eighteenth century, the phylogenetic relationships among the Dracaena species have not been completely clarified and are still unclear. In Dracaena, significant morphological variation has been shown, with species generally3. Until recently, limited number of genetic markers such as chloroplast genes (matK, rbcL and intergenic spacer regions such as rpl32-trnL, trnQ-rps16, psbA-trnH, trnL-trnF etc.) were used to infer the phylogenetic relationships between the various Dracaena species such as D. serrulata, D. cinnabari and D. draco, etc. Therefore, additional genetic markers are required to determine the phylogenetic position of D. cinnabari and D. serrulata. Cp genomes as a super-barcode and concatenated protein coding genes with sufficient informative sites have been proven effective in resolving complicated phylogenetic relationships in various complex plant species19. Therefore, this study determined the phylogenetic dispositions of D. serrulata and D. cinnabari within the subfamily Nolinoideae by analyzing 46 complete cp genomes from subfamily Nolinoideae and four complete cp genomes from subfamily Asparagoidea as outgroups (Fig. 9) and 66 shared protein-coding genes (Fig. S1). Phylogenetic analysis using ML, BI, NJ and MP methods was performed. The phylogenetic analysis of complete genomes and shared protein coding genes revealed almost the same phylogenetic signals. In these phylogenetic trees (Figs. 9, S1), D. serrulata and D. cinnabari formed a single clade with high bootstrap value and BI support.

The phylogenetic tree is based on 46 complete cp genomes from subfamily Nolinoideae and four complete cp genomes from subfamily Asparagoidea as outgroups using neighbor-joining (NJ), maximum likelihood (ML), Bayesian inference (BI) and maximum parsimony (MP) methods.Numbers above the branches represent bootstrap values in NJ, ML, BI and MP trees, respectively. Different colors represent the subfamilies in Asparagaceae family.
Moreover, the tree topology enabled inference of the relationship based on the phylogenetic studies conducted by Durán et al.19. The position of both D. serrulata and D. cinnabari confirms the previously published phylogeny described by Durán et al.19 that D. serrulata is more closely related to D. cinnabari than D. draco (Fig. 9). Furthermore, both trees revealed that D. draco is more closely related to D. cochinchinensis and D. cambodiana. Similar results were reported previously by Durán et al.19on the basis of chloroplast barcode genes such as rbcL and matK genes and intergenic spacer regions(trnQ-rps16 and rpl32-trnL). However, another study by Lu and Morden18 based on combined chloroplast intergenic spacer regions using Bayesian analysis showed contradictory results to our study, where D. serrulata was closely related to D. draco. Furthermore, The earlier finding of Wang et al.95, who placed Liriope and Ophiopogon in the tribe Ophiopogoneae, is also supported by our phylogenetic trees Lun-Kai et al.96, Song‐Yun and Lun‐Kai97, and98 proposed that Ophiopogon and Liriope were closely related based on characteristics of the leaf epidermis and pollen as well as chromosomal counts. Even though our findings, based on the available cp genomes, clarified the phylogenetic relationships of some D. serrulata and D. cinnabari, more complete cp genome sequences are needed to resolve the comprehensive phylogenies of this genus because limited taxon sampling may produce discrepancies in tree topologies as reported earlier99.
Conclusion
In the current study, the complete chloroplast genomes of D. serrulata and D. cinnabari were sequenced and elucidated for the first time. The overall gene order and cp genome organization were similar to nine Dracaena species. Repetitive sequences and SSRs were identified from the sequenced data and nine related cp genomes. In contrast, the highest number of repeats and SSRs were identified in D. terniflora and D. serrulata. Moreover, divergence is detected in intergenic spaces greater than in protein-coding regions of these cp genomes. Current results showed that the D. serrulata and D. cinnabari form a single clade. The whole cp genome sequencing of D. serrulata and D. cinnabari gives exciting insights and valuable data that may facilitate the identification of related species and answer taxonomic questions.
Data availability
All data generated or analyzed during this study are included in this published article. The D. serrulata and D. cinnabari cp genomes were submitted to NCBI with accession numbers MT408026 and OK235335 respectively.
References
Bogawski, P. et al. Current and future potential distributions of three Dracaena Vand. ex L. species under two contrasting climate change scenarios in Africa. Ecol. Evol. 9, 6833–6848 (2019).
Maděra, P. et al. What we know and what we do not know about dragon trees?. Forests 11, 236 (2020).
Marrero, A., Almeida, R. S. & González-Martín, M. A new species of the wild dragon tree, Dracaena (Dracaenaceae) from Gran Canaria and its taxonomic and biogeographic implications. Bot. J. Linn. Soc. 128, 291–314 (1998).
Edwards, H. G., de Oliveira, L. F. & Prendergast, H. D. Raman spectroscopic analysis of dragon’s blood resins—basis for distinguishing between Dracaena (Convallariaceae), Daemonorops (Palmae) and Croton (Euphorbiaceae). Analyst 129, 134–138 (2004).
Gupta, D., Bleakley, B. & Gupta, R. K. Dragon’s blood: botany, chemistry and therapeutic uses. J. Ethnopharmacol. 115, 361–380 (2008).
Agnew, A. AG Miller & M. Morris 1988. Plants of Dhofar (The southern region of Oman; traditional, economic and medicinal uses). The office of the Adviser for Conservation of the Environment, Diwan of Royal Court, Sultanate of Oman. 361 pages. ISBN 07157-0808-2. Price:£ 35.00. Journal of Tropical Ecology 6, 102–102 (1990).
Bos, J. in Flowering Plants· Monocotyledons 238–241 (Springer, 1998).
Al Jabri, Y. et al. in International Symposium on the Role of Plant Genetic Resources in Reclaiming Lands and Environment Deteriorated by Human and 1190. 9–14.
Attorre, F. et al. Will dragonblood survive the next period of climate change? Current and future potential distribution of Dracaena cinnabari (Socotra, Yemen). Biol. Cons. 138, 430–439 (2007).
Vahalík, P. et al. The conservation status and population mapping of the endangered Dracaena serrulata in the Dhofar Mountains Oman. Forests 11, 322 (2020).
Ii, A. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141, 399–436 (2003).
Rudall, P. J., Conran, J. G. & Chase, M. W. Systematics of Ruscaceae/Convallariaceae: a combined morphological and molecular investigation. Bot. J. Linn. Soc. 134, 73–92 (2000).
Brown, N. E. Notes on the genera Cordyline, Dracaena, Pleomele, Sansevieria and Taetsia. Bulletin of Miscellaneous Information (Royal Botanic Gardens, Kew), 273–279 (1914).
Brown, N. E. Sansevieria. A monograph of all the known species. Bulletin of Miscellaneous Information (Royal Botanic Gardens, Kew), 185–261 (1915).
Salisbury, R. A. The genera of plants. (J. Van Voorst, 1866).
Watson, L. & Dallwitz, M. J. The grass genera of the world. (CAB international, 1992).
APG, I. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181(1), 1–20. https://doi.org/10.1111/boj12385 (2016).
Lu, P.-L. & Morden, C. W. Phylogenetic relationships among Dracaenoid genera (Asparagaceae: Nolinoideae) inferred from chloroplast DNA loci. Syst. Bot. 39, 90–104 (2014).
Durán, I. et al. Iconic, threatened, but largely unknown: Biogeography of the Macaronesian dragon trees (Dracaena spp.) as inferred from plastid DNA markers. Taxon 69, 217–233 (2020).
Wilkin, P., Suksathan, P., Keeratikiat, K., van Welzen, P. & Wiland-Szymanska, J. A new species from Thailand and Burma, Dracaena kaweesakii Wilkin & Suksathan (Asparagaceae subfamily Nolinoideae). PhytoKeys 26, 101 (2013).
Celiński, K., Kijak, H. & Wiland-Szymańska, J. Complete chloroplast genome sequence and phylogenetic inference of the Canary Islands Dragon Tree (Dracaena draco L.). Forests 11, 309 (2020).
Daniell, H., Lin, C.-S., Yu, M. & Chang, W.-J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29 (2016).
Weising, K. & Gardner, R. C. A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 42, 9–19 (1999).
Khan, A. L., Asaf, S., Al-Rawahi, A. & Al-Harrasi, A. Decoding first complete chloroplast genome of toothbrush tree (Salvadora persica L.): insight into genome evolution, sequence divergence and phylogenetic relationship within Brassicales. BMC Genomics 22, 1–16 (2021).
Eguiluz, M., Rodrigues, N. F., Guzman, F., Yuyama, P. & Margis, R. The chloroplast genome sequence from Eugenia uniflora, a Myrtaceae from Neotropics. Plant Syst. Evol. 303, 1199–1212 (2017).
Lee, H. J. et al. Authentication of Zanthoxylum species based on integrated analysis of complete chloroplast genome sequences and metabolite profiles. J. Agric. Food Chem. 65, 10350–10359 (2017).
Li, X. et al. Plant DNA barcoding: from gene to genome. Biol. Rev. 90, 157–166 (2015).
Zhang, Z., Zhang, Y., Song, M., Guan, Y. & Ma, X. Species identification of Dracaena using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 10, 1441 (2019).
Song, Y. et al. Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 7, 1–8 (2017).
Dong, W. et al. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 8, 1148 (2017).
Shi, C. et al. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 7, e31468 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Jin, J.-J. et al. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. BioRxiv 4, 256479 (2018).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. https://doi.org/10.1093/bioinformatics/bth352 (2004).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. https://doi.org/10.1093/nar/gki366 (2005).
Lohse, M., Drechsel, O. & Bock, R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. https://doi.org/10.1007/s00294-007-0161-y (2007).
Kurtz, S. et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Katoh, K. & Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900 (2010).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. https://doi.org/10.1093/nar/gkh458 (2004).
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297 (2011).
Kumar, S., Nei, M., Dudley, J. & Tamura, K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9, 299–306 (2008).
Asaf, S., Khan, A. L., Khan, A. & Al-Harrasi, A. Unraveling the chloroplast genomes of two prosopis species to identify its genomic information, comparative analyses and phylogenetic relationship. Int. J. Mol. Sci. 21, 3280 (2020).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Kuan, L., Pratas, F., Sousa, L. & Tomás, P. MrBayes sMC3: Accelerating Bayesian inference of phylogenetic trees. Int. J. High Perform. Comput. Appl. 32, 246–265 (2018).
Swofford, D. L. Phylogenetic analysis using parsimony. (1998).
Gascuel, O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Asaf, S. et al. Expanded inverted repeat region with large scale inversion in the first complete plastid genome sequence of Plantago ovata. Sci. Rep. 10, 1–16 (2020).
Jin, J. et al. Complete chloroplast genome of a medicinal species Polygonatum kingianum in China (Asparagaceae, Asparagales). Mitochondrial DNA Part B 5, 959–960. https://doi.org/10.1080/23802359.2020.1721373 (2020).
Gu, L., Su, T., Luo, G.-L. & Hu, G.-X. The complete chloroplast genome sequence of Heteropolygonatum ginfushanicum (Asparagaceae) and phylogenetic analysis. Mitochondrial DNA Part B 6, 1799–1802. https://doi.org/10.1080/23802359.2021.1933636 (2021).
Jang, J.-H. et al. Characterization of the complete chloroplast genome of Liriope platyphylla (Asparagaceae: Nolinoideae) isolated in Korea. Mitochondrial DNA Part B 5, 2874–2875. https://doi.org/10.1080/23802359.2020.1787898 (2020).
Cay, S. B. et al. Genome skimming approach reveals the gene arrangements in the chloroplast genomes of the highly endangered Crocus L. species: Crocus istanbulensis (B. Mathew) Rukšāns. PLoS ONE 17, e0269747 (2022).
Hishamuddin, M. S. et al. Comparison of eight complete chloroplast genomes of the endangered Aquilaria tree species (Thymelaeaceae) and their phylogenetic relationships. Sci. Rep. 10, 1–13 (2020).
Qian, S.-J., Zhang, Y.-H. & Li, G.-D. The complete chloroplast genome of a medicinal plant, Wikstroemia chamaedaphne (Thymelaeaceae). Mitochondrial DNA Part B 5, 648–649 (2020).
Tao, X., Ma, L., Zhang, Z., Liu, W. & Liu, Z. Characterization of the complete chloroplast genome of alfalfa (Medicago sativa)(Leguminosae). Gene Rep. 6, 67–73 (2017).
Xiang, B. et al. The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II platform. Molecules 21, 1029 (2016).
Duffy, A. M., Kelchner, S. A. & Wolf, P. G. Conservation of selection on matK following an ancient loss of its flanking intron. Gene 438, 17–25 (2009).
Huang, Y.-Y., Matzke, A. J. & Matzke, M. Complete sequence and comparative analysis of the chloroplast genome of coconut palm (Cocos nucifera). PLoS ONE 8, e74736 (2013).
Mason, A. S. in Plant Genotyping 77–89 (Springer, 2015).
Vieira, L. D. N. et al. The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 9, e90618 (2014).
Ebert, D. & Peakall, R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 9, 673–690 (2009).
Khan, A. L., Asaf, S., Lee, I.-J., Al-Harrasi, A. & Al-Rawahi, A. First reported chloroplast genome sequence of Punica granatum (cultivar Helow) from Jabal Al-Akhdar, Oman: phylogenetic comparative assortment with Lagerstroemia. Genetica 146, 461–474 (2018).
Du, X. et al. The complete chloroplast genome sequence of yellow mustard (Sinapis alba L.) and its phylogenetic relationship to other Brassicaceae species. Gene 731, 144340 (2020).
Liu, X., Li, Y., Yang, H. & Zhou, B. Chloroplast genome of the folk medicine and vegetable plant Talinum paniculatum (Jacq.) Gaertn.: gene organization, comparative and phylogenetic analysis. Molecules 23, 857 (2018).
Zhou, J. et al. Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int. J. Mol. Sci. 18, 1839 (2017).
Qian, J. et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8, e57607 (2013).
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. & Rafalski, J. Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. 92, 7759–7763 (1995).
Provan, J., Corbett, G., Powell, W. & McNicol, J. Chloroplast DNA variability in wild and cultivated rice (Oryza spp.) revealed by polymorphic chloroplast simple sequence repeats. Genome 40, 104–110 (1997).
Khan, A. L., Asaf, S., Al-Rawahi, A. & Al-Harrasi, A. Decoding first complete chloroplast genome of toothbrush tree (Salvadora persica L): insight into genome evolution, sequence divergence and phylogenetic relationship within Brassicales. BMC Genomics 22, 312. https://doi.org/10.1186/s12864-021-07626-x (2021).
Ma, Q. et al. Complete chloroplast genome sequence of a major economic species, Ziziphus jujuba (Rhamnaceae). Curr. Genet. 63, 117–129 (2017).
Asaf, S. et al. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 12, e0182281 (2017).
Wu, C.-S. & Chaw, S.-M. Evolutionary stasis in cycad plastomes and the first case of plastome GC-biased gene conversion. Genome Biol. Evol. 7, 2000–2009 (2015).
Khan, A. et al. Comparative chloroplast genomics of endangered Euphorbia species: Insights into hotspot divergence, repetitive sequence variation, and phylogeny. Plants 9, 199 (2020).
Huang, H., Shi, C., Liu, Y., Mao, S.-Y. & Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14, 1–17 (2014).
Asaf, S., Jan, R., Khan, A. L. & Lee, I.-J. Complete chloroplast genome characterization of oxalis corniculata and its comparison with related species from family oxalidaceae. Plants 9, 928 (2020).
Yang, J.-B., Yang, S.-X., Li, H.-T., Yang, J. & Li, D.-Z. Comparative chloroplast genomes of Camellia species. PLoS ONE 8, e73053 (2013).
Deng, N. et al. Complete chloroplast genome sequences and codon usage pattern among three wetland plants. Agron. J. 113, 840–851 (2021).
Zhou, J. et al. Complete chloroplast genomes of papaver rhoeas and papaver orientale: Molecular structures, comparative analysis, and phylogenetic analysis. Molecules 23, 437 (2018).
Yan, M. et al. The complete chloroplast genomes of punica granatum and a comparison with other species in lythraceae. Int. J. Mol. Sci. 20, 2886 (2019).
Somaratne, Y., Guan, D.-L., Wang, W.-Q., Zhao, L. & Xu, S.-Q. The complete chloroplast genomes of two lespedeza species: insights into codon usage bias, RNA editing sites, and phylogenetic relationships in desmodieae (Fabaceae: Papilionoideae). Plants 9, 51 (2020).
Asaf, S. et al. Complete chloroplast genome of nicotiana otophora and its comparison with related species. Front. Plant Sci. https://doi.org/10.3389/fpls.2016.00843 (2016).
Hong, S.-Y. et al. Complete chloroplast genome sequences and comparative analysis of chenopodium quinoa and C. album. Front. Plant Sci. https://doi.org/10.3389/fpls.2017.01696 (2017).
Asaf, S. et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7, 843 (2016).
Zhao, X.-L. & Zhu, Z.-M. Comparative genomics and phylogenetic analyses of christia vespertilionis and urariopsis brevissima in the tribe desmodieae (fabaceae: papilionoideae) based on complete chloroplast genomes. Plants 9, 1116 (2020).
Guo, Y.-Y., Yang, J.-X., Bai, M.-Z., Zhang, G.-Q. & Liu, Z.-J. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 21, 1–14 (2021).
Raubeson, L. A. et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8, 1–27 (2007).
Park, I. et al. The complete chloroplast genome sequences of fritillaria ussuriensis maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other fritillaria species. Molecules 22, 982 (2017).
Shaw, J., Lickey, E. B., Schilling, E. E. & Small, R. L. Comparison of whole chloroplast genome sequences to choose non-coding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288 (2007).
Khakhlova, O. & Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94 (2006).
Wang, G.-Y., Meng, Y., Huang, J.-L. & Yang, Y.-P. Molecular phylogeny of ophiopogon (Asparagaceae) inferred from nuclear and plastid DNA sequences. Syst. Bot. 39, 776–784 (2014).
Lun-Kai, D., Song-Yun, L., Lun-Kai, D. & Song-Yun, L. Epidermal features of leaves and their taxonomic significance in subfamily Ophiopogonoideae (Liliaceae). J. Syst. Evol. 29, 335 (1991).
Song-Yun, L. & Lun-Kai, D. Pollen morphology and generic phylogenetic relationships in Ophiopogonoideae (Liliaceae). J. Syst. Evol. 30, 427 (1992).
Zhang, D. Chromosomal study and an insight into systematics of the tribe Ophiopogoneae (Endl.) Kunth. Ph. D. Dissertation, Inst Bot, Chinese Acad Sci (1991).
Leebens-Mack, J. et al. Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one’s way out of the Felsenstein zone. Mol. Biol. Evol. 22, 1948–1963 (2005).
Author information
Authors and Affiliations
Contributions
A.L.K., A.K. and S.A. performed experiments; A.L.K., S.A. and W.A. wrote the original draft and Bioinformatics analysis: A.R. collected samples, A.L.K. and A.H. supervision and arranging resources. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmad, W., Asaf, S., Khan, A. et al. Complete chloroplast genome sequencing and comparative analysis of threatened dragon trees Dracaena serrulata and Dracaena cinnabari. Sci Rep 12, 16787 (2022). https://doi.org/10.1038/s41598-022-20304-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20304-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
