
Abstract
As a highly infectious epidemic in aquaculture, Pseudomonas plecoglossicida infection results in high mortality of teleosts and serious economic losses. Host–pathogen interactions shape the outcome of an infection, yet we still understand little about the molecular mechanism of these pathogen-mediated processes. Here, a P. plecoglossicida strain (NZBD9) and Epinephelus coioides were investigated as a model system to characterize pathogen-induced host metabolic remodeling over the course of infection. We present a non-targeted metabolomics profiling of E. coioides spleens from uninfected E. coioides and those infected with wild-type and clpV-RNA interference (RNAi) strains. The most significant changes of E. coioides upon infection were associated with amino acids, lysophospatidylcholines, and unsaturated fatty acids, involving disturbances in host nutritional utilization and immune responses. Dihydrosphingosine and fatty acid 16:2 were screened as potential biomarkers for assessing P. plecoglossicida infection. The silencing of the P. plecoglossicida clpV gene significantly recovered the lipid metabolism of infected E. coioides. This comprehensive metabolomics study provides novel insights into how P. plecoglossicida shape host metabolism to support their survival and replication and highlights the potential of the virulence gene clpV in the treatment of P. plecoglossicida infection in aquaculture.
Similar content being viewed by others

Introduction
In aquaculture, the ‘visceral white spot disease’ of teleosts is associated with the infection of a Gram-negative pathogen of Pseudomonas plecoglossicida1. As a highly infectious epidemic, the infection of P. plecoglossicida can cause typical symptoms of numerous white nodules in the spleen, kidney or liver and particularly high mortality in teleosts, resulting in serious economic losses for aquaculture2.
Due to considerable harm to aquaculture, the complex interaction between P. plecoglossicida and host fishes was increasingly noted. In our previous gene expression studies, P. plecoglossicida clpV was identified as a key virulence gene that contributes to the pathogenicity of P. plecoglossicida in both Epinephelus coioides and Larimichthys crocea infections2,3. The ClpV plays an important role in the reassembly of the type VI secretion system (T6SS) machinery, as well as the secretion of T6SS effector proteins4. The T6SS is molecular machine in the secretion of virulence factors, which can directly inject pathogenic effectors into the host cells5. Then, compared with the counterparts infected by the wild-type strain, host fish injected with the clpV-RNA interference (RNAi) strain exhibited a significant improvement in mortality and symptoms2,3. RNAi of clpV resulted in the downregulation of genes in the flagella assembly pathway and a lower pathogen load in host tissues as well as a weaker immune response of the host3. In addition, the diguanylate cyclase gene (L321_RS15240)6, ABC transporter gene (L321_23611)7, secY8, pvdE9, etc., are also virulence candidates for P. plecoglossicida pathogenicity. Although a series of virulence genes associated with P. plecoglossicida pathogenicity continue to be discovered, the mechanism of their actions underlying the complex host–pathogen interactions remains obscure thus far.
Understanding host–pathogen interactions is critical for resolving the outcome of an infection. The host senses the presence of the pathogen through recognition of pathogen-associated molecular patterns, while the pathogen evolves numerous strategies to circumvent host defenses and exploit the host cellular machinery10. Unlike genes and proteins, the functions of which are subject to epigenetic regulation and posttranslational modifications, respectively, metabolites serve as direct signatures of biochemical activity and tightly correlate with phenotype11. These properties make the host cellular metabolome an attractive target for pathogens to introduce phenotypic perturbations that facilitate their survival and replication11. Indeed, there is fierce competition for metabolic precursors between host and pathogen, which may alter both pathogen growth and the nature of the immune response12. The significant alteration in the host metabolome, involving modulation of glucose, fatty acids (FAs), and amino acids, has been emphasized in recent studies regarding host–pathogen interactions10,13. For example, the amino acids arginine (Arg)13,14,15, asparagine (Asn)13,15, and tryptophan (Trp)13,15 were highlighted as central points of competition between the host and pathogen13. Amino acids affect immune responses of the host against a pathogen, such as the function of innate immune cells (e.g., macrophages), the activation and differentiation of T cells and the production of antibodies by B cells15. In addition, amino acids influence the physiology and virulence of pathogens15. Thus, elucidation of these pathogen-induced metabolic changes would provide novel insight into therapeutic approaches to manipulate and prevent progression of an infection. Nevertheless, the exploration of metabolome disturbances associated with ‘visceral white spot disease’ caused by P. plecoglossicida infection has been limited until now.
The emergence of high-throughput metabolomics techniques enables the description of global changes in the host metabolome at the level of individual molecular metabolite species and the identification of metabolite biomarkers16. In this study, to identify metabolome disturbances associated with ‘visceral white spot disease’ caused by P. plecoglossicida infection, a time series spleen sample set from E. coioides injected with PBS, the wild-type strain and the clpV-RNAi strain was collected over the course of infection. We performed metabolomics analysis on this sample set using ultra-high-performance liquid chromatography coupled to mass spectrometry (UPLC-MS). As described in our previous studies2,6,7,8,9, the P. plecoglossicida–E. coioides infection model has been successfully established and confirmed to be a suitable animal model for studying the pathogenic mechanism of P. plecoglossicida in the laboratory. Considering the highest burdens of P. plecoglossicida, the spleen was analyzed in our studies as the representative target organ2,8. Then, the comprehensive rewiring of host metabolic networks was investigated over the course of infection to elucidate the potential mechanism of host–pathogen interactions and evaluate the response of E. coioides to the key virulence gene clpV from the novel perspective of the metabolome.
Results
Characteristics of E. coioides after infection
According to our previous modeling, which traced mortality and symptoms of E. coioides and the abundance of strain over the course of infection2,6,7,8, this P. plecoglossicida–E. coioides infection pattern could be divided into two stages, namely, the developing stage (0–72 hour post injection (hpi)) and the terminal stage (after 72–96 hpi). The dynamic distribution of wild-type P. plecoglossicida in E. coioides showed that P. plecoglossicida yielded the greatest abundance in most organs and blood of E. coioides at 72–96 hpi6. The first mortalities of E. coioides infected by the wild-type strain were at 72 hpi, and the majority of deaths were at 96 hpi. Those E. coioides presented typical symptoms with numerous white splenic nodules at 96 hpi. Compared with the counterparts infected by the wild-type strain, E. coioides injected with the clpV-RNAi strain exhibited a significant improvement in mortality and symptoms. No deaths were observed up to 20 days post injection (dpi) for E. coioides infected by the clpV-RNAi strain. Their spleens failed to develop visible nodules until 5–8 dpi, with the swelling gradually disappearing.
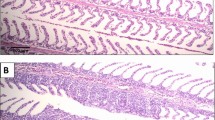
In this study, to explore the molecular mechanisms underlying the complex host–pathogen interaction, time series spleen samples from E. coioides injected with phosphate-buffered saline (PBS group), the wild-type strain (WT group) and the clpV-RNAi strain (clpV-RNAi group), were collected over the course of infection (i.e., 6, 12, 24, 48, 72 and 96 hpi, Fig. 1A). Typical histological microphotographs of spleens are shown in Fig. S1. Compared with the PBS group, the spleens of WT individuals exhibited characteristic histological changes at 72–96 hpi. PBS and clpV-RNAi groups had considerable histological similarity. These histological sections were consistent with our observation of mortality and symptoms, highlighting the effective role of clpV silencing in the improvement of E. coioides infection.

Characteristics of E. coioides upon infection. (A) Scheme of the experimental design. (B,C) Typical metabolite features of pooled tissue extract acquired using LC–MS in ESI positive mode and negative mode, respectively. (D) PCA score plot of the metabolic profile composed of metabolites after UV scaling pretreatment. (E) Metabolic trajectories of the control and different infection groups based on the PCA model. Each point represents the average score values of samples with SEM. (F) Euclidean distance between WT and time-matched PBS (or clpV-RNAi). (G) Venn diagram for overview of univariate statistical analysis and multivariate VIP values. PBS denotes the PBS control group, WT denotes the wild-type strain infection group, and clpV-RNAi denotes the clpV-RNAi strain infection group.
Metabolome of E. coioides spleens
This time series sample set was evaluated by nontargeted metabolomics analysis. The large-scale metabolome profiling of E. coioides spleens revealed approximately 5000 metabolic features for each analysis mode (Fig. 1B,C). A total of 262 metabolites from a wide spectrum of categories were finally identified, involving 45 amino acids, peptides and analogs (10 essential amino acids, 5 non-essential amino acids and 30 analogs), 9 amines, 4 carbohydrates and carbohydrate conjugates, 18 nucleosides, nucleotides and analogs, 13 organic acids and derivatives, 3 alcohols and polyols, and 170 lipids and lipid-like molecules (12 lipid-like molecules, 4 bile acids and derivatives, 11 fatty acyl carnitines, 10 fatty amides, 48 FAs, 75 glycerophospholipids, 6 sphingolipids and 4 glycerolipids). This profiling unveiled a large diversity and complexity of E. coioides splenic metabolome regarding their chemical structures, compositions and polarities. Detailed information on these identified metabolites is listed in Table S1.
The analytical performance of this metabolomics profiling was examined by evaluating quality control (QC) samples and was satisfactory for complex biological samples (Fig. S2). Detailed results of QC evaluation are described in the Supporting Information.
Global profiling of metabolic disturbance
Non-supervised principal component analysis (PCA) was first employed to gain a global overview of the metabolome changes (Fig. 1D). Two types of metabolic disturbance, infection-induced (i.e., PBS vs. WT) and gene silencing-related (i.e., clpV-RNAi vs. WT) changes, were clearly visible on this score scatter plot, as demonstrated by an obvious separation trend among the PBS, WT and clpV-RNAi groups (Fig. 1D). It is clear that individuals in the WT group were more discrete than those in the PBS and clpV-RNAi groups in this PCA profile, implying severe metabolic deregulation of E. coioides upon wild-type strain infection.
The metabolic trajectories of the PBS, WT and clpV-RNAi groups were further depicted based on this PCA score plot (Fig. 1E). The euclidean distance between metabolic trajectories at each time point was analyzed to quantify those two types of metabolic differences over the time course. As shown in Fig. 1F, the disturbance of gene silencing would be more potent than strain infection. Metabolic homeostasis of E. coioides spleen showed interference beginning at the early infection stage of 6 hpi. Both disturbances caused by infection and clpV silencing were noted in the form of more evident metabolic changes at the later infection stage of 72–96 hpi, confirming the discoveries of the E. coioides phenotype (incl. mortality, symptoms and histological examinations).
Based on the metabolic profile across the infection stage, we next determined the significantly disrupted metabolites. Univariate statistical evaluation revealed a total of 157 and 191 metabolites that exhibited significant quantitative changes when PBS and clpV-RNAi were, respectively, compared with WT (p < 0.05 and FDR < 0.05, Fig. 1G). Of note, 124 statistically significant differential metabolites were found in the overlap between these two comparisons. Approximately 80% of significant differential metabolites identified by wild-type strain infection (i.e., PBS vs. WT) showed significant changes by clpV silencing (i.e., clpV-RNAi vs. WT), implying the efficiency of the key virulence gene clpV in this host–pathogen interaction.
Furthermore, two partial least squares discriminant analysis (PLS-DA) models (i.e., PBS vs. WT and clpV-RNAi vs. WT) were developed to evaluate these significant differential metabolites based on variable importance in the projection (VIP) values (Figs. S3, S4). A total of 111 out of 157, and 112 out of 191 significant differential metabolites were identified with VIP > 1 when PBS and clpV-RNAi were compared with WT, respectively (Fig. 1G). Then, we defined the union of 169 metabolites to be the representative differential metabolites with significant disturbance (p < 0.05 FDR < 0.05 and VIP > 1, Table S2).
Comparison between infection-induced and gene silencing-related changes
As identified when PBS and clpV-RNAi were compared with the ‘positive’ WT, both enrichment analyses of significant differential metabolites resulted in the top 3 significant terms associated with amino acids, lysophospatidylcholines (LPCs), and unsaturated FAs (Fig. 2A,B). Most of the top 25 enriched disturbed categories were similar between these two subsets of significant differential metabolites (Fig. 2A,B).

Comparison between infection-induced and gene silencing-related changes. (A,B) Overview of enriched metabolite sets. (C,D) Summary of enriched metabolic pathways. Enriched analyses of all significant differential metabolites were performed based on comparisons between PBS and WT (i.e., infection-induced changes, left panel), and clpV-RNAi and WT (i.e., silencing-related changes, right panel). Analyses of those significant differential metabolites resulted in the top 3 significant metabolite sets (red asterisk*), and revealed the global metabolic disorders of the most relevant pathways.
Linoleic acid metabolism, arginine biosynthesis, alanine, aspartate and glutamate metabolism, glycerophospholipid metabolism, d-glutamine and d-glutamate metabolism, etc., were regarded to be highly responsible for both comparisons (i.e., PBS vs. WT and clpV-RNAi vs. WT, Fig. 2C,D). The similarity in both the most disturbed metabolite sets and metabolic pathways confirmed the prominent role of P. plecoglossicida clpV in the host–pathogen interaction.
These representative differential metabolites were UV-scaled and subjected to hierarchical clustering and were classified into four major groups according to the response pattern (Fig. 3A). In addition to the significant difference between the ‘positive’ WT and ‘control’ groups (i.e., PBS and clpV-RNAi), disparities between PBS and clpV-RNAi also existed due to the complexity of host–pathogen interactions. The change pattern was further specialized, as depicted by correlation networks (Fig. 3B,C). Each point indicates one representative differential metabolite with UV scaling, while the line indicates the correlation with |Cij|> 0.9. We observed a decreased correlation between metabolites (i.e., the number of ‘lines’) in WT, while a reactivated coordination of the metabolome was observed in clpV-RNAi (Fig. 3B). From the perspective of ‘point’, different classes of metabolites exhibited different changing patterns. Most lipids, including FAs and glycerophospholipids (e.g., LPCs) from Group I in the heatmap, sphingolipids and glycerolipids, had a greater response in WT when compared with PBS and clpV-RNAi. Meanwhile, polar and moderately polar metabolites, including amino acids, carbohydrate and carbohydrate conjugates, alcohols and polyols, and organic acids and derivatives, featured a similar decrease after infection (incl. WT and clpV-RNAi). Taken together, the silencing of the P. plecoglossicida clpV gene improved the lipid metabolism of infected E. coioides more significantly.

Response patterns of the metabolome at the terminal stage of 96 hpi. (A) Heatmap. The intersection of representative differential metabolites (p < 0.05, FDR < 0.05 and VIP > 1) from both comparisons (i.e., PBS vs. WT and clpV-RNAi vs. WT) was UV-scaled and subjected to hierarchical clustering. The union of all representative differential metabolites was further specialized by correlation networks. (B) The number of lines in each correlation network. (C) Correlation networks. In correlation networks, each point represents one metabolite with the relative content of UV scaled. Each black dotted (or gray solid) line represents a positive (or negative) correlation with |Cij|> 0.9.
In addition to the analysis of response patterns, we further defined whether there were any alterations associated with the chemical structure (Fig. 4). LPCs and FAs, which were defined as the top significant differential metabolite sets in enrichment analyses, had a greater response and higher degree of unsaturation when positive WT were compared with ‘control’ groups (i.e., PBS and clpV-RNAi). We found a significant positive correlation between the increase in the WT to PBS ratio and the number of double bonds (p < 0.05, R2 = 0.43 for both FAs and LPCs). Meanwhile, their increase in the WT to clpV-RNAi ratio had a similar significant positive correlation with the degree of unsaturation (p < 0.05, R2 = 0.56 for FAs and R2 = 0.54 for LPCs).

Correlation between response increase in WT to PBS ratio (or WT to clpV-RNAi ratio) and the number of double bonds. (A,B) Depict the changes in FAs and LPCs, respectively, at the terminal stage of 96 hpi.
Discovery of potential biomarkers
Potential biomarkers of E. coioides that are sensitive to P. plecoglossicida infection were analyzed. Comparing WT with PBS, a total of 111 representative significant differential metabolites were cross-selected from univariate statistical significance (p < 0.05, FDR < 0.05) and multivariate VIP values (VIP > 1). These significant metabolites were further refined with the criteria of significant changes at the terminal stage (96 hpi, p < 0.05, FDR < 0.05, ratio > 2 or ratio < 0.5), followed by stricter analysis quality (%RSD < 10% in QCs). A total of 27 biomarker candidates were finally retained. They were designated the most representative metabolites, indicating infection-induced disturbances of the host. These candidates were subjected to receiver operating characteristic curves (ROCs) analysis to quantize their discrimination performance. The closer the AUC (area under the ROC curve) value approaches 1, the better the model provides discrimination potential. Then, candidates with the top AUC value, namely, dihydrosphingosine and FA 16:2, were assumed to be potential biomarkers (Fig. 5A,B, Fig. S5A,B). The combination of these two potential biomarkers achieved an AUC value of 0.889 in the discrimination of all WT samples (including different stages of WT samples) and PBS, and the sensitivity and specificity were 82.9% and 88.1%, respectively (Fig. 5C, Table S3).

Evaluation of potential biomarkers. (A,B) Are the response trajectories of dihydrosphingosine and FA 16:2, respectively. Each point in the trajectory is presented as the mean ± SE. (C,D) ROC curves for the combination of biomarkers of dihydrosphingosine and FA 16:2. Diagnostic potential was evaluated based on binary logistic regression.
These 27 representative infection features were further evaluated in the clpV-RNAi group. Comparing clpV-RNAi with WT, a total of 21 out of 27 candidates (77.8%) were validated with significant changes (p < 0.05 and FDR < 0.05). Satisfactory ROC results were also acquired in the application of those two potential biomarkers (Fig. S5C,D), resulting in an AUC value of 0.931 and sensitivity and specificity of 85.4% and 92.9%, respectively (Fig. 5D, Table S3). These analyses confirmed our previous finding that clpV is a key virulence gene during in vivo P. plecoglossicida infection. The combination of potential biomarkers dihydrosphingosine and FA 16:2 provided an effective model for discrimination of P. plecoglossicida infection.
Discussion
P. plecoglossicida is the pathogen of ‘visceral white spot disease’ for marine teleosts, which can result in serious economic losses for aquaculture1. The P. plecoglossicida-E. coioides infection model has been confirmed to be a suitable animal model for studying the pathogenic mechanism of P. plecoglossicida in the laboratory2,6,7,8,9. Our previous gene expression studies revealed that clpV is a key virulence gene during P. plecoglossicida infection2,3. Compared with the counterparts infected by the wild-type strain, E. coioides injected with the clpV-RNAi strain exhibited a significant improvement in mortality and symptoms2. The histological examination confirmed the efficiency of clpV silencing on the improvement of infection symptoms (Fig. S1). To trace the interaction process between host and pathogen, a non-targeted metabolomics analysis was further performed in this study (Fig. 1A).
Global perturbations of E. coioides metabolome upon wild-type strain infection were clearly depicted by PCA (i.e., WT vs. PBS, Fig. 1D). The active changes could be noted by metabolic profiles from the early infection stage (6 hpi, Fig. 1E,F), confirming the efficacy of the current metabolomics study. Metabolic homeostasis showed increased disturbances across the course of the infection process, consistent with the increasingly severe symptoms of E. coioides (Fig. 1E,F). A decreased correlation between metabolites was also observed in WT when compared with control PBS (Fig. 3B). WT infection resulted in the most significant differences associated with amino acids, LPCs, and unsaturated FAs (Fig. 2A). Linoleic acid metabolism, arginine biosynthesis, alanine, aspartate and glutamate metabolism, glycerophospholipid metabolism, d-glutamine and d-glutamate metabolism, etc., were highly responsible for infection by the wild-type strain. The combination of potential biomarkers dihydrosphingosine and FA 16:2 provided an effective model for discrimination of P. plecoglossicida infection (Fig. 5, Table S3).
This metabolomics profiling highlighted the silencing of P. plecoglossicida clpV on improving infection. Metabolome changes associated with the influence of clpV were identified from the differences between clpV-RNAi and WT along the direction of the first principal component in the PCA score plot (i.e., clpV-RNAi vs. WT, Fig. 1D). Specifically, approximately 80% of significant differential metabolites identified by WT infection showed significant changes by clpV silencing (Fig. 1G). The coordination of metabolites was also reactivated in the clpV-RNAi group (Fig. 3B). Both the most enriched disturbed metabolites and metabolic pathways were similar when WT was compared with control PBS and clpV-RNAi. Comparing clpV-RNAi with WT, satisfactory ROC results were also acquired in the application of potential biomarkers dihydrosphingosine and FA 16:2 (Fig. 5, Table S3). This metabolomics results confirmed our findings regarding phenotype (incl. mortality, symptoms and histological examinations of E. coioides) and gene expression2,3, highlighting that clpV is a key virulence gene during in vivo P. plecoglossicida infection.
In this infection model, P. plecoglossicida clpV silencing greatly recovered the lipid metabolism of E. coioides (Fig. 3C). Although the improvement of amino acid metabolism was weaker than that of lipid metabolism after clpV silencing (Fig. 3C), the recovery of central points of competition between the host and pathogen, i.e., Arg & ornithine, glutamate (Glu) & glutamine (Gln), and Asn, could not be ignored.
Amino acid metabolism
Our study revealed a decrease in most amino acids (incl. essential and nonessential amino acids) at the terminal infection stage (Fig. 6A,B). Amino acids affect the host’s physiology by serving as an energy source for cells, a basic substrate for protein synthesis and a regulator of cell signaling pathways15. Pathogens also show potent nutritional versatility in the assimilation of various amino acids that are present in their hosts15. Thus, the downregulation of amino acids after infection could potentially serve as a pathogenic strategy responsible for the competition of nutritional utilization by pathogens and the inhibition of host physiology.

Profile of metabolite response at the terminal stage of 96 hpi. (A,B) Present the mapping of key pathways of amino acid metabolism. (C) Metabolism of lipids. All data are presented as the mean ± SE. The black asterisk indicates statistical significance (p < 0.05).
Arg-ornithine metabolism
When compared with control PBS and clpV-RNAi, ornithine was observed as an exception with a significant increase upon WT infection, while its substrate Arg was significantly depleted in WT (Fig. 6B).
There are two predominant pathways for host Arg metabolism, i.e., via NO synthase (NOS) for the production of NO, and via arginase for the production of ornithine15. As an antimicrobial molecule, NO can protect the host against pathogens17. Conversely, ornithine can be used to produce polyamines (putrescine, spermidine, and spermine), which are essential for the proliferation of pathogens18. Arginase competes with NOS for Arg depletion during infections13,15. Then, the metabolic signature of ornithine and substrate Arg discovered in this study might suggest the promotion of ornithine synthesis, attributed to the extensive competition for Arg utilization between the host and pathogen. Pathogens alter the metabolic flux of host Arg from producing antimicrobial NO to producing polyamines by increasing the expression of arginase to facilitate their growth. Other mechanisms, including Arg transport and the Arg deiminase (ADI) pathway to deplete Arg and/or divert Arg away from host cells that produce NO, have also been identified in previous studies of host–pathogen interactions19. Moreover, in the clpV-RNAi group, the upregulation of Arg and downregulation of ornithine confirmed the efficacy of clpV silencing on the reduction of P. plecoglossicida virulence.
Glu metabolism (GM)
Glu is crucial for pathogens due to its involvement in a wide range of metabolic processes15,20. In addition to being transported from the extracellular milieu, Glu can be enzymatically generated by cytosolic Gln (via glutaminase) and aspartate (Asp) (via aspartate transpeptidase)21. Furthermore, Glu is the precursor to synthesize glutathione (GSH)20,21.
In this study, GM-related metabolites, including Glu, Gln, Asp, GSH and glutathione disulfide (GSSG), were significantly decreased upon WT infection (Fig. 6B). Glu is an important nutritional source for pathogens20. In WT, the depletion of Glu and its precursors Gln and Asp might be attributed to the direct competition between pathogens and host cells for the nutrient pools. In addition, as an important product of Glu, GSH contributes to oxidative stress defense by playing a critical role as a redox buffer to detoxify noxious oxygen species21. The downregulation of GSH and Glu could attenuate the antioxidant capacity of the host tissue.
In clpV-RNAi, GM-related metabolites were significantly recovered (Fig. 6). The silencing of the key virulence gene clpV weakens the pathogenic competitive utilization of GM-related nutrient substrates and facilitates the improvement of the oxidative stress defense of the host.
Asn metabolism
Asn is another central point of interaction13,15, and was significantly decreased upon infection with a slight rebound in clpV-RNAi.
Asn has diverse roles in bacterial physiology. Asn represents an important nitrogen source for pathogens and is also essential for an intracellular pathogen to resist acidic stress in a phagosome by serving as the substrate for the production of the weak base ammonia22. In addition to its role in pathogen survival and infection, Asn is necessary for lymphocyte activation and protective immunity of the host23. Asn may affect T cell functions via mTORC1 which is critically important for T cell activation and differentiation15. Thus, the pathogen could inhibit the immune response of the host by inducing starvation of Asn in host cells. The depletion of Asn could be responsible for the inhibition of immune responses in the host.
FA and LPC metabolism
Defined as the top significant differential metabolite classes in enrichment analyses, FAs and LPCs had a greater response and a higher degree of unsaturation in WT compared with the control PBS and clpV-RNAi (Fig. 6C). Of note, we found a significant positive correlation between the increase in the WT to PBS ratio and the number of double bonds (p < 0.05, Fig. 4A,B). Their increase in the WT to clpV-RNAi ratio had a similar significant positive correlation with the degree of unsaturation (p < 0.05, Fig. 4A,B).
The modulation of lipids in content and composition by virulent pathogens is an effective strategy to facilitate their invasion and propagation in host cells10,11. FAs and lyso-compounds tend to be used as building blocks for this complex lipidome modulation11. As key players in inducing inflammation, lipid mediators of prostaglandins and leukotrienes are synthesized from phospholipid-derived polyunsaturated FAs (PUFAs)24. Phospholipase, hydrolyzes phospholipids into fatty acids and other lipophilic substances (e.g., lyso-phospholipids), leading to membrane dysfunction or even disruption of the cell11. Then, cyclooxygenase COX-2 converts the PUFA of arachidonic acid to prostaglandin endoperoxide H211. To promote the synthesis of prostaglandins and leukotrienes, both Gram-negative and Gram-positive bacteria can trigger signal transduction pathways that enhance the activities of phospholipase and/or cyclooxygenase COX-2 in targeted cells25. Salmonella can deliver the effector protein SpiC to the cytosol of infected macrophages to alter host cell signaling and then promote an immunosuppressive phenotype that impairs bacterial killing26. Similar mechanisms are used by Pseudomonas to promote prostaglandin production for their own benefit11. Thus, the changes in those lipids with a complex structure of polyunsaturated hydrocarbon chains produced from the pools of FAs and LPCs confirmed the inflammation induction in WT and the significant improvement of inflammation in clpV-RNAi.
Taken together, as the direct link of the metabolome with cellular phenotype, the cellular metabolome would be an attractive target for pathogens to modulate host cell processes to facilitate their survival and replication15,27. Metabolic crosstalk between host and pathogen profoundly shapes the pathogenesis of an infection15. Currently, research in the area of cell biology has yielded a series of findings regarding P. plecoglossicida infection2,3,6,7,8,9; however, the analysis of metabolic crosstalk between host and pathogen is limited and thereby has gained interest recently. Our metabolomics study revealed global metabolic changes associated with wild-type strain infection, indicating key disturbances in host nutritional utilization and immune responses. From this perspective of the metabolome, our study highlighted the efficiency of the key virulence gene clpV, consistent with our previous gene expression discoveries. Further research will be performed involving a more comprehensive lipidomics study.
In this study, a P. plecoglossicida strain (NZBD9) and the economic fish E. coioides were investigated as an attractive model system to characterize pathogen-induced host metabolic remodeling over the course of infection using nontargeted metabolomics approach. This comprehensive metabolomics study revealed how strains shape host metabolism to support their survival and replication and highlighted the potential of the virulence gene clpV in the treatment of an infection. Then, this study would expand our understanding of biochemical mechanism for host–pathogen interactions and provide novel clues for therapeutic approaches to manipulate and prevent progression of an infection in aquaculture.
Materials and methods
Bacterial strains
The wild-type P. plecoglossicida strain (NZBD9) was isolated from the spleen of naturally infected Larimichthys crocea28. According to previous methods with minor modification2,29, the P. plecoglossicida RNAi strain with optimal silencing efficiency targeting clpV was constructed. Details of the wild-type strain and clpV-RNAi strain are provided in our previously published studies2.
E. coioides infection and sampling
All E. coioides (about 100 g) were obtained from Zhangzhou (China). The protocol of the fish experiment was approved by the Animal Ethics Committee of Jimei University (Acceptance NO. JMULAC201159, Xiamen, China), complying with the ‘Guide for the Care and Use of Laboratory Animals’ set by the National Institutes of Health. The study is also in accordance with ARRIVE guidelines (https://arriveguidelines.org).
The P. plecoglossicida–E. coioides infection model was established as described in our previous reports2,7,8,9. The scheme of this fish experiment is provided in Fig. 1A. Briefly, after 1 week of adaptive inhabitation at 18 °C under specific pathogen-free laboratory conditions, all size-matched E. coioides were randomly divided into three groups: PBS control group (PBS group), wild-type strain infection group (WT group) and clpV-RNAi strain infection group (clpV-RNAi group). E. coioides of the infection group were intrapleurally injected with 105 colony-forming units per fish (cfu/fish) of P. plecoglossicida (wild-type strain or clpV-RNAi strain), while control E. coioides received an equivalent volume of PBS. The temperature of the water was maintained at 18 ± 1 °C during the infection experiment. The spleens of E. coioides were collected at 6, 12, 24, 48, 72 and 96 hpi. Seven independent biological replicates were prepared for each group at each time point. Finally, all 126 spleen specimens from 6 time points were stored immediately at − 80 °C.
Non-targeted metabolomics analysis
The metabolite extract (cf. Supplement) from each biological replicate was analyzed to conduct a non-targeted metabolomics study using an ACQUITY UPLC system (Waters, USA) coupled with Q-Exactive HF MS (Thermo Fisher Scientific, USA). Details of the metabolomics analysis including sample preparation, equipment, and methods are provided in the Supporting Information.
QC samples were obtained from pooled metabolic extracts and prepared as real samples30. These QC samples were analyzed every 10 injections during the entire run to monitor the robustness of the analysis.
Data processing and statistics
Approximately 2000 metabolite standards were preanalyzed by our collaborator to develop an in-house database31. This in-house database was used for peak identification based on accurate m/z, MS/MS fragmentation patterns, and retention time31. The original intensities of metabolites were normalized to the weight of spleen tissue, followed by the intensity of internal standards to eliminate systematic bias. Then, this processed dataset was employed for subsequent statistical analysis. Details of the data processing and statistics are provided in the Supporting Information.
Data availability
The datasets generated and/or analysed during the current study are available in the Metabolomics Workbench (https://www.metabolomicsworkbench.org/) repository (Data Track ID 3186).
References
Zhang, J. T., Zhou, S. M., An, S. W., Chen, L. & Wang, G. L. Visceral granulomas in farmed large yellow croaker, Larimichthys crocea (Richardson), caused by a bacterial pathogen, Pseudomonas plecoglossicida. J. Fish Dis. 37, 113–121. https://doi.org/10.1111/jfd.12075 (2014).
Luo, G. et al. clpV is a key virulence gene during in vivo Pseudomonas plecoglossicida infection. J. Fish Dis. 42, 991–1000. https://doi.org/10.1111/jfd.13001 (2019).
Tang, Y. et al. Mechanistic insight into the roles of Pseudomonas plecoglossicida clpV gene in host-pathogen interactions with Larimichthys crocea by dual RNA-seq. Fish Shellfish Immunol. 93, 344–353. https://doi.org/10.1016/j.fsi.2019.07.066 (2019).
Pietrosiuk, A. et al. Molecular basis for the unique role of the AAA(+) chaperone ClpV in type VI protein secretion. J. Biol. Chem. 286, 30010–30021. https://doi.org/10.1074/jbc.M111.253377 (2011).
Ho, B. T., Dong, T. G. & Mekalanos, J. J. A view to a kill: The bacterial type VI secretion system. Cell Host Microbe 15, 9–21. https://doi.org/10.1016/j.chom.2013.11.008 (2014).
Luo, G. et al. Integrated dual RNA-seq and dual iTRAQ of infected tissue reveals the functions of a diguanylate cyclase gene of Pseudomonas plecoglossicida in host-pathogen interactions with Epinephelus coioides. Fish Shellfish Immunol. 95, 481–490. https://doi.org/10.1016/j.fsi.2019.11.008 (2019).
Tang, R. Q. et al. Dual RNA-Seq uncovers the function of an ABC transporter gene in the host pathogen interaction between Epinephelus coioides and Pseudomonas plecoglossicida. Fish Shellfish Immunol. 92, 45–53. https://doi.org/10.1016/j.fsi.2019.05.046 (2019).
Luo, G. et al. Time-resolved dual RNA-seq of tissue uncovers Pseudomonas plecoglossicida key virulence genes in host-pathogen interaction with Epinephelus coioides. Environ. Microbiol. 22, 677–693. https://doi.org/10.1111/1462-2920.14884 (2020).
Xin, G. et al. Integration of RNA-seq and RNAi provides a novel insight into the effect of pvdE gene to the pathogenic of Pseudomonas plecoglossicida and on the immune responses of orange-spotted grouper (Epinephelus coioides). Aquaculture 529, 735695. https://doi.org/10.1016/j.aquaculture.2020.735695 (2020).
van der Meer-Janssen, Y. P. M., van Galen, J., Batenburg, J. J. & Helms, J. B. Lipids in host-pathogen interactions: Pathogens exploit the complexity of the host cell lipidome. Prog. Lipid Res. 49, 1–26. https://doi.org/10.1016/j.plipres.2009.07.003 (2010).
Helms, J. B. et al. Targeting of the hydrophobic metabolome by pathogens. Traffic 16, 439–460. https://doi.org/10.1111/tra.12280 (2015).
Lotscher, J. & Balmer, M. L. Sensing between reactions—How the metabolic microenvironment shapes immunity. Clin. Exp. Immunol. 197, 161–169. https://doi.org/10.1111/cei.13291 (2019).
Olive, A. J. & Sassetti, C. M. Metabolic crosstalk between host and pathogen: Sensing, adapting and competing. Nat. Rev. Microbiol. 14, 221–234. https://doi.org/10.1038/nrmicro.2016.12 (2016).
Gogoi, M., Datey, A., Wilson, K. T. & Chakravortty, D. Dual role of arginine metabolism in establishing pathogenesis. Curr. Opin. Microbiol. 29, 43–48. https://doi.org/10.1016/j.mib.2015.10.005 (2016).
Ren, W. et al. Amino acids as mediators of metabolic cross talk between host and pathogen. Front. Immunol. 9, 319. https://doi.org/10.3389/fimmu.2018.00319 (2018).
Dai, W. et al. N-ethyl-2-pyrrolidinone-substituted flavan-3-Ols with anti-inflammatory activity in lipopolysaccharide-stimulated macrophages are storage-related marker compounds for green tea. J. Agric. Food Chem. 68, 12164–12172. https://doi.org/10.1021/acs.jafc.0c03952 (2020).
Nathan, C. & Shiloh, M. U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848. https://doi.org/10.1073/pnas.97.16.8841 (2000).
Iniesta, V., Gomez-Nieto, L. C. & Corraliza, I. The inhibition of arginase by N-omega-hydroxy-L-arginine controls the growth of Leishmania inside macrophages. J. Exp. Med. 193, 777–783. https://doi.org/10.1084/jem.193.6.777 (2001).
Cusumano, Z. T., Watson, M. E. & Caparon, M. G. Streptococcus pyogenes arginine and citrulline catabolism promotes infection and modulates innate immunity. Infect. Immun. 82, 233–242. https://doi.org/10.1128/iai.00916-13 (2014).
Seifi, H. S., Van Bockhaven, J., Angenon, G. & Hofte, M. Glutamate metabolism in plant disease and defense: Friend or foe? Mol. Plant-Microbe Interact. 26, 475–485. https://doi.org/10.1094/mpmi-07-12-0176-cr (2013).
Ramond, E. et al. Glutamate utilization couples oxidative stress defense and the tricarboxylic acid cycle in Francisella phagosomal escape. PLoS Pathog. 10, e1003893. https://doi.org/10.1371/journal.ppat.1003893 (2014).
Gouzy, A. et al. Mycobacterium tuberculosis nitrogen assimilation and host colonization require aspartate. Nat. Chem. Biol. 9, 674. https://doi.org/10.1038/nchembio.1355 (2013).
Kullas, A. L. et al. L-asparaginase II produced by Salmonella typhimurium inhibits T cell responses and mediates virulence. Cell Host Microbe 12, 791–798. https://doi.org/10.1016/j.chom.2012.10.018 (2012).
Tam, V. C. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin. Immunol. 25, 240–248. https://doi.org/10.1016/j.smim.2013.08.006 (2013).
Agard, M., Asakrah, S. & Morici, L. A. PGE(2) suppression of innate immunity during mucosal bacterial infection. Front. Cell. Infect. Microbiol. 3, 11. https://doi.org/10.3389/fcimb.2013.00045 (2013).
Uchiya, K., Groisman, E. A. & Nikai, T. Involvement of Salmonella pathogenicity island 2 in the up-regulation of interleukin-10 expression in macrophages: Role of protein kinase a signal pathway. Infect. Immun. 72, 1964–1973. https://doi.org/10.1128/iai.72.4.1964-1973.2004 (2004).
Pernas, L. Cellular metabolism in the defense against microbes. J. Cell Sci. 134, 9. https://doi.org/10.1242/jcs.252023 (2021).
Hu, J. et al. Isolation, identification and virulence of the pathogen of white-spots disease in internal organs of Pseudosciaena crocea. Oceanol. Limnol. Sin. 45, 409–417 (2014).
Darsigny, M. et al. Hepatocyte nuclear factor-4 alpha promotes gut neoplasia in mice and protects against the production of reactive oxygen species. Cancer Res. 70, 9423–9433. https://doi.org/10.1158/0008-5472.Can-10-1697 (2010).
Zeng, J. et al. Lipidome disturbances in preadipocyte differentiation associated with bisphenol A and replacement bisphenol S exposure. Sci. Total Environ. 753, 10. https://doi.org/10.1016/j.scitotenv.2020.141949 (2021).
Zhao, X. J. et al. Comprehensive strategy to construct in-house database for accurate and batch identification of small molecular metabolites. Anal. Chem. 90, 7635–7643. https://doi.org/10.1021/acs.analchem.8b01482 (2018).
Acknowledgements
The study has been supported by National Key R&D Program of China (2019YFD0901704), Cultivation Plan for Distinguished Young Scholars in Fujian Province University (KL41843) and Natural Science Foundation of Fujian Province of China (2022J01330).
Author information
Authors and Affiliations
Contributions
J.Z. and Z.Y. collected the data, wrote the manuscript, G.L. provided samples, Z.Y., Y.Z., Y.Z. and J.H. analyzed and discussed the raw data, J.Z. and Q.Y. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, J., Yang, Z., Zhong, Y. et al. Metabolomics insights into the interaction between Pseudomonas plecoglossicida and Epinephelus coioides. Sci Rep 12, 13309 (2022). https://doi.org/10.1038/s41598-022-17387-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17387-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
