
Abstract
Omobranchus punctatus is native to the Indo-Pacific region and invasive in the Atlantic region, currently being considered one of the most widely distributed blenny species. However, recent molecular studies indicated that O. punctatus is a complex of species, with three divergent mtDNA lineages identified to date, stressing the need for a taxonomic revision. In this study, we used an integrative approach, combining morphological and genetic data, to shed light on the taxonomy and distribution of O. punctatus. Moreover, we provide the first genetic records of introduced populations in Brazil and discuss the introduction pattern of this species in this region. Morphological data shows that O. punctatus consists of at least five distinct and geographically restricted species: O. punctatus sensu stricto, O. dispar, O. sewalli, O. cf. kochi, and O. cf. japonicus. Species delimitation analyses performed using the mtDNA data available confirmed that O. punctatus sensu stricto, O. dispar and O. sewalli correspond to different species that started to diverge about 2.6 Mya. Furthermore, O. sewalli was identified as the invasive species colonizing Atlantic shores. The existence of historical oceanographic barriers, such as the emergence of the Sunda Shelf in the Eastern Indian Ocean during the Pleistocene, and the biological traits of these blennies are the most likely factors responsible for their genetic differentiation and subsequent speciation.
Similar content being viewed by others

Introduction
Invasive alien species (IAS) constitute one of the major threats to marine biodiversity and ecosystems worldwide1. IAS can out-compete native species, act as facilitators of hosts and/or vectors of parasites and pathogens, change the community structure, and alter ecosystem processes, thereby impairing the associated ecosystem function. Ultimately, IAS can lead to significant economic impacts with its associated detrimental effects on human well-being2.
Fishes are among the most commonly introduced organisms globally3, with documented increases over recent decades due to globalization, changes in seawater temperatures, and the ever-increasing magnitude of shipping, aquaculture, fisheries, aquarium trade, and habitat modification (e.g., dams, canals or waterways, urbanization and deforestation)1,4,5. Nevertheless, most genetic and genomic studies on marine alien fishes have been mainly focused on a relatively small number of taxa6. In this context, blennies (Blenniidae) are among the most neglected groups of reef vertebrates7, despite their high invasive potential8,9 and being one of the most diverse families of teleost fishes (with at least 405 species widely distributed in tropical and subtropical habitats)10,11. As far as we know, a total of 11 blenny species have so far been introduced far from their native range (Supplementary Table S1): e.g., Petroscirtes breviceps12 to Papua New Guinea13, Omobranchus anolius12 to New Zealand14, or Parablennius thysanius (Jordan & Seale 1907) to the Hawaiian Islands and the Mediterranean Sea15,16.
Omobranchus punctatus12 is thought to be one of the most widely distributed blenny species, largely due to transport associated with ship’s ballast water and biofouling, which favour its long-distance dispersal17,18,19,20,21. It is considered native to the Indo-Pacific region, ranging from the Persian Gulf, in the Western Indian Ocean (WIO) and the Arabian Sea, to South-East Asia, Japan, Australia and the Fiji Islands, in the Western Pacific Ocean (WPO) (Fig. 1 and Supplementary Table S2, and references therein). The first occurrences of O. punctatus outside its native range were recorded from 1930 to 1963 in five localities on the island of Trinidad (Republic of Trinidad and Tobago) and one in Venezuela, in the Western Atlantic Ocean (WAO)17,22,23 (Supplementary Table S3). Since then, O. punctatus has been identified in another 39 localities on the Atlantic coast of Central and South America, including Panama (1966–1974, three localities), Colombia (1989, one locality), Venezuela (1978–2009, 14 localities), and Brazil (2002–2019, 21 localities) (Fig. 1 and Supplementary Table S3). Moreover, it has also been introduced to the Suez Canal, the Mediterranean and Red Sea, and along the eastern coast of Africa (Fig. 1 and Supplementary Table S4, and references therein). According to historical and morphological analyses, it seems that O. punctatus was first introduced to the WAO on slave boats from the Bay of Bengal (Madras or, more probably, Calcutta) to Trinidad, secondarily spreading to Venezuela, Panama and Colombia17,21,24,25,26, and then to different Brazilian localities4,17,19,27,28,29 (Supplementary Table S3).

Current global distribution of Omobranchus punctatus group including its native (in yellow) and introduced range (in red). See Tables S2–S4 for further details for each location (year of the first record, reference collection, sources, and genetic data availability). Records of introduced populations in the Western Atlantic Ocean (WAO) would correspond to Omobranchus sewalli23, and that from the Western Indian Ocean (WIO) and the Mediterranean Sea to Omobranchus cf. sewalli (see Table 2).
Although there is no genetic data for this exotic blenny in the Bay of Bengal (the putative origin of invasive populations), and only one mitochondrial sequence is available from its introduced range (Venezuela)30 (Supplementary Table S3), recent molecular analyses showed the existence of several highly divergent genetic lineages across the species’ distribution. First, the study of Gibbs et al.31 showed the existence of three deeply divergent lineages within O. punctatus: two lineages in the Eastern Indian Ocean (EIO), separated by the Sunda Shelf Barrier (SSB), were confirmed by both mitochondrial and nuclear data; a third lineage was identified in Japan with nuclear markers, with no mtDNA data currently available for the species in this region. Later, Mehraban et al.32, found an additional highly divergent mtDNA lineage in the Oman Sea, which the authors suggested could correspond to the originally described O. punctatus, due to the proximity to the species type locality (Mumbai = Bombay, India). Recently, a phylogenetic study showed that populations outside O. punctatus’ native range, specifically those from the Gulf of Paria in Venezuela, clustered within the clade of individuals from the west of the SSB30. These data, together with the fact that the species is continuingly spreading in the Atlantic coast, highlight the urgent need to resolve O. punctatus’ taxonomy.
In order to continue to shed light on O. punctatus’ taxonomy and distribution, we conducted an integrative study of both invasive and native populations of this species, using morphological data and molecular species delimitation methods. Specifically, we aimed to: 1) confirm whether this exotic blenny represents a complex of undetected cryptic species (taxa that are morphologically similar but genetically divergent)33,34; and if so, 2) clarify whether they can be distinguished based on morphological characters; 3) examine their phylogenetic relationships and estimate their divergence times; and 4) unveil their distribution. In addition, we provided the first genetic records for introduced Omobranchus in Brazil.
Results
Phylogenetic data and species delimitation
The COI dataset included a total of 44 sequences, 28 from this study and 16 available from GenBank, originating from previous publications (Table 1). After quality filtering, the length of the final alignment was 560 bp. No stop codons, insertions, or deletions were observed.
Phylogenetic analyses based on BI and ML approaches, rendered trees with similar overall topologies, with main clades receiving high bootstrap and posterior probabilities (> 90% and 0.99 respectively, Fig. 2). Both analyses supported the monophyly of O. punctatus group and the existence of three highly divergent lineages: the first one including Taiwan, China, and the Gulf of Thailand populations (Clade A), the second one restricted to the Oman Sea (Clade B), and the third one including Western Atlantic (Venezuela and Brazil) and Andaman Sea (Thailand) populations (Clade C). Mean K2P distance between these clades ranged from 4.9 to 6.0%, with the lowest value found between Clades A and C, and the highest between Clades B and C. Genetic divergence between O. punctatus clades and the congeneric species O. woodi was on average 11.8% (ranging from 10.8 to 12.4%).

Bayesian consensus tree of Omobranchus punctatus group, based on COI sequences. Bayesian posterior probabilities (BPP) over 0.99 are represented by red circles at nodes and values correspond to bootstrap support (>75%) given by the maximum likelihood analyses. Clades A-C are identified. The tree was rooted with O. woodi (sequences available in GenBank: JF494019, HQ561537, HQ561538). Vertical black bars represent results from the species delimitation analyses: Barcode Index Number (BIN), Assemble Species by Automatic Partitioning (ASAP) and Bayesian Poisson Tree Process model (bPTP).
The minimum spanning network also supported the existence of three divergent groups within O. punctatus (Fig. 3), which matched the distinct clades observed in the phylogenetic analyses (Fig. 2). A total of 14 haplotypes were identified: 8 in Clade A, two in Clade B and four in Clade C. From these haplotypes, 10 were unique and only four (H1-H3 and H8) were shared by more than one individual. Within each clade, most haplotypes were separated by few mutational steps. However, clades were separated by at least 21 mutational steps (Fig. 3). Overall, 11 haplotypes (H4-H14), most of them restricted to a single location, were identified in the native range at the Indo-Pacific region (WIO, EIO, WPO); meaning that only 3 haplotypes were found among introduced populations of the Atlantic coast of Central and South America (Fig. 3, Table 1). No haplotypes were shared between native and introduced populations. Nevertheless, the haplotype from the Andaman Sea population (H4) was separated by only one nucleotide substitution from the most abundant haplotype found in Brazil (H3). On the other hand, within the three haplotypes observed in the introduced populations, only the haplotype H2 was shared between Brazilian and Venezuelan individuals.

Median-joining network of all COI sequences for the Omobranchus punctatus group. Localities are coded by filling patterns (see legend). Each circle represents a haplotype, and its size is proportional to the observed haplotype frequency. Non-observed haplotypes are represented by small white circles. Every crossbeam on the connecting lines between haplotypes represents a single mutational step. Distinct clades (A-C) are depicted as dashed-lines circles.
Finally, all species delimitation analyses (BIN, ASAP and bPTP) clustered the sequences of O. punctatus in 3 distinct MOTUS (Fig. 2). These results are in agreement with those from phylogenetic analysis and therefore, strongly support the existence of three distinct species.
Evolutionary divergence
The MCC tree (Fig. 4) recovered a sister clade relationship of Western Atlantic haplotypes (and Andaman Sea) (Clade C) to all other haplotypes (Clades A and B) and was highly similar to the BI topology (Fig. 2). Posterior probabilities higher than 98% were recovered for all but one main node. Based on a mutation rate of 1.2% per Mya, which is commonly used to date divergence between fish35,36,37, the estimated divergence times between Clade C and rest of haplotypes was 2.6 Mya (1.7–3.6, 95% HPD). The estimated divergence time between the Clade B, including Oman Sea populations, and the rest of the native haplotypes (Clade A) was of 2.1 Mya (1.3–2.9, 95% HPD). Divergence time between populations from the Western Atlantic (haplotypes H1, H2 and H3) and the Andaman Sea (haplotype H4) was estimated at 180,000 years ago (0.05–0.39, 95% HPD), within the late Pleistocene, whereas divergences times within western Atlantic haplotypes occurred within the early Holocene. Time to most recent common ancestor for haplotypes belonging to Clades A and B was estimated at 0.45 Mya (0.22–0.75, 95% HPD) and 0.33 Mya (0.1–0.68, 95% HPD), respectively.

Bayesian time tree for Omobranchus punctatus group as inferred by BEAST. Scale bar in Mya. The green clade represents the samples sequenced in this study. Clades A-C are identified. Bayesian posterior probabilities are represented by colour and node size (red values by nodes are strongly supported). Values by nodes indicate the estimated age of the split event and horizontal blue bars represent 95% of the highest posterior density (HPD) interval. H denotes Holocene. Photo of O. punctatus from Venezuela by James Van Tassell (American Museum of Natural History) and Ross Robertson (Smithsonian Tropical Research Institute).
Morphological analysis and species nomination
In the PCA based on 10 meristic characters of 584 individuals identified as O. punctatus, the first two principal components (PC1 – 49.09%; PC2 – 20.44%) accounted for nearly 70% of the total variance (Supplementary Table S5). This analysis clustered the individuals into 5 morphogroups (Fig. 5), 3 of them matching the genetic clades observed in the phylogenetic analyses. Morphogroup 1 included all individuals from Papua New Guinea, and Salomon and Moluccas islands, all of them in the WPO (Fig. 5, Supplementary Table S6). Morphogroup 2, the biggest one and which corresponds to the genetic Clade C (Fig. 2), grouped individuals from 21 locations belonging to the 4 regions analysed in the present study (WIO, EIO, WPO and WAO) (Fig. 5, Supplementary Table S6). Interestingly, all Western Atlantic introduced populations (Panama, Trinidad, Venezuela, and Brazil) were included in this group, which also included native populations from the Andaman Sea (Nicobar Islands and west coast of Thailand) and the Gulf of Bengal (Sri Lanka and Vizagapatam) in the EIO (Fig. 5, Supplementary Table S6). Morphogroup 3 consisted of individuals from the WIO, specifically from the Gulf of Oman, the Arabian Sea, and the type locality of Bombay (Fig. 5, Supplementary Table S6). This group corresponds to the Clade B in the phylogenetic analyses (Fig. 2). Morphogroup 4 included individuals from two Australian populations together with those from Japan and the Fiji Islands (Fig. 5, Supplementary Table S6). This morphogroup could correspond to the clade that included an individual from Japan recovered by Gibbs et al.31 based on nuclear genetic data. Finally, the last morphogroup, which corresponds to the genetic Clade A (Fig. 2), included Chinese populations (Hong Kong and Zhoushan Island) located at the Fujian Coast (Fig. 5, Supplementary Table S6).

Principal Component Analysis (PCA) based on 10 meristic characters analysed in 36 populations (localities) of Omobranchus punctatus group. 1: Omobranchus cf. kochi, 2: Omobranchus sewalli; 3: Omobranchus punctatus sensu stricto, 4: Omobranchus cf. japonicus, and 5: Omobranchus dispar. Populations analysed are listed in Table S6. First (Dim 1) and second (Dim 2) principal components accounted for nearly 70% of the total variance.
Both genetic and morphological results supported the existence of at least three distinct species within the O. punctatus group. Thus, following the Priority Principle of the ICZN and considering the existent synonyms and their corresponding type locality (Supplementary Table S7), the different clades/morphogroups were identified as follow (Table 2). The genetic Clade A or morphogroup 5 corresponds to Omobranchus dispar38, whose type locality is Amoy (now Xiamen) at the Fujian coast (Supplementary Table S7). The Clade B or morphogroup 3 should be considered the true Omobranchus punctatus, hereafter O. punctatus12 sensu stricto (Table 2), since it included the Bombay population, the type locality of this nominal species (Supplementary Table S7). Finally, Clade C or morphogroup 2 corresponds to Omobranchus sewalli23 (Table 2), a species first described in Trinidad (Supplementary Table S7). Although further molecular analyses should be conducted to confirm the species status of the remaining morphogroups (1 and 4), they could correspond to Omobranchus cf. kochi39 (type locality = mouth of the Meranke River at Papua New Guinea) (Supplementary Table S7) and Omombranchus cf. japonicus40 (type locality = Tokyo, Japan) (Supplementary Table S7), respectively.
Discussion
In this study we used an integrative approach, using morphological and the most comprehensive genetic dataset available to date (mtDNA), to shed light on the taxonomy and worldwide distribution of the Omobranchus punctatus group. Our morphological analysis suggests the existence of five species within this group, three of which could be genetically confirmed by the present study, and another one supported by the genetic data of Gibbs et al.31. Although further molecular and morphological analysis are required to fully understand the distribution of these species, genetic and morphological data were generally congruent. The present results also suggest that only Omobranchus sewalli is found beyond the Indo-West Pacific region, the native range of the genus.
Species delimitation
Although Omobranchus Valenciennes12 is the most species-rich genus in the Omobranchini tribe of the Blenniidae, with 23 valid species24,41, its actual diversity remains underestimated7,30,31,32.
Our phylogenetic analyses are in agreement with those of Gibbs et al.31, Cabezas et al.30 and Mehraban et al.32, recovering three well differentiated and supported mitochondrial lineages within O. punctatus group (Clades A, B and C) (Figs. 2 and 3), which were also confirmed by three different approaches of species delimitations methods (Fig. 2). The genetic divergences recovered were slightly lower as compared to what has been established in earlier studies on blennies species32,42. However, they were greater or equal tenfold to the mean intraspecific distance established in the seminal work by Hebert et al.43 and exceed the threshold value of 3% established for species delimitation in fishes44. On the other hand, morphological analyses based on meristic characters discriminated five distinct morphogroups (Fig. 5), three of them corresponding to the genetic lineages recovered by the phylogenetic analyses. Unfortunately, for the remaining two groups no mitochondrial data were available. As in previous studies24, no single character could be discerned to distinguish between morphogroups, something that seems to be common in small, cryptobenthic fishes where most species can only be distinguished through combinations of different morphological traits7,42. To further understand which characters could be used in species diagnosis, a further extensive and in-deep morphological analysis, with more specimens, would be needed. Overall, the concordance between molecular and morphological groups, strongly supports that the muzzled blenny Omobranchus punctatus is a species complex consisting of at least three distinct species, which are geographically restricted.
The use of integrative taxonomic approaches (combining morphological, genetic, and ecological data, among other characters) has shown to be the best strategy to produce well-supported species delimitations45,46. However, most studies concentrate exclusively on documenting current species diversity or identifying independent lineages without naming them46, as is also the case for previous studies on O. punctatus30,31,32. The proper naming of the detected lineages is essential in biodiversity assessments and for its subsequent conservation planning45, especially when dealing with IAS47. Therefore, following the ICZN and considering the list of synonymies attributed to O. punctatus24,41 (Supplementary Table S7), we were able to identify each phylogenetic/morphological group (Table 2). According to our results, specimens from the first genetic Clade (Clade A) and the morphogroup 5, mainly distributed in China (Figs. 2, 3 and 5), can be attributed to Omobranchus dispar38 (Table 2), a species originally described as Petroscirtes dispar Günther, 1861 from the Chinese locality of Amoy (Xiamen) (Supplementary Table S7). As observed in previous molecular studies30,32, Clade A also included specimens sequenced from Gulf of Thailand and Taiwan localities (Figs. 2 and 3). The analyses of meristic characters, however, placed them in the morphogroup 2 (Fig. 5; Supplementary Table S6), probably due to morphological stasis48. For this reason, although Taiwan and Thailand specimens could belong to O. cf. dispar (see Supplementary Table S7), such taxonomical arrangement will require an additional morphological and genetic analysis.
The second clade (Clade B) and its corresponding morphogroup 3 were restricted to the WIO (Figs. 2, 3 and 5). Since these included the specimens from Bombay (type locality) and nearby localities from the Oman Sea, we designated it as Omobranchus punctatus sensu stricto (hereafter O. punctatus s.s.) (Table 2), in accordance with what was concluded in the study by Mehraban et al.32. Although genetic data for Bombay is lacking, this locality could be the source population for O. punctatus in the Oman Sea, due to the ocean currents pattern in this region, and which could explain the morphological affinities found between these populations (Fig. 5). The complex oceanographical conditions in the region49,50 would allow the connectivity between O. punctatus populations along the WIO (North Arabian Sea), ensuring a significant level of gene flow and, therefore, preventing speciation. In fact, other records of this blenny species have been documented from the Arabian Sea (Fig. 1, Supplementary Table S2), such as in the Gulf of Kutch (India) and the coast of Karachi (Pakistan), where it was described as Salarias sindensis (Day, 1888), currently considered a synonym of O. punctatus s.s. (Supplementary Table S7).
The Clade C, and its corresponding morphogroup 2, showed a wider distribution (Figs. 2, 3 and 5; Supplementary Table S6). All populations from the WAO, including those from Brazil, sequenced and morphologically analysed for the first time in the present study, were included in this third lineage. The Andaman Sea (Thailand) population from the EIO (Bay of Bengal) was also included in this clade (Figs. 2 and 3), thus agreeing with recent phylogenetic studies30,32. Morphogroup 2 included a larger number of populations from all the four oceanic regions (WIO, EIO, WPO and WAO) (Fig. 5; Supplementary Table S6). The population of Trinidad, the first record of “O. punctatus” in WAO24, was also included in this group, therefore supporting the morphological affinity previously found between WAO and EIO populations24. Considering all the above, we assigned Clade C and morphogroup 2 to the species Omobranchus sewalli23 (Table 2), originally described as Poroalticus sewalli by Fowler23 from tide pools of the west coast of Trinidad in the WAO (Supplementary Table S7). Interestingly, O. sewalli is the only species of the genus that has been described based on an introduced population23. To the best of our knowledge, this could be one of few cases where an introduced fish was described as a new species23, later synonymized (Supplementary Table S7)24,41, and posteriorly confirmed as a species through to morphological and genetic data (present study).
Finally, morphological analyses retrieved two additional groups (morphogroups 1 and 4), for which no mitochondrial data are available (Fig. 5; Supplementary Table S6). Morphogroup 1 included populations from Papua New Guinea and nearby islands, and could correspond to Omobranchus kochi39 (referred herein as Omobranchus cf. kochi, Supplementary Table S7), a species first described from the Meranke River in southern New Guinea (Supplementary Table S7). On the other hand, morphogroup 4 included Japan, Australia, and Fiji Islands populations (Fig. 5; Supplementary Table S6). Because this group may correspond in part, to the highly divergent genetic lineage found by Gibbs et al.31 based on nuclear data from individuals collected in Kagoshima, Japan, we assigned these specimens to Omobranchus cf. japonicus40 (Supplementary Table S7). However, further molecular analyses are necessary to confirm the taxonomic status of fishes from these two regions.
Phylogeographic and ecological insights
The fact that most benthic marine species have planktotrophic (self-feeding) larvae that can spend days to months in the water column, has led marine ecologists to presume that most marine populations are demographically “open” and therefore naturally highly connected51,52. However, many studies in the last decade have confirmed this perception to be inaccurate42,53, with oceanographic processes (e.g., oceanographic barriers, currents, habitat discontinuities) and the biological traits of species (e.g., dispersal abilities, larval duration) being the main likely mechanisms responsible of population differentiation and their subsequent speciation52,53.
In the present study, the geographic distribution of the three lineages reported for the O. punctatus group, now recognized as three distinct species (O. dispar, O. punctatus s.s., and O. sewalli), indicates that their limited dispersal abilities but also the existence of oceanographic barriers played an important role in the differentiation of these species. In the EIO and WPO, the separation between O. sewalli and O. dispar, based on a mutation rate of 1.2% per Mya35,36,37, seems to have occurred at the beginning of the Pleistocene (~ 2.6 Mya; Fig. 4), coinciding with the emergence of the SSB, a phylogenetic break located at the Thai-Malay Peninsula (TMP)54. The emergence of the SSB due to sea-level lowering during Plio-Pleistocene glaciations restricted gene flow between the tropical Indian Ocean and the WPO, which possibly led to isolation among populations and their further differentiation55. Therefore, allopatric speciation seems to be the most likely scenario to explain the separation between O. sewalli and O. dispar in this region. In fact, the presence of this phylogeographic break separating populations from the Andaman Sea from those located West of the TMP has been suggested in previous studies of these species30,31,32. Moreover, it has also been documented for other fishes56, including other Omobranchus species31, as well as for many other marine taxa, including sharks57, crustaceans58, and molluscs59. Interestingly, specimens from the Gulf of Thailand were morphologically more closely related to those on the west side of the TMP than to WPO individuals (Fig. 5). Long-lasting extreme environmental conditions, as those from estuaries, intertidal areas, and tide pools that Omobranchus species inhabit, may have prevented morphological differentiation by imposed stabilizing selection on morphology48. Further molecular and morphological analyses including additional populations from both regions are necessary to resolve the inconsistency found between both type of analyses, which could indicate the possible coexistence of both lineages and/or the existence of hybrids.
On the other hand, as suggested by Mehraban et al.32, different hydrological and ecological characteristics prevalent in the Arabian Sea and the Bay of Bengal (Andaman Sea) could explain the differentiation between O. punctatus s.s and O. sewalli ( Figs. 2, 3, 4 and 5, Table 2 and Supplementary Table S6), inhabiting the Indian Ocean, and that occurred at approximately 2.6 Mya (Fig. 4). Populations inhabiting divergent environments deal with different selection pressures during their evolution, which determine their geographic distribution, and enhance ecological speciation60.
In addition, the life history traits of Omobranchus species could also have favoured the differentiation of O. punctatus s.s, O. dispar, and O. sewalli. The species of this genus are small, benthic inhabitants of intertidal zones and tide pools in coastal marine and estuarine ecosystems17,20,24,61,62. They are considered as permanent residents, showing a strong site fidelity during most of their life20,21,27,61,63,64. Moreover, their fertilized eggs are adhesive and demersal, and larvae are planktonic, settling about 3–7 weeks after hatching65 usually in protected areas near to the coast (OML-A and JLSN personal observations). All these factors suggest that Omobranchus species have limited dispersal capabilities (by natural means)64,66, which could have affected the connectivity among populations, and, thus, promoted their genetic differentiation and subsequent speciation.
Introduction pattern of Omobranchus sewalli
Of the three species proposed in the present study, only O. sewalli occurs outside the Indo-West Pacific region (Fig. 5; Supplementary Tables S3, S6, and S7), the natural distribution range considered for Omobranchus species24,41. Indeed, O. sewalli is the only species of the genus recorded in the WAO (Supplementary Table S7)24,41. Considering the limited natural dispersal capabilities of this species, as discussed above, the presence of O. sewalli in this very distant geographical area could be explained as a result of human-mediated activities, which, intentionally or unintentionally, transport species beyond their natural ranges67 (see Supplementary Tables S1 and S3).
Supported by historical, morphological, and recent molecular analyses17,24,26,30,31,32, the Andaman Sea has been suggested as the most likely source population of O. sewalli in the WAO. The present results (Figs. 2, 3 and 5) also confirm this. In Brazil, the first occurrences of O. sewalli were registered in the states of Rio de Janeiro, Bahia, and Santa Catarina (Supplementary Table S3). Since then, the species spread rapidly to many other northern and southern localities, mainly by ballast water, biofouling, oil rigs and larval dispersal on nearshore ocean currents (see Supplementary Table S3and references herein). Nevertheless, due to the lack of molecular data, the source population and introduction pattern of O. sewalli in this region remained unknown until now. In the present study, we provided the first genetic records of this species for Brazil. Based on mitochondrial data, three haplotypes (H1–H3) were observed in this region (Table 1, Fig. 3). The presence of haplotype H2 in São Marcos, Barra Grande and Jericoacoara, which is also present in Venezuela (Table 1, Fig. 3), indicate that: 1) O. sewalli could have been introduced to Brazil directly from a Venezuelan population; or 2) the same pathway may have been responsible for the introduction of O. sewalli in Venezuela and Brazil. In addition, the exclusive presence of haplotypes H1 and H3 in all Brazilian populations, except for Jericoacoara (Table 1, Fig. 3), suggest that more than one introduction pathway may been operating in this region. Unfortunately, due to the limited genetic data available for the WAO no robust conclusions can be reached.
Human-mediated dispersal also seems to be responsible for the introduction of this blenny in the east coast of Africa (Mozambique, Madagascar, Tanzania, Kenya, and South Africa (Fig. 1, Supplementary Table S4) 9,17. The main arguments are: 1) the limited natural dispersal capabilities of this species; 2) the ocean currents pattern existing in the region; and 3) the remoteness from the closest native populations (likely the Andaman Sea – 7000 km). The morphological analyses conducted in the present study grouped Mozambique, Andaman Sea and WAO populations together (morphogroup 2, Fig. 5; Supplementary Table S6), suggesting a close relationship between them and indicating that they could be the same species. For this reason, we suggest that Mozambique (previously described as Omobranchus japonicus scalatus by Smith 1959) and the remaining African populations of this blenny should be considered as Omobranchus cf. sewalli (Supplementary Table S7), pending further confirmation.
Conclusions
This is the first study to perform an integrative analysis combining morphological and genetic data on the Omobranchus punctatus group. Our data suggests the existence of five species within this group, with O. sewalli identified as the invasive species colonising Atlantic shores. Considering the association of O. sewalli with man-made vectors, its high tolerance to a wide range of salinity levels and adverse environmental conditions9,17,18,21, and its capability of self-recruitment19,64, further introductions of this species are likely expected. Recent records of O. sewalli in Brazil (see Supplementary Table S3), confirm this assumption.
Methods
Sample collection
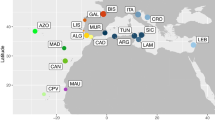
Between 2012 and 2021, a total of 28 specimens of the putative species O. punctatus, were collected, using hand nets and anaesthetic clove oil63, from intertidal flat reefs at six localities along the Brazilian coast (Fig. 6, Table 1 and Supplementary Table S3). For each specimen, a small fragment of muscle and fin tissue was removed and stored in 96% ethanol for the molecular analyses. Voucher specimens were fixed in 10% formalin, and later stored in 70% ethanol and deposited at the Fish Collection of the Universidade Federal de Maranhão (CPUFMA-UFMA), under the numbers CPUFMA 3476–3478, and at the Universidade Federal do Pará (Aquatic Ecology Group – GEA.ICT) under the numbers GEA.ICT#241, 242, 253, 04214, 04222 and 12001 (Table 1). Samples were collected with the permission of the Instituto Chico Mendes de Conservacão da Biodiversidade (ICMBio) and the Sistema de Autorizacão e Informacão em Biodiversidade (SISBIO), license numbers 67481 and 35625, respectively. The care and use of experimental animals complied with Brazilian animal welfare laws, guidelines, and policies. No surgical procedures were performed, and no procedures that cause lasting harm to the fish were carried out. All methods conducted were in accordance with ARIIVE guidelines.

Sampling locations of the putative species Omobranchus punctatus (species considered in this work as Omobranchus sewalli) along the Atlantic coast of South America. See Table 1 for additional information.
DNA extraction, PCR amplification, and sequencing
Total genomic DNA was extracted from a small amount of alcohol-preserved muscle tissue using the Purelink Genomic DNA Mini Kit (Invitrogen, Paisley, UK), according to the manufacturer’s protocol. A fragment (~ 670 bp) of the mitochondrial cytochrome c oxidase subunit I (COI, standard barcoding fragment) was amplified by polymerase chain reaction (PCR) using the M13 tailed primer cocktail C_FishF1t1 and C_FishR1t168. PCR amplifications consisted of 25 µl reaction volumes containing 3–5 µl of template DNA, 10 × buffer MgCl2 free (Invitrogen), 2 mM MgCl2, 0.2 mM dNTPs, 1 µM of each primer, 0.3 U Platinum Taq DNA polymerase (Invitrogen), and double-distilled H2O to volume. PCR conditions used were as described in Cabezas et al.30.
The resulting PCR products were purified and bidirectionally sequenced at GENEWIZ (Leipzig, Germany).
Sequence analysis and phylogenetic reconstructions
All newly obtained sequences were edited with Sequencher v5.4.6 (Gene Codes Corporation, Ann Arbor, MI, USA), and checked for potential contaminations using GenBank’s BLASTn search69. They were thereafter deposited in GenBank (see Table 1).
All COI sequences of O. punctatus (N = 13) available from GenBank (November 2021) were included in the phylogenetic analysis. Additionally, COI sequences of the closely related species O. woodi (Gilchrist & Thompson, 1908) were used as outgroups (see Table 1). Sequences were aligned using MUSCLE70 as implemented in MEGA X71. The final dataset was checked for the presence of pseudogenes by translating sequences into amino acids.
Phylogenetic tree reconstructions were performed using maximum likelihood (ML) and Bayesian inference (BI), through Garli v2.0.172 and MrBayes v3.2.673, respectively. Only one individual (or sequence) per haplotype was included in the phylogenetic analyses to reduce redundancy. Analyses were conducted using data partitions by codons (1 + 2 + 3) to minimize saturation effects of codon positions and to account for different rates of evolution of each one. The Akaike Information Criterion (AICc)74 implemented in PartitionFinder v2.1.175 was used to select the best fit evolutionary model for each partition. The resulting models were SYM (1st position), F81 + I (2nd position) and GTR + G (3rd position). ML analysis was performed using 10 independent searches and 1,000 bootstrap replicates. The evaluation of log-likelihood values across searches allowed to check the convergence between the topologies of the trees generated. The SumTrees command from the package DendroPy76 was used to summarize non-parametric bootstrap support values for the best tree, after generating a majority-rule consensus tree. For the BI analysis, two independent runs (each with four Markov chains for 2 × 107 generations) were performed. Trees and parameters were sampled every 1,000 generations, with the heating parameter set to 0.25. The convergence of the analyses was validated by the standard deviation of split frequencies being lower than 0.01 and by graphical monitoring of the likelihood values over time using Tracer v1.7.177. The majority-rule consensus tree was estimated combining results from duplicated analyses, after discarding 25% of the total samples as burn-in. Clades with bootstrap support or BI posterior probability (BPP) greater than 90% or 0.9, respectively, were considered well supported. The consensus tree inferred for each phylogenetic approach was visualized and rooted using FigTree v1.4.478, and later prepared as a graphic with the software Inkscape v1.0.1 (http://www.inkscape.org). Pairwise nucleotide distances among clades were calculated using the Kimura-2-Parameter model (K2P)79 implemented in MEGA X. In addition, relationships among haplotypes were further examined by building a median-joining network using the PopART v1.7 software80.
Molecular species delimitation
Analyses of species delimitation were performed on the COI dataset using three different approaches: two distance-based methods, the Barcode Index Number (BIN) system81 and the Assemble Species by Automatic Partitioning (ASAP)82; and one tree-based method, the Bayesian Poisson Tree Process (bPTP) model83. Using the BIN system of the Barcode of Life Data Systems (BOLD)84, COI sequences were clustered into molecular operational taxonomic units (MOTUs) independent of any prior taxonomic assignment, and then assigned to a unique alphanumeric code or BIN. This method provides a means of confirming the concordance between barcode sequence clusters and species designations81. The ASAP method was implemented on a web interface (https://bioinfo.mnhn.fr/abi/public/asap/asapweb.html), and it was applied with default settings using the K2P distance matrix. By building partitions from single locus sequence alignments, it provides a score for each defined partition and sorts the sequences into putative species82. Finally, the bPTP model was performed on the PTP species delimitation web server (https://species.h-its.org/ptp/), using the Bayesian tree as input, running 100,000 MCMC generations, and with the burn-in set to 25%. In contrast to BIN and ASAP, bPTP infers putative species based on a non-ultrametric phylogenetic tree, mainly by identifying the transition points between inter- and intraspecific branching events83.
Estimation of divergence times
Divergence times of the Omobranchus lineages were computed in BEAST v2.6.385 together with the bModelTest package86. For this analysis, the dataset was reduced to unique haplotypes and a constant coalescent model was used. We used a 1.2% divergence per million years, as previously estimated for the fish COI locus35, and which is commonly used in marine fish studies36,37. Two BEAST MCMC chains were run independently with 40 M generations each, sampling every 4,000 states, and discarding the first 10% of samples as burn-in. Convergence and parameter mixing were verified with Tracer v1.7.177, ensuring consistency across runs and that most parameters had sufficient effective sample sizes (ESS > 200). Trees and logfiles of both runs were then combined using LogCombiner v2.6.3, and TreeAnnotator v2.6.3 was used to summarize estimates into a maximum-clade-credibility (MCC) tree. The blenny O. woodi was used as an outgroup in all analyses. Trees were visualized and edited in FigTree v.1.4.478. All analyses were performed on CIPRES87.
Morphological analysis
For morphological analysis, previously published morphological data on 444 specimens from 29 populations of the putative species O. punctatus, from both its natural and introduced distribution range (see Table 13 in24), were combined with data collected for the present study on 140 specimens from Venezuela (N = 75) and Brazil (N = 65) (Supplementary Table S6). The specimens examined in the present study included fish that were genetically analysed (see Table 1), and others deposited in the following collections: Museo de Historia Natural La Salle (MHNLS), Museo Oceanológico Hermano Benigno Román (MOBR-EDIMAR), and Museo de Ciencias Naturales de la UNELLEZ (MCNG) in Venezuela; and CPUFMA-UFMA and GEA.ICT in Brazil. For each specimen, and following the procedure established by Springer and Gomon24, the 10 most important meristic characters (counts of body structures) of the genus were assessed: number of dorsal-fin spines, number of segmented rays in the dorsal and anal fins, sum of the unsegmented and segmented rays (i.e., the total dorsal-fin elements), sum of dorsal and ventral procurrent rays in the caudal fins, number of precaudal and caudal vertebrae, total number of vertebrae, number of lateral-line tubes, and the position of the last lateral-line tube relative to a dorsal-fin spine (Supplementary Table S6).
To determine the relationships among individuals from the different populations, a principal component analysis (PCA) was performed in R88, using the FactoMineR89 and factoextra90 packages. Prior to analysis, the frequencies (62 variations) of the 10 meristic characters analysed (discrete data) were transformed to weighted averages.
Species nomination
The genetic lineages and associated morphological groups found in the present work (see “Results” Section) were named according to the 12 currently available synonyms of O. punctatus24,41 (Supplementary Table S7), following the Priority Principle established in article 23, Chapter 6, of the International Code of Zoological Nomenclature91. For this, geographical proximity between the type localities of the species described and the currently synonyms available within the group attributed to O. punctatus (Supplementary Table S7) is required.
Data availability
The dataset generated during the current study are available in the GenBank database (Accession Numbers: OM056858—OM056885).
References
Molnar, J. L., Gamboa, R. L., Revenga, C. & Spalding, M. D. Assessing the global threat of invasive species to marine biodiversity. Front. Ecol. Environ. 6, 485–492 (2008).
Pyšek, P. et al. Scientists’ warning on invasive alien species. Biol. Rev. 95, 1511–1534 (2020).
Rius, M., Turon, X., Bernardi, G., Volckaert, F. A. M. & Viard, F. Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol. Invasions 17, 869–885 (2015).
Doria, C. R. D. C. et al. The silent threat of non-native fish in the Amazon: ANNF database and review. Front. Ecol. Evol. 9, 646702 (2021).
Katsanevakis, S., Zenetos, A., Belchior, C. & Cardoso, A. C. Invading European seas: assessing pathways of introduction of marine aliens. Ocean Coast. Manag. 76, 64–74 (2013).
Chiesa, S., Azzurro, E. & Bernardi, G. The genetics and genomics of marine fish invasions: a global review. Rev. Fish Biol. Fish. 29, 837–859 (2019).
Brandl, S. J., Goatley, C. H. R., Bellwood, D. R. & Tornabene, L. The hidden half: ecology and evolution of cryptobenthic fishes on coral reefs. Biol. Rev. 93, 1846–1873 (2018).
Patzner, R. A., Gonçalves, E., Hastings, P. & Kapoor, B. The Biology of Blennies (CRC Press, 2009).
Wonham, M. J., Carlton, J. T., Ruiz, G. M. & Smith, L. D. Fish and ships: relating dispersal frequency to success in biological invasions. Mar. Biol. 136, 1111–1121 (2000).
Nelson, J. S., Grande, T. C. & Wilson, M. V. H. Fishes of the World: Nelson/Fishes of the World (John Wiley & Sons, Inc, 2016). https://doi.org/10.1002/9781119174844.
Fricke, R., Eschmeyer, W. N. & Fong, J. D. Eschmeyer’s Catalog of Fishes - Genera/Species by Family/Subfamily. (http://researcharchive.calacademy.org/research/ichthyology/catalog/SpeciesByFamily.asp). Electronic version accessed 28 April 2022. (2022).
Valenciennes, A. Histoire naturelle des poissons. Tome onzième. Livre treizième. De la famille des Mugiloïdes. Livre quatorzième. De la famille des Gobioïdes. 11 (i-xx) 1–506pp. Pls. 307–343 (1836).
Eldredge, L. G. Perspectives in aquatic exotic species management in the Pacific Islands: introductions of commercially significant aquatic organisms to the Pacific Islands (South Pacific Commission, 1994).
Francis, M. P., Smith, P. J., Walsh, C. & Gomon, M. F. First records of the Australian blenny, Omobranchus anolius, from New Zealand. N. Z. J. Mar. Freshw. Res. 38, 671–679 (2004).
Özbek, E. Ö., Özkaya, M., Öztürk, B. & Golani, D. First record of the blenny Parablennius thysanius (Jordan & Seale, 1907) in the Mediterranean. J. Black Sea Mediterr. Environ. 20, 53–59 (2014).
Randall, J. E. Reef and shore fishes of the Hawaiian Islands (University of Hawaiʻi, 2007).
Lasso-Alcalá, O. et al. Invasion of the Indo-Pacific blenny Omobranchus punctatus (Perciformes: Blenniidae) on the Atlantic Coast of Central and South America. Neotrop. Ichthyol. 9, 571–578 (2011).
Contente, R. F., Brenha-Nunes, M. R., Siliprandi, C. C., Lamas, R. A. & Conversani, V. R. M. Occurrence of the non-indigenous Omobranchus punctatus (Blenniidae) on the São Paulo coast, South-Eastern Brazil. Mar. Biodivers. Rec. 8, 941–946 (2015).
Gerhardinger, L. C., Freitas, M. O., Andrade, Á. B. & Rangel, C. A. Omobranchus punctatus (Teleostei: Blenniidae), an Exotic Blenny in the Southwestern Atlantic. Biol. Invasions 8, 941–946 (2006).
Lasso, C., Lasso-Alcalá, O., Pombo, C. & Smith, M. Ictiofauna de las aguas estuarinas del delta del río Orinoco (caños Pedernales, Manamo, Manamito) y golfo de Paria (río Guanipa): Diversidad, distribución, amenazas y criterios para su conservación. in A Biological Assessment and Socio Economical Aspects of the Aquatic Ecosystems of the Gulf of Paria and Orinoco Delta, Venezuela 70–84 (RAP Bulletin of Biological Assessment N° 37, 2004).
Lasso-Alcalá, O. & Lasso, C. Blenio hocicudo: Omobranchus punctatus. in Los peces del delta del Orinoco: diversidad, bioecología, uso y conservación. 500 (Fundación La Salle de Ciencias Naturales y Chevron C.A., 2011).
Cervigón, F. Los peces marinos de Venezuela. (Estación de Investigaciones Marinas de Margarita, Fundación La Salle de Ciencias Naturales, 1966).
Fowler, H. W. Fishes obtained by the Barber Asphalt company in Trinidad and Venezuela in 1930. Proc. Acad. Nat. Sci. Phila. 83, 391–410 (1931).
Springer, V. G. & Gomon, M. F. Revision of the blenniid fish genus Omobranchus, with descriptions of three new species and notes on other species of the tribe Omobranchini. Smithson. Contrib. Zool. 177, 1–135 (1975).
Golani, D. First record of the muzzled blenny (Osteichthyes: Blenniidae: Omobranchus punctatus) from the Mediterranean, with remarks on ship-mediated fish introduction. J. Mar. Biol. Assoc. U. K. 84, 851–852 (2004).
Lasso-Alcalá, O., Lasso, C. A. & Posada, J. M. Especies Exóticas en el Mar Caribe: Introducción de Omobranchus punctatus (Valenciennes, 1836) (Perciformes, Blennidae) en las Costas de Venezuela. in Proceedings of the Sixty First Annual Gulf and Caribbean Fisheries Institute. Gulf and Caribbean Fisheries Institute 391–395 (Gulf and Caribbean Fisheries Institute, 2008).
de Paula Costa, M., Souza-Conceicao, J., Schwingel, P. & Louis, H. Assessment of larval distribution of invasive. Aquat. Invasions 6, S33–S38 (2011).
Loebmann, D., Mai, A. C. G. & Lee, J. T. The invasion of five alien species in the Delta do Parnaíba environmental protection area, Northeastern Brazil. Rev. Biol. Trop. 58, 909–923. https://doi.org/10.15517/rbt.v58i2.5254 (2010).
Soares, B., Raiol, R. & Montag, L. Occurrence of the non-native blenny Omobranchus punctatus (Valenciennes, 1836) (Perciformes: Blenniidae) in the Amazon coastal zone, Brazil. Aquat. Invasions 6, S39–S43 (2011).
Cabezas, M. P., Lasso-Alcalá, O. M., Xavier, R. & Jowers, M. J. First genetic record of the non-native muzzled blenny Omobranchus punctatus (Teleostei: Blenniidae) in the Atlantic Coast of Central and South America. J. Fish Biol. 96, 841–846 (2020).
Gibbs, S. et al. Systematics of the combtooth blenny clade Omobranchus (Blenniidae: Omobranchini), with notes on early life history stages. Zootaxa 4369, 270–280 (2018).
Mehraban, H., Zarei, F. & Esmaeili, H. R. A prelude to the molecular systematics and diversity of combtooth blennies (Teleostei: Blenniidae) in the Persian Gulf and Oman Sea. Syst. Biodivers. 19, 438–452 (2021).
Singhal, S., Hoskin, C. J., Couper, P., Potter, S. & Moritz, C. A framework for resolving cryptic species: a case study from the lizards of the Australian wet tropics. Syst. Biol. 67, 1061–1075 (2018).
Korshunova, T. et al. Multilevel fine-scale diversity challenges the ‘cryptic species’ concept. Sci. Rep. 9, 6732 (2019).
Bermingham, E., McCafferty, S. S. & Martin, A. P. Fish biogeography and molecular clocks: perspectives from the Panamanian Isthmus. In Molecular Systematics of Fishes (eds Kocher, T. D. & Stepien, C. A.) 113–128 (Academic Press, 1997).
Delrieu-Trottin, E. et al. Population expansions dominate demographic histories of endemic and widespread Pacific reef fishes. Sci. Rep. 7, 40519 (2017).
Reece, J. S., Bowen, B. W., Joshi, K., Goz, V. & Larson, A. Phylogeography of two moray eels indicates high dispersal throughout the Indo-Pacific. J. Hered. 101, 391–402 (2010).
Günther, A. Catalogue of the fishes in the British Museum. Catalogue of the acanthopterygian fishes in the collection of the British Museum. Gobiidae, Discoboli, Pediculati, Blenniidae, Labyrinthici, Mugilidae, Notacanthi. London. 3, 1–586 (1861).
Weber, M. Süsswasserfische von Neu-Guinea. Ein Beitrag zur Frage nach dem früheren Zusammenhang von Neu-Guinea und Australien. in Nova Guinea. Résultats de l'expédition scientifique Néerlandaise à la Nouvelle-Guinée en 1903 (ed. Wichmann, A.), 201–267 (1907).
Bleeker, P. Neuvième notice sur la faune ichthyologique du Japon. Verslagen en Mededeelingen der Koninklijke Akademie van Wetenschappen. Afdeeling Natuurkunde. 3, 237–252 (1869).
Fricke, R., Eschmeyer, W. N. & Van der Laan, R. Eschmeyer’s Catalog of Fishes: Genera, Species, Reference. (2022). Electronic version. Accessed 28 April 2022.
Araujo, G. S. et al. Phylogeny of the comb-tooth blenny genus Scartella (Blenniiformes: Blenniidae) reveals several cryptic lineages and a trans-Atlantic relationship. Zool. J. Linn. Soc. 190, 54–64 (2020).
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S. & Francis, C. M. Identification of Birds through DNA Barcodes. PLOS Biol. 2, e312 (2004).
Ward, R. D. DNA barcode divergence among species and genera of birds and fishes. Mol. Ecol. Resour. 9, 1077–1085 (2009).
Sheth, B. P. & Thaker, V. S. DNA barcoding and traditional taxonomy: an integrated approach for biodiversity conservation. Genome 60, 618–628 (2017).
Carstens, B. C., Pelletier, T. A., Reid, N. M. & Satler, J. D. How to fail at species delimitation. Mol. Ecol. 22, 4369–4383 (2013).
Mačić, V. et al. Biological invasions in conservation planning: a global systematic review. Front. Mar. Sci. 5, 178 (2018).
Bickford, D. et al. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 22, 148–155 (2007).
Ayouche, A. et al. Structure and dynamics of the Ras al Hadd oceanic dipole in the Arabian Sea. Oceans 2, 105–125 (2021).
Tsang, L. M., Achituv, Y., Chu, K. H. & Chan, B. K. K. Zoogeography of intertidal communities in the west Indian Ocean as determined by ocean circulation systems: Patterns from the Tetraclita barnacles. PLoS One 7, e45120 (2012).
Caley, M. J. et al. Recruitment and the local dynamics of open marine populations. Annu. Rev. Ecol. Syst. 27, 477–500 (1996).
Palumbi, S. R. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Evol. Syst. 25, 547–572 (1994).
Pascual, M., Rives, B., Schunter, C. & Macpherson, E. Impact of life history traits on gene flow: a multispecies systematic review across oceanographic barriers in the Mediterranean Sea. PLoS One 12, e0176419 (2017).
Voris, H. K. Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J. Biogeogr. 27, 1153–1167 (2000).
Gaither, M. R. & Rocha, L. A. Origins of species richness in the Indo-Malay-Philippine biodiversity hotspot: evidence for the centre of overlap hypothesis. J. Biogeogr. 40, 1638–1648 (2013).
Kakioka, R. et al. Cryptic genetic divergence in Scolopsis taenioptera (Perciformes: Nemipteridae) in the western Pacific Ocean. Ichthyol. Res. 65, 92–100 (2018).
Lim, K. C. et al. Brown banded bamboo shark (Chiloscyllium punctatum) shows high genetic diversity and differentiation in Malaysian waters. Sci. Rep. 11, 14874 (2021).
Alam, M. M. M. Recent phylogeographic studies revealed distinct lineages in penaeid shrimps. Oceanogr. Fish. Open Access J. 7, 555721 (2018).
Simmonds, S. E. et al. Evidence of host-associated divergence from coral-eating snails (genus Coralliophila) in the Coral Triangle. Coral Reefs 37, 355–371 (2018).
Savolainen, O., Lascoux, M. & Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820 (2013).
Ismail, W. & Clayton, D. Biology of Omobranchus punctatus (Blenniidae) on rocky shores in Kuwait. Cybium 14, 285–293 (1990).
Egan, J. P., Buser, T. J., Burns, M. D., Simons, A. M. & Hundt, P. J. Patterns of body shape diversity and evolution in intertidal and subtidal lineages of combtooth blennies (Blenniidae). Integr. Org. Biol. Oxf. Engl. 3, obab004 (2021).
Andrades, R., Machado, F. S., Reis-Filho, J. A., Macieira, R. M. & Giarrizzo, T. Intertidal biogeographic subprovinces: local and regional factors shaping fish assemblages. Front. Mar. Sci. 5, 412: 1–14 (2018).
Macieira, R. M., Simon, T., Pimentel, C. R. & Joyeux, J.-C. Isolation and speciation of tidepool fishes as a consequence of Quaternary sea-level fluctuations. Environ. Biol. Fishes 98, 385–393 (2015).
Watson, W. Larval development in blennies. in The Biology of Blennies pp. 309–350 (Science Publishers, 2009). ( Eds R. A Patzner, E. J. Gonçalves & P.A. Hastings)
Sponaugle, S. et al. Predicting self-recruitment in marine populations: Biophysical correlates and mechanisms. Bull. Mar. Sci. 70, 341–375 (2002).
Bullock, J. M. et al. Human-mediated dispersal and the rewiring of spatial networks. Trends Ecol. Evol. 33, 958–970 (2018).
Ivanova, N. V., Zemlak, T. S., Hanner, R. H. & Hebert, P. D. N. Universal primer cocktails for fish DNA barcoding. Mol. Ecol. Notes 7, 544–548 (2007).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Zwickl, D. J. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Dissertation, University of Texas (2006).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 19, 716–723 (1974).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2017).
Sukumaran, J. & Holder, M. T. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 17. Syst. Biol. 67, 901–904 (2018).
Rambaut, A. FigTree-version 1.4.4, a graphical viewer of phylogenetic trees. (2018). http://tree.bio.ed.ac.uk/software/figtree/
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Leigh, J. W. & Bryant, D. PopART: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Ratnasingham, S. & Hebert, P. D. N. A DNA-Based registry for all animal species: The Barcode Index Number (BIN) System. PLoS One 8, e66213 (2013).
Puillandre, N., Brouillet, S. & Achaz, G. ASAP: assemble species by automatic partitioning. Mol. Ecol. Resour. 21, 609–620 (2021).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876 (2013).
Ratnasingham, S. & Hebert, P. D. N. BOLD: the Barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364 (2007).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLOS Comput. Biol. 15, e1006650 (2019).
Bouckaert, R. R. & Drummond, A. J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 17, 42 (2017).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. in 2010 Gateway Computing Environments Workshop (GCE) pp. 1–8 (2010). doi:https://doi.org/10.1109/GCE.2010.5676129.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (2020).
Lê, S., Josse, J. & Husson, F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25, 1–18 (2008).
Kassambara, A. and Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.7. https://CRAN.R-project.org/package=factoextra (2020).
ICZN (International Code of Zoological Nomenclature). International Code of Zoological Nomenclature. (2021).
Xu, C. et al. Species identification of partial fish larvae and juveniles in Fujian coastal areas based on cytochrome C oxidase subunit I barcoding. J. Fish. Sci. China 24, 1176 (2017).
Chang, C.-H. et al. DNA barcodes of the native ray-finned fishes in Taiwan. Mol. Ecol. Resour. 17, 796–805 (2017).
Steinke, D., Connell, A. D. & Hebert, P. D. N. Linking adults and immatures of South African marine fishes. Genome 59, 959–967 (2016).
Acknowledgements
O.M.L-A. and E.Q.-T. specially thank the managers of the Museo de Historia Natural La Salle (Ramón Varela) and Fundación La Salle de Ciencias Naturales (Horacio Morales), in Venezuela, for the support in carrying out this work. The authors also thank J.D. Carrillo Briceño (University of Zurich, Switzerland) for his collaboration in the shipping of some of the samples studied. J.L.S.N. thanks the Fundação de Amparo á Pesquisa e ao Desenvolvimento Científico e Tecnológico do Maranhão for his productivity scholarship (BEPP 03654/15). Special thanks to James Van Tassell (American Museum of Natural History) and Ross Robertson (Smithsonian Tropical Research Institute) for providing the photo of Omobranchus used in Fig. 4. M.J.J. is supported by the fellowship from the Portuguese Foundation for Science and Technology (FCT, SFRH/BPD/109148/2015). Work supported by the European Union’s Horizon 2020 Research and Innovation Programme under the Grant Agreement Number 857251.
Author information
Authors and Affiliations
Contributions
O.M.L-A., M.J.J., R.X. and M.P.C. conceived the study. O.M.L-A., E.Q.-T., T.G., J.L.S.N., F.S.M. and W.S.P. conducted the fieldwork. M.P.C. performed the DNA extraction and PCR amplification of all samples. M.P.C., R.X., and M.J.J. conducted the phylogenetic analyses. O.M.L-A., E.Q.-T. and J.L.S.N. performed the morphological analyses. J.G., O.M.L-A. and T.G. conducted the statistical analyses. M.P.C. wrote the final manuscript with input from all coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cabezas, M.P., Lasso-Alcalá, O.M., Quintero-T, E. et al. Clarifying the taxonomy of some cryptic blennies (Blenniidae) in their native and introduced range. Sci Rep 12, 9514 (2022). https://doi.org/10.1038/s41598-022-12580-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-12580-z
This article is cited by
-
First Molecular Identification of a Goussia Parasite from a New World Invasive Blenny
Acta Parasitologica (2023)
-
Ecology, evolution and conservation of tidepool fishes of the Americas
Reviews in Fish Biology and Fisheries (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
