
Abstract
Antibody mediated rejection is a major cause of renal allograft loss. Circulating preformed donor specific antibodies (DSA) can result as a consequence of blood transfusion, pregnancy or prior transplantation. Current treatment strategies are limited due to partial or transient efficacy, adverse side-effects or patient unsuitability. Previous in vivo studies exploring autoimmune diseases have shown that spleen tyrosine kinase (SYK) signalling is involved in the development of pathogenic autoantibody. The role of SYK in allogenic antibody production is unknown, and we investigated this in a rodent model of sensitization, established by the transfusion of F344 whole blood into LEW rats. Two-week treatment of sensitized rats with selective SYK inhibitor fostamatinib strongly blocked circulating DSA production without affecting overall total immunoglobulin levels, and inhibition was sustained up to 5 weeks post-completion of the treatment regimen. Fostamatinib treatment did not affect mature B cell subset or plasma cell levels, which remained similar between non-treated controls, vehicle treated and fostamatinib treated animals. Our data indicate fostamatinib may provide an alternative therapeutic option for patients who are at risk of sensitization following blood transfusion while awaiting renal transplant.
Similar content being viewed by others

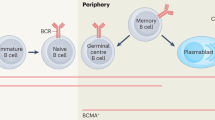

Introduction
Antibody mediated rejection (AMR) represents a significant barrier to allograft survival. The presence of preformed alloreactive donor specific antibodies (DSA) can arise from exposure to cells originating from other individuals via pregnancy, prior transplantation and blood transfusion1.
There is abundant research implicating the pathogenic role of DSA directed against polymorphic human leukocyte antigen class I (HLA I) and HLA class II (HLA II) in multiple organs. Non-HLA targets also play a crucial role in allograft rejection and DSA targets include minor histocompatibility molecules such as major histocompatibility complex (MHC) class I chain-related protein A (MICA) and major histocompatibility complex (MHC) class I chain-related protein B (MICB)2. Other commonly reported non-HLA targets are expressed on epithelial cells and endothelial cells3, 4.
There are two main mechanisms of allograft damage by DSA. The classical complement pathway can be activated by DSA binding to target antigen on endothelium, with generation of complement components which recruit inflammatory cells to the graft or directly damage the graft through the formation of the membrane attack complex5. In the process of antibody-dependent cell mediated cytotoxicity (ADCC), effector cells bearing Fcγ receptors (FcγR) can interact with the crystalline fragment (Fc) of bound DSA on endothelium and trigger lysis of target cells6.
Over 40% of patients awaiting kidney transplant in the United Kingdom are presensitized, with a reported median wait time of 6.1 years, which is double that of non-sensitized patients7. Current desensitization strategies include plasmapheresis and intravenous immunoglobulin, with or without immunosuppression using the B cell depletion-agent rituximab8, 9. However, these strategies are limited and have partial or transient efficacy, in addition to their adverse effects and unsuitability for patients with certain comorbidities10. It is crucial therefore to identify new, safer and more effective therapeutic targets for desensitization of patients.
Spleen tyrosine kinase (SYK) is a cytosolic non-receptor tyrosine kinase mainly expressed in haemopoietic cells. Activation of SYK and subsequent signal transduction is initiated downstream of classical immunoreceptors including FcγR and the B cell receptor (BCR). In the B cell, SYK signalling plays a critical role in B cell maturation and effector functions11,12,13,14. Increasing numbers of studies have targeted SYK for the treatment of immune and inflammatory diseases, and it has potential efficacy in the treatment of presensitized patients15,16,17,18. Fostamatinib is a small molecule SYK inhibitor, and through its active metabolite R406 has previously shown efficacy in the treatment of experimental autoimmune glomerulonephritis in rats, where treatment resulted in attenuation of autoantibody production19.
Here, in a rat model of sensitization, we demonstrate that fostamatinib treatment is able to prevent the production of allogenic antibody, even after cessation of treatment, maintaining overall non-allogenic immunoglobulin levels, whilst having no depletory effect on the levels of plasma cell or B cell populations.
Materials and methods
Animals and transfusions
Eight-week old LEW.Crl RT1I (LEW) and F344/DUCrl RT1lv (F344) male rats were purchased from Charles River UK Ltd (Margate, UK) and maintained in a pathogen-free animal facility at the Central Biomedical Services unit, Hammersmith Hospital Campus, Imperial College London. LEW received 800 µL heparinised whole blood from F344 via an intravenous route. All animal studies were licensed by the Home Office Science Unit. Studies and procedures were approved by Imperial College London Research Ethics committee and carried out in accordance with the regulations of the UK Animals (Scientific Procedures) Act (1986) and ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Treatments
SYK specific inhibitor fostamatinib was provided by Rigel Pharmaceuticals (South San Francisco, CA, USA). Fostamatinib or vehicle (0.1% carboxymethylcellulose) was administered in sensitized animals by oral gavage twice daily for a period of 14 days at 40 mg/kg from either 24 h or 7 days post-transfusion.
Detection of total serum immunoglobulin
Serum levels of total IgG and IgM were detected with Enzyme Linked Immunosorbent Assay (ELISA) from ThermoFischerScientific. Serum was diluted by 1:10,000 (IgM) or 1:50,000 (IgG).
Flow cytometry
Samples were analysed on a BD LSR Fortessa™ X-20 and data analysed on FlowJo 9.6.6 software.
Flow crossmatch analysis
IgG and IgM alloantibodies were detected by flow cytometry. 1 × 106 F344l splenocytes were suspended in RPMI-1640 medium containing 10% FCS and incubated with diluted recipient LEW (1/50) sera at 37 °C for 30 min. Splenocytes were incubated with the following anti-rat antibodies for 20 min in 0.5% BSA: anti-IgM (MRM-47 Biolegend), anti-IgG (Poly4054 Biolegend), anti-IgG1 (MARG1-2 AbCam), anti-IgG2a (MRG2A-83 Biolegend), anti-IgG2b (MRG2b-85 Biolegend), IgG2c (R2C-23A3 eBioscience), anti-CD3 (14F Biolegend) and anti-CD45 (OX-1 Biolegend). Alloreactivity was analysed by determining the mean fluorescence intensity (MFI) on gated live CD45+ (OX-1 Biolegend) CD3+ (14F Biolegend) cells.
B cell and plasma cell subset analysis
Splenocytes were isolated from LEW rats and incubated with the following antibodies: anti-CD3 (1F4 Biolegend), anti-CD45 (OX1 Biolegend), anti-CD45R (HIS24 BDBiosciences), anti-CD27 (LG.3A10 Biolegend), anti-CD138 (DL-101 Santa Cruz Biotechnology), anti-IgD (MARD1 ThermoFisher Scientific), anti-IgM (MRM-47 Biolegend), anti-CD3 (14F Biolegend), and anti-CD45RA (OX-33 Biolegend).
Cells were gated to select singlets then CD3- cells. Following this, plasma cells were selected by gating on IgD−CD45R−IgM−CD138+, memory cells by CD45R+CD27+, switched cells by CD45R+CD27+IgD− and non-switched cells by CD45R+CD27+IgD+. Precision count beads (Biolegend) were used to obtain absolute cell counts.
Statistical analyses
Data were analysed with GraphPad Prism 8.0 (GraphPad Software, Inc, La Jolla, CA) and are displayed as the mean ± SEM using a Mann–Whitney U test as appropriate. A P < 0.05 was defined as significant.
Results
Transfusion results in the production of donor specific antibody
A rat model of sensitization was established by whole blood transfusion between F344 and LEW rats. T lymphocyte (CD3+) flow crossmatch analysis showed that transfusion between these strains elicited an alloantibody response. An initial peak in allogenic IgM levels 7 days post-transfusion (Fig. 1A), was followed by isotype switching to IgG (Fig. 1B) where levels peaked at day 17. Additionally, all DSA IgG isotypes IgG1 (Fig. 1C), IgG2a (Fig. 1D), IgG2b (Fig. 1E), IgG2c (Fig. 1F) increased following transfusion.

Whole F344 blood transfusion to LEW rats induces an alloantibody response. Flow cytometry T lymphocyte crossmatch was performed to detect allogenic (A) IgM levels, (B) IgG levels and IgG subsets (C) in sensitized LEW rats (n = 4).
Treatment with fostamatinib inhibits allogenic antibody production
Following establishment of a sensitized model, the role of fostamatinib in DSA production was investigated. Sensitized rats were treated with fostamatinib 24 h after blood transfusion. Flow crossmatch analysis demonstrated that SYK inhibitor treatment was able to block production of significant levels of IgM (Fig. 2A), IgG (Fig. 2B) and IgG subsets IgG1 (Fig. 2C), IgG2a (Fig. 2D), IgG2b (Fig. 2E) and IgG2c (Fig. 2F) throughout the course of treatment.

Fostamatinib treatment prevented production of donor specific antibody. T Lymphocyte crossmatch was performed to detect DSA levels in sensitized fostamatinib treated and vehicle treated LEW rats. (A) IgM, (B) IgG and (C–F) IgG subset production was inhibited by fostamatinib treatment at all time points measured (n = 6 rats/group). *P ≤ 0.05, **P ≤ 0.01. Blue Filled circle—vehicle, red filled triangle—fostamatinib.
Total circulating IgG and IgM levels are not affected by fostamatinib
As fostamatinib prevented the formation of an alloantibody response, serum was analysed for total circulating IgM (Fig. 3A) and IgG levels (Fig. 3B). These levels remained comparable between treatment groups.

Fostamatinib treatment had no effect on total circulating IgM and IgG. Serum was analysed by ELISA for (A) total IgM and (B) total IgG levels. There was no significant difference at any measured time point between fostamatinib treated rats and vehicle treated rats for both IgM and IgG levels (n = 6 rats/group). Blue filled circle—vehicle, red filled triangle—fostamatinib.
Fostamatinib treatment did not affect mature B lymphocyte subsets or plasma cell numbers
Splenic mature B lymphocyte populations were evaluated. Memory B lymphocytes (CD45R+CD27+) (Fig. 4A,B), switched cells (CD45R+CD27+IgD−) (Fig. 4C,E) and non-switched cells (CD45R+CD27+IgD+) (Fig. 4D,E), were detected at similar levels in both treatment groups compared to control rats. Given that DSA production was profoundly inhibited by fostamatinib, effects on numbers of splenic plasma cells were evaluated between treatment groups and compared to control non-sensitized LEW rats (Fig. 4F,G). Unlike DSA production, plasma cell numbers (CD138+CD45R−) were not affected by fostamatinib therapy and both treatment groups remained at similar levels compared to non-treated non-sensitized animals.

Fostamatinib treatment did not affect B cell or plasma cell populations. Flow cytometry was used to measure mature B lymphocytes populations in fostamatinib treated and vehicle treated sensitized LEW rats. Each graph shows number of cells per gram of spleen. Mature B lymphocyte numbers remained similar between treatment groups. (A) Memory cells were defined as CD45R+CD27+, (C) switched cells as CD45R+CD27+IgD−, (D) non-switched cells CD45R−CD27+ IgD+, (F) plasma cells as IgD−CD45R−IgM−CD138+. (n = 6 rats/group). Graphs show cell numbers per gram of spleen (n = 6 rats/group). Representative flow cytometry data of (B) memory B lymphocytes (E) switched and non-switched B lymphocytes (G) plasma cells. Number shown represents percentage of cells in gate. Blue filled circle—vehicle, red filled triangle—fostamatinib.
Fostamatinib prevented DSA levels rising 5 weeks after termination of treatment
Following potent inhibition of DSA levels with fostamatinib treatment, we wanted to evaluate if this suppression was transient or maintained. Sensitized rats were treated with fostamatinib for the 2-week regimen as described previously and, upon completion of the treatment course, rats were left untreated for 5 weeks with weekly venesection for measurement of DSA levels. T lymphocyte crossmatch analysis of serum showed that 2-week fostamatinib treatment was able to maintain suppression of formation of DSA IgM (Fig. 5A) and IgG (Fig. 5B) up to 5 weeks after completion of the treatment regimen.

Fostamatinib prevents DSA production up to 5 weeks post completion of treatment. Treatment was given from day 1 to day 14 after blood transfusion. Then, the antibody levels were monitored until day 49. T lymphocyte crossmatch analysis was performed to detect DSA levels in sensitized fostamatinib and vehicle dosed LEW rats. (A) IgM and (B) IgG production was inhibited up to 5 weeks post-termination of the fostamatinib treatment regimen (n = 6 rats/group) (*P ≤ 0.05, **P ≤ 0.01). Blue filled circle—vehicle, red filled triangle—fostamatinib.
Delayed fostamatinib treatment prevented IgG DSA reaching levels found in vehicle treated rats
To assess the efficacy of fostamatinib treatment initiated at a later timepoint in sensitized rats, the fostamatinib treatment regimen was delayed until 7 days post-transfusion. Results showed that fostamatinib did not block allogenic IgM production and circulating levels were comparable between treatment groups (Fig. 6A). However allogenic IgG levels were significantly reduced from day 14 in fostamatinib treated rats (Fig. 6B). Investigating this further, IgG subsets were measured, demonstrating variable results (Fig. 6C–F). Levels of IgG2b, IgG1 and IgG2a showed significant blocking of alloantibody production from days 14, 17 and 21 respectively. IgG2c showed no difference between treatment groups at any time point.

Delayed fostamatinib treatment reduces levels of DSA IgG in sensitised rats. T lymphocyte crossmatch analysis was performed to detect DSA levels in sensitized fostamatinib and vehicle treated LEW rats. Treatment initiation was delayed until 7 days post-transfusion. (A) IgM levels remained the same in both treatment groups. (B) Circulating allogenic IgG levels were significantly lower in fostamatinib treated rats at days 14, 17 and 21 post-transfusion. (C) From 17 days onwards IgG1 levels were significantly lower in fostamatinib treated animals, from 14 days onwards IgG2a (D) levels were significantly lower in fostamatinib treated animals, and by 21 days post-transfusion IgG2b (E) levels were significantly lower in fostamatinib treated animals. No differences were observed in IgG2c levels at any measured time point (F) (n = 6 rats/group) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). Blue filled circle—vehicle, red filled triangle—fostamatinib.
Discussion
In this study, we demonstrate for the first time that SYK inhibition with fostamatinib significantly prevents allogenic DSA production in a rat model of sensitization. Fostamatinib treatment initiated 24 h post-transfusion for the treatment regimen of 2 weeks was effective at preventing production of both allogenic IgG and IgM, and had no depletory effects on total IgG and IgM levels. Fostamatinib treatment remained effective even 5 weeks post-termination of treatment, with significantly lower levels of IgG and IgM detected by the T cell crossmatch test at this time point. Fostamatinib was also able to partially block DSA production when treatment was implemented 7 days post-sensitisation, after the initiation of an allogenic antibody response. Our study shows that fostamatinib had a direct effect on inhibition of alloantibody without adversely affecting B cell survival. Splenic plasma cell numbers were comparable between both treatment groups. In addition to plasma cells, we examined the effect of SYK blockade on other mature B lymphocyte populations. Memory, switched and non-switched populations of B cells in the spleen remained similar between both treatment groups. Normal B cell activation and development is therefore not being affected by fostamatinib treatment, rather the production of allogenic antibody is.
SYK has a well-defined role in signal transduction downstream of immunoreceptors and is critical in mediating BCR responses and FCR responses in mast cells20, dendritic cells20, macrophages21 and neutrophils22. The SYK homologue zeta chain-associated protein kinase 70 (ZAP-70)23 is the predominant signalling molecule downstream of the TCR in T cells and natural killer cells. Studies involving SYK knockout murine models have shown the critical role of SYK signalling for B cell development and maturation, where B cell maturation is arrested from progressing at the early pro-B-cell state in murine SYK knockout models11, 24, 25. Early mechanistic data has demonstrated that, in vitro fostamatinib was able to inhibit BCR responses in primary human B cells, where CD69 cell surface upregulation induced by BCR crosslinking with IgM was inhibited with fostamatinib treatment26.
The role of SYK in antibody production in plasma cells is unclear, as B cells from SYK knockout mice are unable to mature beyond the pro-B-cell stage25. In vivo models have provided some insight and indications that SYK inhibition may be an effective target for the treatment of antibody-mediated diseases. In a rodent model of anti-glomerular basement membrane disease, R406 treatment prevented the induction of disease, and inhibited circulating and deposited autoantibody production in established disease19. Interestingly in this model, CD45RA+ cell numbers remained the same despite decrease of autoantibody. Non-obese diabetic mice were protected against developing diabetes with fostamatinib treatment in a prevention setting. Treatment was also able to delay disease progression in glucose intolerant mice, with a measured reduction in anti-glutamic-acid-decarboxylase anti-islet antibodies27.
Currently there are no reported data on the efficacy of fostamatinib in reduction of alloantibody production, and to our knowledge this is the first report of its kind. In contrast to our experiments, early immunotoxicology studies where rats were immunised with KLH-Ribi antigen and treated with fostamatinib, did not show reduced KLH specific IgM and IgG28. Differences between the effects of fostamatinib on production of alloantibody compared to autoantibody are intriguing and require further investigation. The mechanism of inhibition of DSA production by fostamatinib in our experiments is unclear. Due to the functional action of fostamatinib, it is likely that downstream signalling molecules have been blocked from phosphorylation, which in-turn has impacted DSA production. Fostamatinib inhibition of SYK prevents phosphorylation of downstream molecules, which include phospholipase Cγ1, Akt/protein kinase B, c-Jun N-terminal kinase, p38 and extracellular signal-regulated kinase26. Exactly which signals contribute to immunoglobulin production requires further investigation. It is possible in our experiments that fostamatinib treatment is blocking antigen presentation to B cells via follicular dendritic cells, a process required for the progression of the B cell response. Studies have also established a crucial role for SYK signalling in the induction of the antigen presentation machinery in both B cells27 and dendritic cells29. In dendritic cells, SYK signalling has shown to be crucial for immunocomplex uptake and antigen presentation20.
In our model, blood transfusion initially induced IgM production, with peak titres approximately 7 days post sensitization. Traditionally in the context of AMR, preformed IgM antibodies have been perceived to be non-pathogenic. However emerging studies suggest that anti-HLA IgM may make important contributions to the pathogenesis of allograft loss30,31,32,33. In our experiments where treatment was implemented 24 h post sensitization, DSA IgM production was blocked, indicating this might be beneficial for human presensitization. Fostamatinib did not block IgM levels when treatment was initiated at a later time-point, as class switching had converted production to the IgG class.
Rodents have four IgG subclasses IgG1, IgG2a, IgG2b and IgG2c34. IgG2a and IgG2b have been reported to have the strongest complement fixing ability35. In humans four IgG subclasses exist, IgG1, IgG2, IgG3 and IgG4. It is widely accepted that IgG1 and IgG3 are the most pathogenic subclasses in AMR due to their ability to activate the complement cascade36. IgG3 has the highest binding efficiency to complement component C1q, and IgG1 is highly effective at complement dependent cell lysis37. The presence of these subclasses as a result of sensitization is associated with poor graft outcome33. In early treatment experiments we have shown the efficacy of fostamatinib in blocking production of all IgG subtypes, thereby resulting in prevention of downstream effects of humoral immunity, suggesting this approach could be suitable in prevention of presensitization when given concomitantly to blood transfusions or possibly de novo DSA during transplantation38. In addition to DSA level reduction, utilizing fostamatinib to inhibit the effector phase and ultimately influencing the pathogenicity of the DSA underpins the rationale for using fostamatinib in AMR.
Fostamatinib treatment initiated 7 days post transfusion was able to significantly reduce the levels of DSA IgG detected in the CD3+ crossmatch assay at days 14, 17 and 21. Study of IgG subsets provided variable results. IgG2c levels were similar between groups, but IgG1 and the most pathogenic subsets IgG2a and IgG2b demonstrated significantly lower levels in treated rats from at day 14. It is probable that fostamatinib treatment was inhibitory soon after administration, as demonstrated by early time point experiments, however fostamatinib is unlikely to inhibit DSA IgG that has already been made. The circulating half-life of IgG is 7–25 days and it is likely that an extended follow-up time would continue to show more pronounced repression of DSA IgG levels. The ability of fostamatinib to block de novo DSA IgG production but not existing DSA IgG could explain the substantially higher DSA IgG levels measured in late treatment experiments compared to earlier treatment experiments.
In the context of blood transfusion as a sensitizing event, there is an abundance of literature associating allosensitization with higher rates of graft rejection and lower rates of graft survival39. Chronic anaemia is prevalent in patients with chronic kidney disease or end-stage kidney disease and avoiding transfusion is not always feasible40. A causal link between blood transfusions and DSA production has been identified by Hassan et al.41. In patients receiving blood transfusion after renal allograft transplantation, this study found direct evidence of a de novo HLA alloimmune response elicited against the blood donor, and enhanced risk of transplant specific antibody development within these patients. These factors were significantly associated with the increased risk of AMR and allograft failure. Current treatment options including early post-transplant erythropoietin therapy and cell salvage have limited efficacy42.
In a pre-transplant setting, current treatment strategies for sensitized patients have partial or transient efficacy and include the use of plasma exchange, anti-CD20 monoclonal antibody therapy with rituximab, or blocking macrophage FcγR with high-dose intravenous immunoglobulin43. Fostamatinib treatment presents a potential alternative strategy in preventing allosensitization in both pre-transplant and post-transplant blood transfusions. In our experiments, a relatively transient period of orally given treatment was able to inhibit DSA levels even up to 5 weeks post sensitization, which is significant in a clinical context as it is likely to improve patient compliance compared to more regular or invasive treatment. Our data are limited by the moderately short follow up time, and the long-term impact of SYK inhibition on plasma cell and B cell function is unknown. Further work will be needed to investigate whether treatment with fostamatinib may have a role in treatment of sensitized rats in models of experimental transplant rejection.
LEW and F344 rats are weakly histocompatible due to differing partially at both MHC I & II and non-MHC loci44. Transplantation of an F344 kidney into a LEW rat is a well-characterized model of chronic antibody mediated rejection, where rejection develops over the course of a few months45. It will be crucial to investigate the protective potential of fostamatinib in sensitized transplanted rats from developing chronic AMR. In a rat model of acute rejection, involving the Dark Agouti (DA/Arc, RT1av1) kidney transplanted into the LEW, SYK inhibition demonstrated the ability to reduce acute injury in transplanted renal allografts in sensitized recipients46. This study explored the efficacy of SYK inhibitor GS-492429 in acute rejection, with a splenocyte transfusion for the sensitization method. The protective results are promising for our future studies in a chronic AMR rat model. Fostamatinib is currently FDA approved for the treatment of thrombocytopenia in adults with chronic ITP with insufficient response to other treatment. Appropriate dosing, PKA data and a good safety profile for fostamatinib are available47. The data presented in this study suggest that fostamatinib should be investigated for reduction of circulating allogenic antibodies in patients with chronic kidney disease needing blood transfusion and highly sensitized patients with end-stage renal disease awaiting transplant.
Data availability
The data supporting the findings of this study is available from the corresponding author upon reasonable request.
References
Lefaucheur, C. et al. Determinants of poor graft outcome in patients with antibody-mediated acute rejection. Am. J. Transplant. 7, 832–841. https://doi.org/10.1111/j.1600-6143.2006.01686.x (2007).
Zhang, Q. et al. HLA and MICA: Targets of antibody-mediated rejection in heart transplantation. Transplantation 91, 1153–1158. https://doi.org/10.1097/TP.0b013e3182157d60 (2011).
Hilbrands, L., Hoitsma, A. & Wetzels, J. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med. 352, 2027–2028. https://doi.org/10.1056/NEJM200505123521921 (2005).
Jaramillo, A. et al. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: A possible mechanism for bronchiolitis obliterans syndrome. Hum. Immunol. 64, 521–529. https://doi.org/10.1016/s0198-8859(03)00038-7 (2003).
Cernoch, M. & Viklicky, O. Complement in kidney transplantation. Front. Med. (Lausanne) 4, 66. https://doi.org/10.3389/fmed.2017.00066 (2017).
Stegall, M. D., Chedid, M. F. & Cornell, L. D. The role of complement in antibody-mediated rejection in kidney transplantation. Nat. Rev. Nephrol. 8, 670–678. https://doi.org/10.1038/nrneph.2012.212 (2012).
Pruthi, R. et al. UK Renal Registry 16th annual report: Chapter 4 demography of patients waitlisted for renal transplantation in the UK: National and centre-specific analyses. Nephron Clin. Pract. 125, 81–98. https://doi.org/10.1159/000360023 (2013).
Ahmed, A. R. & Kaveri, S. Reversing autoimmunity combination of rituximab and intravenous immunoglobulin. Front. Immunol. 9, 1189. https://doi.org/10.3389/fimmu.2018.01189 (2018).
Jordan, S. C., Choi, J. & Vo, A. Achieving incompatible transplantation through desensitization: Current perspectives and future directions. Immunotherapy 7, 377–398. https://doi.org/10.2217/imt.15.10 (2015).
Marfo, K. et al. Lack of effect in desensitization with intravenous immunoglobulin and rituximab in highly sensitized patients. Transplantation 94, 345–351. https://doi.org/10.1097/TP.0b013e3182590d2e (2012).
Cornall, R. J., Cheng, A. M., Pawson, T. & Goodnow, C. C. Role of Syk in B-cell development and antigen-receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 97, 1713–1718. https://doi.org/10.1073/pnas.97.4.1713 (2000).
Baba, Y. & Kurosaki, T. Role of calcium signaling in B cell activation and biology. Curr. Top. Microbiol. Immunol. 393, 143–174. https://doi.org/10.1007/82_2015_477 (2016).
Liu, D. & Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 10, 145. https://doi.org/10.1186/s13045-017-0512-1 (2017).
McAdoo, S. P. et al. Spleen tyrosine kinase inhibition is an effective treatment for established vasculitis in a pre-clinical model. Kidney Int. 97, 1196. https://doi.org/10.1016/j.kint.2019.12.014 (2020).
Pine, P. R. et al. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin. Immunol. 124, 244–257. https://doi.org/10.1016/j.clim.2007.03.543 (2007).
Pamuk, O. N. et al. Spleen tyrosine kinase inhibition prevents tissue damage after ischemia-reperfusion. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G391–G399. https://doi.org/10.1152/ajpgi.00198.2010 (2010).
Bussel, J. et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 93, 921–930. https://doi.org/10.1002/ajh.25125 (2018).
Braegelmann, C. et al. Spleen tyrosine kinase (SYK) is a potential target for the treatment of cutaneous lupus erythematosus patients. Exp. Dermatol. 25, 375–379. https://doi.org/10.1111/exd.12986 (2016).
McAdoo, S. P. et al. Spleen tyrosine kinase inhibition attenuates autoantibody production and reverses experimental autoimmune GN. J. Am. Soc. Nephrol. 25, 2291–2302. https://doi.org/10.1681/ASN.2013090978 (2014).
Sedlik, C. et al. A critical role for Syk protein tyrosine kinase in Fc receptor-mediated antigen presentation and induction of dendritic cell maturation. J. Immunol. 170, 846–852. https://doi.org/10.4049/jimmunol.170.2.846 (2003).
Crowley, M. T. et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 186, 1027–1039. https://doi.org/10.1084/jem.186.7.1027 (1997).
Kiefer, F. et al. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell Biol. 18, 4209–4220. https://doi.org/10.1128/mcb.18.7.4209 (1998).
Mocsai, A., Ruland, J. & Tybulewicz, V. L. The SYK tyrosine kinase: A crucial player in diverse biological functions. Nat. Rev. Immuno. 10, 387–402. https://doi.org/10.1038/nri2765 (2010).
Turner, M. et al. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 378, 298–302. https://doi.org/10.1038/378298a0 (1995).
Cheng, A. M. et al. Syk tyrosine kinase required for mouse viability and B-cell development. Nature 378, 303–306. https://doi.org/10.1038/378303a0 (1995).
Braselmann, S. et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 319, 998–1008. https://doi.org/10.1124/jpet.106.109058 (2006).
Lankar, D. et al. Syk tyrosine kinase and B cell antigen receptor (BCR) immunoglobulin-alpha subunit determine BCR-mediated major histocompatibility complex class II-restricted antigen presentation. J. Exp. Med. 188, 819–831. https://doi.org/10.1084/jem.188.5.819 (1998).
Zhu, Y. et al. Immunotoxicity assessment for the novel Spleen tyrosine kinase inhibitor R406. Toxicol. Appl. Pharmacol. 221, 268–277. https://doi.org/10.1016/j.taap.2007.03.027 (2007).
Colonna, L. et al. Therapeutic targeting of Syk in autoimmune diabetes. J. Immunol. 185, 1532–1543. https://doi.org/10.4049/jimmunol.1000983 (2010).
Everly, M. J. et al. Impact of IgM and IgG3 anti-HLA alloantibodies in primary renal allograft recipients. Transplantation 97, 494–501. https://doi.org/10.1097/01.TP.0000441362.11232.48 (2014).
Dodd, P. et al. Significance of HLA IgM and IgM donor specific antibodies in renal transplantation: Abstract# A123. Transplantation 98, 435 (2014).
Calligaro, G. L. et al. (190) Acute antibody mediated rejection due to preformed donor-specific IgM HLA antibodies: Two case reports. J. Heart Lung Transplant. 36, S80–S81. https://doi.org/10.1016/j.healun.2017.01.201 (2017).
Lefaucheur, C. et al. IgG donor-specific anti-human HLA antibody subclasses and kidney allograft antibody-mediated injury. J. Am. Soc. Nephrol. 27, 293–304. https://doi.org/10.1681/ASN.2014111120 (2016).
Bazin, H., Beckers, A. & Querinjean, P. Three classes and four (sub)classes of rat immunoglobulins: IgM, IgA, IgE and IgG1, IgG2a, IgG2b, IgG2c. Eur. J. Immunol. 4, 44–48. https://doi.org/10.1002/eji.1830040112 (1974).
Medgyesi, G. A. et al. Classes and subclasses of rat antibodies: Reaction with the antigen and interaction of the complex with the complement system. Immunology 43, 171–176 (1981).
Griffiths, E. J., Nelson, R. E., Dupont, P. J. & Warrens, A. N. Skewing of pretransplant anti-HLA class I antibodies of immunoglobulin G isotype solely toward immunoglobulin G1 subclass is associated with poorer renal allograft survival. Transplantation 77, 1771–1773. https://doi.org/10.1097/01.tp.0000129408.07168.40 (2004).
Bruggemann, M. et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. Med. 166, 1351–1361. https://doi.org/10.1084/jem.166.5.1351 (1987).
Jordan, S. C. Donor-specific HLA antibody IgG subclasses are associated with phenotypes of antibody-mediated rejection in sensitized renal allograft recipients. J. Am. Soc. Nephrol. 27, 6–8. https://doi.org/10.1681/ASN.2015060608 (2016).
Scornik, J. C. et al. An update on the impact of pre-transplant transfusions and allosensitization on time to renal transplant and on allograft survival. BMC Nephrol. 14, 217. https://doi.org/10.1186/1471-2369-14-217 (2013).
Afshar, M. & Netzer, G. Update in critical care for the nephrologist: Transfusion in nonhemorrhaging critically ill patients. Adv. Chronic Kidney Dis. 20, 30–38. https://doi.org/10.1053/j.ackd.2012.10.007 (2013).
Hassan, S. et al. Shared alloimmune responses against blood and transplant donors result in adverse clinical outcomes following blood transfusion post-renal transplantation. Am. J. Transplant. 19, 1720–1729. https://doi.org/10.1111/ajt.15233 (2019).
Vella, J. P., O’Neill, D., Atkins, N., Donohoe, J. F. & Walshe, J. J. Sensitization to human leukocyte antigen before and after the introduction of erythropoietin. Nephrol. Dial. Transplant. 13, 2027–2032. https://doi.org/10.1093/ndt/13.8.2027 (1998).
Garnier, A. et al. Rituximab for second desensitization in patients with rebound of donor-specific anti-HLA antibodies before T-replete haplo-transplant using high-dose post-transplant cyclophosphamide. Bone Marrow Transplant. 53, 1044–1047. https://doi.org/10.1038/s41409-018-0107-7 (2018).
Shrestha, B. & Haylor, J. Experimental rat models of chronic allograft nephropathy: A review. Int. J. Nephrol. Renovasc. Dis. 7, 315–322. https://doi.org/10.2147/IJNRD.S65604 (2014).
White, E., Hildemann, W. H. & Mullen, Y. Chronic kidney allograft reactions in rats. Transplantation 8, 602–617. https://doi.org/10.1097/00007890-196911000-00007 (1969).
Ramessur Chandran, S. et al. Inhibition of spleen tyrosine kinase reduces renal allograft injury in a rat model of acute antibody-mediated rejection in sensitized recipients. Transplantation 101, e240–e248. https://doi.org/10.1097/TP.0000000000001826 (2017).
Fostamatinib (Tavalisse) for ITP. Med. Lett. Drugs Ther. 61, 28–30 (2019).
Acknowledgements
This work was funded by Making Every Kidney Count Research Grant from Kidney Research UK. FWKT is supported by the Ken and Mary Minton Chair of Renal Medicine. A selection of this research was presented in abstract form at the American Society of Nephrology Kidney Week conference in Washington DC, USA 2019.
Author information
Authors and Affiliations
Contributions
S.T.-R. planned, performed and analysed all experiments and wrote the manuscript. M.P., C.C. and T.T.-S. assisted with in vivo experiments. F.T. supervised the project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
FWKT has received research project grants from AstraZeneca Limited, Baxter Biosciences, Boehringer Ingelheim, MedImmune and Rigel Pharmaceuticals, and has consultancy agreements with Rigel Pharmaceuticals, Novartis and Baxter Biosciences, and is the Chief Investigator of an international clinical trial of a SYK inhibitor in IgA nephropathy (ClinicalTrials.gov NCT02112838) funded by Rigel Pharmaceuticals and a phase 2 clinical trial of fostamatinib in the treatment of chronic active antibody mediated rejection in renal transplantation, funded by Rigel Pharmaceuticals and Auchi Charitable Fund (EudraCT Number: 2018-000027-14). CDP has received a research project grant from GlaxoSmithKline and has a consultancy agreement with Genzyme. EM owns stock and is employed by Rigel Pharmaceuticals, Inc. CR reports personal fees from UCB, personal fees from Achillion Pharmaceutical, personal fees from Rigel Pharmaceuticals outside the submitted work. All other authors do not have competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tempest-Roe, S., Prendecki, M., McAdoo, S.P. et al. Inhibition of spleen tyrosine kinase decreases donor specific antibody levels in a rat model of sensitization. Sci Rep 12, 3330 (2022). https://doi.org/10.1038/s41598-022-06413-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06413-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
