
Abstract
Ionome contributes to maintain cell integrity and acts as cofactors for catalyzing regulatory pathways. Identifying ionome contributing genomic regions provides a practical framework to dissect the genetic architecture of ionomic traits for use in biofortification. Meta-QTL (MQTL) analysis is a robust method to discover stable genomic regions for traits regardless of the genetic background. This study used information of 483 QTLs for ionomic traits identified from 12 populations for MQTL analysis in Arabidopsis thaliana. The selected QTLs were projected onto the newly constructed genetic consensus map and 33 MQTLs distributed on A. thaliana chromosomes were identified. The average confidence interval (CI) of the drafted MQTLs was 1.30 cM, reduced eight folds from a mean CI of 10.88 cM for the original QTLs. Four MQTLs were considered as stable MQTLs over different genetic backgrounds and environments. In parallel to the gene density over the A. thaliana genome, the genomic distribution of MQTLs over the genetic and physical maps indicated the highest density at non- and sub-telomeric chromosomal regions, respectively. Several candidate genes identified in the MQTLs intervals were associated with ion transportation, tolerance, and homeostasis. The genomic context of the identified MQTLs suggested nine chromosomal regions for Zn, Mn, and Fe control. The QTLs for potassium (K) and phosphorus (P) were the most frequently co-located with Zn (78.3%), Mn (76.2%), and Fe (88.2% and 70.6%) QTLs. The current MQTL analysis demonstrates that meta-QTL analysis is cheaper than, and as informative as genome-wide association study (GWAS) in refining the known QTLs.
Similar content being viewed by others

Introduction
Nutrient elements contribute to the plant’s tolerance to abiotic stresses and detoxification of reactive oxygen species (ROS) by altering metabolic pathways involved in gene expression; biosynthesis of proteins, carbohydrates, and lipids; and production of phytohormones that protect plants from ROS-induced injury1,2,3. In crops, essential nutrient concentration affects the quality and quantity of crop production1 and a nutrient imbalance result in various disorders in the world’s population especially for plant-based dietary societies. Among nutrient elements, the essential trace elements such as zinc (Zn), iron (Fe) and manganese (Mn) play crucial role in human health, immune system and life longevity4,5. Trace elements are involved in psychomotor development, maintenance of physical activity, and resistance to infection5,6. The deficiency of trace elements causes several health problems including retarded growth, skeletal abnormalities, increased abortion risk, anemia, diarrhea, impaired mental development, and learning capacity7,8. An inadequate intake of essential nutrients like iron and zinc which is known as “hidden hunger” affects around two billion people globally9. Today, agriculture is undergoing a shift from producing more quantity of food crops to producing higher quality such as nutrient-rich food crops in sufficient quantities.
All mineral nutrient, trace elements (including both essential and non-essential elements, metals and non-metals), and the inorganic component of cellular and organismal systems constitutes the “Ionome”10. It is an important determinant of the physiological state of an organism. For example, iron deficiency in a plant can be identified by looking at a number of other elements, rather than iron itself. The ionomics is a powerful approach for analysis of dynamic network of elements that are controlled by the genome of the plant in response to the environmental conditions11. Understanding the ionome function and dynamics is also vital to uncover how living organisms work, especially for plants that depend on external sources for the uptake of almost all elements except oxygen and carbon10,11. The complex nature of the molecular mechanism of ionomic variation requires detailed information illustrated at the OMICs (biological sciences ends with omics such as genomics, transcriptomics, proteomics, or metabolomics) levels and highlights critical concepts for assessment of the ionome12,13. Thus, ionomics should depict the functional status of a complex biological organism in both a quantitative and qualitative pattern of elements in various components of the organism10.
The ionomics method refers to quantitative analysis of ionomic traits and their changes in living organisms in response to developmental state, physiological stimuli and genetic modifications. In addition to ionomics, the application of practical genetic analysis such as the genetic mapping approach leads to an understanding of the genetic architecture of ionomic traits. The use of molecularly well-characterized and diverse germplasms of model species like Arabidopsis thaliana expands the knowledge about ionomic variation and their associated genomic regions. Various QTLs have been identified using linkage mapping approaches for numerous important ionomic traits in plants14,15,16,17,18,19. Traditional QTL mapping has been successful in identifying genomic regions controlling the variation of ionomic traits. In traditional QTL mapping models, the genetic effects of QTL identified in an individual population may not be validated in other genetic backgrounds or environments and more often, only a limited number of traits can be tested in a single study20. However, meta- quantitative trait loci (Meta-QTL or MQTL) overcomes the limitation posed by traditional QTL analysis21,22. In the meta-QTL model, QTL data from independent studies and populations are used to detect consensus QTL regions known as Meta-QTL23. Further, the information on QTLs derived from various population structure, origin and sizes can be used to refine the known QTLs regardless of the genetic backgrounds, marker density and phenotypic variations21,24,25. In this way, a meta-QTL analysis helps to narrow down the confidence interval (CI) of the initial QTLs identified in independent populations and lays the foundation for a better understanding of traits underlying a QTL region than what is possible in independent QTL mapping studies26. Moreover, the MQTL analysis helps to validate the genetic association of loci identified by genome-wide association study (GWAS) approach27,28,29.
Despite the wide use of meta-QTL analysis in plant species30,31,32,33,34,35,36,37,38,39, little is known about ionomic meta- QTL analysis in A. thaliana. This study aims to perform a meta-QTL analysis to uncover genomic control of ionome related traits in A. thaliana. Due to the importance of essential elements in biofortification studies, the emphasis of the current study is placed on the assessment of genomic regions controlling trace elements, especially Zn, Fe, and Mn. In addition, the ability of MQTL analysis to validate the loci associated with micronutrients identified by GWAS40 was assessed in our study.
Materials and methods
Development of QTL database
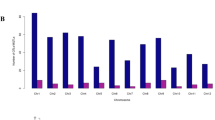
A database containing information on 483 QTLs for 38 different traits (Fig. 1) derived from 12 segregating populations was developed. The number of QTL per study ranged from 5 to 83 QTLs that were used to find the most stable genomic regions controlling ionomic quantitative traits in A. thaliana (Table 1). Except for those lacking proper genetic map and QTL-related information, QTL mapping studies including important quantitative traits with different markers in A. thaliana were used for the identification of MQTLs. The information of populations used in the meta-QTL analysis is summarized in Table 1. The number of QTLs for each ionomic trait and their distribution on A. thaliana chromosomes are presented in Fig. 1 and Supplementary Figure S1. The assessed ionomic QTLs were distributed across different chromosomes (χ(4)2 = 17.40, P = 0.0016) with chromosome 1 showing the highest number of QTLs (119 QTLs), followed by chromosome 5 (117 QTLs). Chromosomes 4 and 2 had the lowest number of QTLs (Fig. 1). The chromosomes 1 and 5 with 9 and 7 QTLs possessed the highest QTL numbers for Mn and Fe, respectively (Fig. 1). The highest number of QTLs for Zn were located on chromosome 5 with 14 QTLs. Details regarding the position, the proportion of phenotypic variance (% Expl.), and the log of odds ratios (LOD score) for QTLs were used for the analysis of meta-QTLs.

Distribution of QTL associated with different ionomic traits across the A. thaliana chromosomes. The numbers inside parentheses represent the number of QTL in each trait.
Constructing consensus genetic map and QTL projection
A consensus genetic map was constructed by integrating 12 map files. The constructed map file for each population included information about the type of cross, population size, map function, map units and the position of DNA markers used in different linkage groups. For map comparisons, the genome lengths from the individual and consensus genetic maps were calculated according to Hubert and Hedgecock45. First, the average spacing (χ) between markers was calculated by dividing the total length of all linkage groups by the number of intervals (number of markers minus the number of linkage groups). Then, the genome lengths for individual and consensus linkage maps were calculated by adding 2χ to the length of each linkage group to account for terminal chromosome regions46. After the development of the final consensus genetic map, the individual QTLs obtained from each population were projected onto the consensus genetic map in BioMercator v 4.224,25.
Meta-QTL analysis
The BioMercator v4.2 was24,25 used to perform the meta-QTL analysis (available at https://urgi.versailles.inra.fr/Tools/BioMercator-V4). The Veyrieras et al.22 approach was used for meta-QTL analysis where the number of QTLs in each chromosome was higher than 10 and the best QTL model was selected based on Akaike information criterion (AIC)22, corrected AIC criterion (AICc)22, AIC 3 candidate (AIC3)22, Bayesian information criterion (BIC)22, and Approximate Weight of Evidence (AWE)22. The best QTL model shows lower values in at least three of these five criterions. Consensus QTLs from the optimum model was regarded as meta-QTLs (MQTLs). The MQTL position and distribution in each linkage group were presented as a heatmap using the RIdeogram R package47 (Fig. 2).

Position of identified MQTLs on Arabidopsis genome associated with ionomic quantitative traits with 95% confidence interval. Each color in different linkage groups indicates the number of initial QTLs involved in each MQTL. The flanking markers for each MQTL are presented on the left side of linkage groups (cM).
The variation of QTLs of traits over genetic consensus map and the MQTLs density in both genetic and physical maps regions were estimated following the study by Martinez et al.32. The QTL and MQTL densities on the genetic map were evaluated by counting the number of QTLs and MQTLs in every 50 cM interval across the A. thaliana genome, starting from the centromeric region towards the ends of the chromosome (Fig. 3). The same procedure was followed for estimating MQTLs density in 10 Mb intervals across the A. thaliana physical map. The centromere position for genetic and physical maps of A. thaliana was retrieved from The Arabidopsis Information Resource (TAIR) database [https://www.arabidopsis.org/]. The distribution of QTLs and MQTLs was also compared in perspective with the gene density calculated with a bin size of in 0.1 Mb distance in each chromosome using the gff file of TAIR10. The projection of gene densities in comparison with the MQTLs was generated using the RIdeogram R package47 (Fig. 4).

Number and distribution of QTLs and MQTLs over Arabidopsis genome. (a) comparison between QTLs and MQTLs number on five chromosomes of Arabidopsis thaliana; (b) Distribution of assessed QTLs for ionomic traits over genetic consensus map; (c) Projection of the detected MQTLs over Arabidopsis genome based on genetic map; (d) Distribution of MQTLs for ionomic quantitative traits on physical map of Arabidopsis thaliana. The projection of QTLs and MQTLs on genetic map were illustrated as number per 50 cM distance. For physical map 10 Mb was considered. The distance was started from the centromeric region of each chromosome where it was considered as position 0 cM or Mb. Dots represent the location of QTLs and MQTLs.

The gene density of Arabidopsis thaliana chromosomes based on heat map and bar plot illustrations. The position of proved genes (detected in MQTLs interval) for different ionomic traits are presented in the right side of chromosomes.
The genetic loci highly associated with grain Zn, Fe, and Mn (as the most targeted mineral elements in biofortification studies) in the A. thaliana GWAS study was retrieved and also compared with the MQTLs and the results of GWAS based on SNP markers40. The genomic positions of MQTLs and associated loci reported in the A. thaliana GWAS study40 were compared based on the A. thaliana genome.
Functional candidate genes (CGs) in MQTLs interval
To uncover functional CGs controlling the ionomic traits (especially for grain Zn and Fe content), the sequences of the flanking markers for each MQTL were used to identify the corresponding genomic interval on the A. thaliana genome. The sequences of flanking markers were retrieved from the TAIR10 genome and BLAST was used to identify the precise genomic location. For those flanking markers lacking a definite position on the A. thaliana genome, the closest marker on the consensus map was selected to identify the MQTL position on the genome. The corresponding gene annotation was retrieved from the gff file containing gene coordinates for TAIR10 in addition to the Uniprot ID corresponding to the A. thaliana reference genome (TAIR10).
Trait analysis within MQTL
To identify ionomic traits within MQTL regions, the MQTLs result was converted into binary scores (0 or 1) based on the absence/presence of an individual trait QTL within an MQTL region. Hence, the number of times a trait was present within an MQTL, the number of times more than one QTL for a trait was present within MQTL (implying confirmation of the QTL), and the number of times traits co-localized within an MQTL were calculated. A chi-squared test with a degree of freedom (DF) = 1 was performed to determine traits showing significant co-localization with grain Zn, Fe, and Mn contents beyond what would be expected for a random distribution of QTL within MQTL throughout the genome. The expected number of each Zn, Fe, and Mn MQTL co-localized with each tested ionomic trait was calculated using Eq. (1) that was 0.7, 0.52, and 0.64 for Zn, Fe, and Mn, respectively.
Traits within MQTL regions were further analyzed using IBM SPSS Statistics v.24.
Results
Genetic consensus map and meta-QTL analysis
The original linkage map consisted of five linkage groups with an average of 118 DNA markers and covered 485.8 cM of the A. thaliana genome (Table 2). The constructed consensus genetic map contained 725 markers that covered 514.01 cM of the A. thaliana genome (Table 2).
All the 483 initial QTLs were successfully projected onto the consensus genetic map and used for meta-QTL analysis. The meta-QTL analysis confined these QTLs into 33 MQTLs with QTLs originating from at least two populations for all the ionomic traits. The distribution of MQTLs for each ionomic trait on each chromosome is presented in Table 3, Fig. 2, and Supplementary Figure S1. Of these MQTLs, 22 MQTLs (60.67%) were obtained from at least six independent populations (Table 3; Supplementary Figure S1).
Chromosome 5 harbored the largest number (9 MQTLs) and chromosomes 2 and 4 had the lowest number of MQTLs (Table 3, Fig. 2). The MQTLs with the highest initial QTLs could be considered as the more stable regions which control ionomic traits independent of genetic background and environment. Among the identified MQTLs in our study, MQTL-3/Chr2 had the highest number of initial QTLs (42 QTLs) derived from 10 studies followed by MQTL-7/Chr1, MQTL-1/Chr3, and MQTL-3/Chr5 with 32, 28 and 28 initial QTLs derived from nine, seven, and nine independent population studies, respectively (Table 3). The results showed that the “CH.192L-Col”, “HH.357L”, “AD.191L-Col”, “SNP135”, “SNP105”, “DF.252L”, “m291” and “BH.127L” markers were linked to these stable MQTLs.
The current meta-QTL analysis reduced the average confidence interval (CI) up to eight folds with an average of 1.30 cM in MQTLs in comparison to the mean CI of the original QTLs (10.88 cM). Among the detected MQTLs, the CI of 12 MQTLs (36.4%) was reduced to < 1 cM (Table 3).
Gene density and distribution of MQTLs and QTLs on chromosomes
The distribution patterns of QTLs on the consensus genetic map and MQTLs on the genetic and physical maps were investigated and compared with gene density (Figs. 3, 4). The overview of the distribution of the assessed QTLs for different ionomic traits exhibited that the non-telomeric regions possessed majority of QTLs and MQTLs in their intervals. Chromosomes 2, and 4 harbored relatively all of the QTLs on their non-telomeric regions (Figs. 2, 3b). A large number of QTLs were projected over the sub-telomeric region of chromosomes 1, 3, and 5 (Figs. 2, 3b). Similarly, the same pattern was observed for MQTLs distribution over the genetic consensus map. Chromosomes 2 and 4 possessed the majority of MQTLs in their non-telomeric intervals and chromosomes 1, 3, and 5 harbored several MQTLs in their sub-telomeric regions (Figs. 2, 3c). In contrast, the result of MQTLs distribution over A. thaliana physical map indicated a relatively high number of MQTLs in sub-telomeric regions (Fig. 3d).
The lowest number of QTLs and MQTLs were detected at the centromeric intervals for the ionomic traits (Figs. 2, 3). The MQTL-1/Chr2, MQTL-5/Chr3, and MQTL-5/Chr5 were located near the centromeric region of chromosomes 2, 3, and 5 of the physical and genetic consensus maps, respectively (Figs. 2, 3c,d). Furthermore, there was a significant correlation between the number of QTLs and MQTLs over the A. thaliana chromosomes (r = 0.97, P = 0.007) (Fig. 3a). The gene density over A. thaliana chromosomes showed a similar pattern with QTLs and MQTLs distributions (Fig. 4).
Functional candidate genes within MQTL intervals
The number of identified candidate genes in the detected MQTL intervals are reported in Table 3. All the annotated genes located in each MQTL interval and the functions of the potential candidate are presented in Supplementary Table S1. The highest number of candidate genes in the identified meta-QTLs was observed in MQTL-2/Chr4 (774 genes) followed by MQTL-4/Chr2 and MQTL-2/Chr5 with 693 and 692 gene number, respectively (Table 3). The lowest number of candidate genes belongs to MQTL-5/Chr3 with nine genes.
Several well-known candidate genes including bHLH-encoding genes family (e.g. bHLH34 (At3g23210)), WRKY (e.g. WRKY12 (At2g44745), WRKY13 (At4g39410)), MHX (At2g47600), NRAMP3 (At2g23150), ZIP4 (At5g48390), MTP11 (At2g39450), IRT3 (At1g60960), and FIT (At2g28160) were identified in the MQTLs regions of our study. The CGs have shown diverse functions for micronutrients transportation and homeostasis (Supplementary Table S1).
Trait analysis within MQTLs
The results of trait –MQTL analysis showed the unequal presence of individual QTLs across the confined MQTLs. Individual QTLs for potassium (K) were present in 26 of the 33 MQTL regions (78.8%), the most for any ionomic trait, followed by Zn (23 MQTL), P (22 MQTL), Mn (21 MQTL), Mg (20 MQTL) and Ca (20 MQTL). The Fe was presented in 17 MQTL regions. The meta-analysis was able to confirm at least one QTL for all assessed ionomic traits in the detected MQTLs intervals (Table 4).
The association between the individual ionomic QTLs and target QTLs (i.e. Zn, Fe, and Mn) was assessed by analyzing the co-localization frequency. There was 65.2% and 43.5% co-localization frequency between QTLs for Fe and Mn with Zn QTLs, 58.8% and 70.6% co-localization between Zn and Mn QTLs with Fe QTLs. The Zn and Fe QTLs represent 71.4% and 57.1% co-localization with Mn QTLs, respectively (Table 4). Among the identified MQTLs, the MQTL-1/Chr1, MQTL-3/Chr2, MQTL-1/Chr3, MQTL-3/Chr3, MQTL-5/Chr3, and MQTL-3/Chr5 harbored all Zn, Fe, and Mn QTLs. Besides, the highest QTLs number for Zn (5 QTL) was located on MQTL-3/Chr5 whilst the MQTL-3/Chr2 includes the highest QTLs number for Fe (5) and Mn (4) QTLs. Association between ionomic QTLs with Zn and Mn QTLs did not differ from the expectations. However, the association between ionomic (especially Mn and Zn QTLs) and Fe QTLs was statistically significant suggesting their association was more frequent than expected (Table 4).
Comparison MQTLs and GWAS results
Location of the identified MQTLs was compared with the SNPs linked to Zn, Fe, and Mn traits in a GWAS study for A. thaliana43. This comparison led to the identification of nine highly associated loci for Zn, Fe, and Mn elements that illustrated more reliable MQTLs. In fact, two, three, and four highly associated loci from the GWAS study were co-located with our identified MQTLs for Zn, Fe, and Mn, respectively (Table 5). These loci were distributed on chromosomes 3 and 4. In addition, 104 common genes between GWAS study and the current MQTL analysis were located in the MQTL-3/Chr1 (11 genes), MQTL-3/Chr3 (16 genes), MQTL-4/Chr3 (10 genes), MQTL-5/Chr3 (2 genes), MQTL-2/Chr4 (14 genes), MQTL-3/Chr4 (19 genes), MQTL-4/CHr4 (9 genes), MQTL-2/Chr5 (12 genes) and MQTL-7/Chr5 (11 genes) intervals. Among the detected common genes between GWAS study and the current MQTL analysis, the well-known At4g08170 gene annotated as ITPK3 was located in the MQTL-2/Chr4 interval (Supplementary Table S2).
Discussion
The construction of a new consensus genetic map is a prerequisite for the projection of QTLs in the QTL database and running meta-QTL analysis. The results of the current MQTL analysis suggested that the MQTL-3/Chr2, MQTL-7/Chr1, MQTL-1/Chr3, and MQTL-3/Chr5 with the highest number of initial QTLs identified in independent populations were the most viable, stable, and robust QTLs under different experimental conditions. Identification of meta-QTLs significantly increases the power and precision of our ability to map important traits that will provide greater precision for future fine mapping and marker development48. In the current MQTL analysis, the average confidence interval (CI) for QTLs of ionomic traits reduced up to eight folds compared to the mean CI of the original QTLs. The ability of the MQTL analysis methods to reduce the QTL confidence interval by taking advantage of pooling QTLs helps to increase the resolution of selected candidate genes22 and consequently increases the precision of detecting functional candidate genes. Two complementary approaches for genetic mapping, linkage mapping (QTL mapping) and association mapping (LD mapping) have led to the successful dissection of complex traits in many plant species by detecting QTLs and identifying marker-trait associations49. Often, the genetic and environmental background, number of traits, phenotypic variation, marker density, and map regulation are the most limiting factors in linkage mapping approaches. However, the meta-QTL analysis helps to identify the most stable QTLs regardless of limitations in traditional QTL mapping studies49. The LD mapping uncovers genomic regions underlying quantitative traits with higher accuracy compared to the linkage mapping that is particularly efficient in species with low linkage disequilibrium (LD)50,51. Besides, the population structure of the studied panel in LD mapping can lead to false-positive association discovery52,53. Despite the limitations of QTL- and LD-mapping methods, the statistical power to detect QTLs through the use of these approaches is particularly affected by the sample size54. To overcome these limitations, meta- analysis has been used to combine results from multiple studies and increase the power of genetic mapping49. The QTL meta-analysis method is a horizontal way to integrate information for the same trait from different experiments and populations. Thus, a meta-analysis allows for comparative genomics and is a valuable tool to complement information obtained from vertical integration55.
Our meta-QTL analysis over the A. thaliana chromosomes revealed that chromosomes 4 and 2 possessed a low number of QTLs and MQTLs, which could be attributed to fewer markers on these chromosomes. This result was in line with the results of the Serin et al.56 study. Our results revealed a similar pattern for the distribution of QTLs and MQTLs over the A. thaliana chromosomes based on the genetic map used. Besides, the projection of QTLs and MQTLs across A. thaliana genome was in accordance with the constructed gene density map. It has been shown that QTLs result from the genetic segregation of sequence polymorphisms at functional elements such as regulatory sequences upstream of genes and/or coding sequences57,58. Therefore, it is expected that QTL density is related to the gene density, polymorphism rate at functional sites in genic regions and the frequency of recombination in the genome32. These outcomes showed for the first time that majority of QTLs and MQTLs controlling ionomic traits are located in the non-telomeric regions of the A. thaliana genetic map. However, higher QTL density at the sub-telomeric regions was apparent when physical distances were considered for meta-QTL analysis (Figs. 2, 3)32. Particularly, the distribution of MQTLs was higher in the gene dense regions located in non-telomeric and sub-telomeric regions in the A. thaliana genome. This result was in accordance with the result of the Martinez et al.32 study showing QTLs appeared to more densely map on non-telomeric regions of genetic maps and sub-telomeric regions of A. thaliana physical map.
Identifying genes for ionomic traits control is a fundamental step for better understanding of ionome transportation, accumulation and mineral element changes in plants. Results of our meta-QTL analysis for ionomic traits showed that several important genes identified in the MQTLs were associated with micronutrient homeostasis. A better understanding of responsible genes involved in micronutrient homeostasis in plants is among the most important proceedings for breeding programs to combat “micronutrient malnutrition”6. A useful gene can be used to target multiple crops through genomic engineering approaches. The bHLH-encoding genes family including bHLH34 (8283003–8285081 bp) was identified in the MQTL-4/Chr3 of our meta-QTL study. Several members of bHLH-encoding genes are associated with the Fe homeostasis control59. The bHLH34 belonging to the clade IVc involved in A. thaliana Fe deficiency response59. The MQTL-2/Chr2 contains the NRAMP3 (9856149–9858778 bp) gene that is associated with iron deficiency in both roots and aerial parts of A. thaliana and localize to the vacuolar membrane, indicating its contribution to intracellular metal homeostasis60,61,62. The MQTL-3/Chr2 harbored another important gene, FIT (12004658–12006276 bp), which plays a predominant role in the regulation of iron mobilization63,64,65,66. The role of FIT in Fe acquisition had been documented67. The ZIP4 (19612020–19615383 bp) and IRT3 (22445299–22447299 bp) genes that located in the MQTL-7/Chr5 and MQTL-7/Chr1 intervals of our study are involved in Zn, Fe, Mn, and Cd transport68. Further, ZIP proteins that express in shoot tissues contribute to Zn uptake from the soil translocation of Zn throughout the plant69,70. In A. thaliana, the ZIP4 is highly expressed under zinc deficiency conditions68,71. The expression of AtIRT3 can respond to Zn72, which implies a possible role for IRT3 in Zn transport and homeostasis73. Overexpressing of AtIRT3 in A. thaliana increases accumulation of Zn in the shoot and Fe in the root of transgenic lines suggesting the role of IRT3 as a Zn and Fe‐uptake transporter73. Detection of the MTP11 (16471560–16474060 bp) gene in the MQTL-5/Chr2 intervals in the current meta-analysis provides an evidence for Mn2+-specific transport activity of AtMTP11 that implicate the pre-vacuolar compartments in both Mn2+ tolerance and Mn2+ homeostasis mechanisms74. Our meta-analysis confirmed the presence of WRKY12 (18447203–18449093 bp) and WRKY13 (18332606–18334893 bp) genes located in the MQTL-4/Chr2 and MQTL-5/Chr4 intervals, respectively. The AtWRKY1275 gene regulates cadmium (Cd) accumulation and AtWRKY1376 enhances tolerance to Cd. MHX (19524117–19527580 bp) located in the identified MQTL-4/Chr2 was another identified gene associated with concentration and transportation of Mg14,77. Electrophysiological analysis and overexpression studies have shown that the A. thaliana MHX gene encodes Mg2+/H+ exchanger, which exchange protons with Mg2+, Zn2+, Cd2+, and possibly Fe2+ ions across the vacuolar membrane78,79.
The results of our meta-QTL analysis suggested that QTLs for K, Zn, P, Mn, Mg, and Ca were the most frequent distributed QTLs in the detected MQTLs regions that could be due to high heritability and the role of these ions in physiological pathways80,81,82,83. Our MQTL-analysis revealed co-localization between QTLs for Zn, Mn, and Fe ions. The co-localization of QTLs may reflect the correlations observed between the ionomic traits. Based on A. thaliana QTL analysis, different ionomic traits demonstrated significant correlation which most robust was the correlations between Zn, Mn, and Fe traits16,42. The result of co-localization frequency of ionomic QTLs on detected MQTLs regions in this study revealed the possible signs of MQTLs interaction, which impacts the association of ionomic traits at the genomic level. Co-localized QTLs or pleiotropic genes are of great significance for breeders and geneticists, for assessing the effects of selective genetic improvement programs, or for understanding pathways of genetic activity84. For biofortification, especially with Zn and Fe, large-effect and consistent QTLs are of particular importance and merit further examination85. Furthermore, the co-localization of QTLs of different traits in the same chromosomal regions suggests the existence of physiological and/or genetic relationships between traits86. Thus, the co-localized QTLs are important for breeding programs, QTL pyramiding, and simultaneous improvement of several ionomic traits.
Association analysis helps to unravel the genetic control of quantitative complex traits and validation of their corresponding genes in independent backgrounds87,88. In our study, four MQTLs (MQTL-3/Chr3, MQTL-4/Chr3, MQTL-3/Chr5, and MQTL-3/Chr4) were collinear with nine highly associated SNPs responsible for Zn, Fe, and Mn quantitative traits. Further, the ITPK3 (5163220–5167192 bp) gene, which involved in micronutrients chelating and accumulation, was among the identified functional candidate genes detected through the current MQTL analyses and the GWAS study47,89. The ability of the MQTL analysis method to validate identified loci/QTLs using GWAS have been previously documented27,28,29. Results of our meta-QTL analysis represent the coherence and consistency of MQTL analyses and GWAS for the identification of genomic regions corresponding to the studied ionomic traits.
Conclusions
To our knowledge, this is the first MQTL analysis report that identified several major genomic regions associated with ionomic traits in A. thaliana. This analysis defines a genome-wide landscape on the most stable genomic regions (MQTL-3/Chr2, MQTL-7/Chr1, MQTL-1/Chr3, and MQTL-3/Chr5) along with reliable genetic markers (CH.192L-Col, HH.357L, AD.191L-Col, SNP135, SNP105, DF.252L, m291, and BH.127L) that provide a robust tool for breeding ionomic traits through marker-assisted selection. Measuring trace elements in large sample size population is costly for breeders and identifying linked DNA markers accelerates pre-breeding and variety screening for biofortification programs and improving quality traits in plants. Results of our study suggested that the genomic positions of QTLs and MQTLs were mainly located in non-telomeric regions based on the newly constructed genetic consensus map, whilst the physical map revealed the projection of higher MQTLs in sub-telomeric regions. This was in line with the results of gene density in sub-telomeric and sub-centromeric regions of the Arabidopsis chromosomes. The results of the analysis for co-localization frequency revealed co-map QTLs controlling ionomic traits suggesting the existence of physiological and/or genetic relationships between these traits which lead to possible improvement of multiple ionome traits simultaneously. Further, the current study demonstrates that meta-QTL analysis method is cheaper than, and as informative as GWAS and then is powerful enough to provide greater resolution for future fine mapping without GWAS. Overall, the identified candidate genes at the detected MQTLs regions will provide a better understanding of ionomic variation in the A. thaliana genome. Besides their function can be generalized to other plant species which provides the raw material for breeders in various breeding programs such as biofortification.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Singh, S. B., Singh, K., Butola, S. S., Rawat, S. & Arunachalam, K. Determination of macronutrients, micronutrients and heavy metals present in Spilanthes acmella Hutch and Dalz: possible health effects. Nat. Prod. Sci. 26(1), 50–58 (2020).
Singh, U. M., Sareen, P., Sengar, R. S. & Kumar, A. Plant ionomics: a newer approach to study mineral transport and its regulation. Acta Physiol. Plant. 35(9), 2641–2653 (2013).
Khan, M. et al. Trace elements in abiotic stress tolerance. In Plant Nutrients and Abiotic Stress Tolerance (ed. Hasanuzzaman, M., Fujita, M., Oku, H., Nahar, K. & Hawrylak-Nowak, B.) 137–151 (Springer, Singapore, 2018).
Narwal, R. P., Malik, R. S., Malhotra, S. K. & Singh, B. R. Micronutrients and human health. In Encyclopedia of Soil Science (ed. Lal, R.) 1443–1448 (CRC Press, 2017).
Pecora, F., Persico, F., Argentiero, A., Neglia, C. & Esposito, S. The role of micronutrients in support of the immune response against viral infections. Nutrients 12(10), 3198 (2020).
Shariatipour, N. & Heidari, B. Genetic-based biofortification of staple food crops to meet zinc and iron deficiency-related challenges. In Plant Micronutrients: Deficiency and Toxicity Management (ed. Aftab, T. & Hakeem, K.R.) 173–223 (Springer, Cham, 2020).
Salgueiro, M. J. et al. Zinc status and immune system relationship. Biol. Trace Elem. Res. 76, 193–205 (2000).
Maxfield, L., & Crane, J. S. Zinc deficiency. In StatPearls. Treasure Island (FL: StatPearls Publishing, 2020).
Hodge, J. Hidden hunger: approaches to tackling micronutrient deficiencies. In Nourishing Millions: Stories of Change in Nutrition (ed. Gillespie, S., Hodge, J., Yosef, S. & Pandya-Lorch, R.) 35–43 (Washington: International Food Policy Research Institute (IFPRI), 2016).
Salt, D. E., Baxter, I. & Lahner, B. Ionomics and the study of the plant ionome. Annu. Rev. Plant. Biol. 59, 709–733 (2008).
Baxter, I. Ionomics: the functional genomics of elements. Brief. Funct. Genomics 9(2), 149–156 (2010).
Barh, D. OMICS Applications in Crop Science (CRC Press, 2013).
Borém, A. & Fritsche-Neto, R. Omics in plant breeding. (Wiley Blackwell, 2014).
Vreugdenhil, D., Aarts, M. G., Koornneef, M., Nelissen, H. & Ernst, W. H. Natural variation and QTL analysis for cationic mineral content in seeds of Arabidopsis thaliana. Plant Cell Environ. 27(7), 828–839 (2004).
Norton, G. J. et al. Genetic mapping of the rice ionome in leaves and grain: identification of QTLs for 17 elements including arsenic, cadmium, iron and selenium. Plant Soil 329(1–2), 139–153 (2010).
Ghandilyan, A., Kutman, U. B., Kutman, B. Y., Cakmak, I. & Aarts, M. G. Genetic analysis of the effect of zinc deficiency on Arabidopsis growth and mineral concentrations. Plant Soil 361(1–2), 227–239 (2012).
Gu, R. et al. Comprehensive phenotypic analysis and quantitative trait locus identification for grain mineral concentration, content, and yield in maize (Zea mays L.). Theor Appl Genet 128(9), 1777–1789 (2015).
Liu, J., Wu, B., Singh, R. P. & Velu, G. QTL mapping for micronutrients concentration and yield component traits in a hexaploid wheat mapping population. J. Cereal Sci. 88, 57–64 (2019).
Wang, C. et al. Genetic mapping of ionomic quantitative trait loci in rice grain and straw reveals OsMOT1; 1 as the putative causal gene for a molybdenum QTL qMo8. Mol. Genet. Genom. 295(2), 391–407 (2020).
Acuña-Galindo, M. A., Mason, R. E., Subramanian, N. K. & Hays, D. B. Meta-analysis of wheat QTL regions associated with adaptation to drought and heat stress. Crop Sci. 55(2), 477–492 (2015).
Goffinet, B. & Gerber, S. Quantitative trait loci: a meta-analysis. Genetics 155(1), 463–473 (2000).
Veyrieras, J. B., Goffinet, B. & Charcosset, A. MetaQTL: a package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinform. 8(1), 49 (2007).
Hanocq, E., Laperche, A., Jaminon, O., Lainé, A. L. & Le Gouis, J. Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis. Theor. Appl. Genet. 114(3), 569–584 (2007).
Arcade, A. et al. BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20(14), 2324–2326 (2004).
Sosnowski, O., Charcosset, A. & Joets, J. BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 28(15), 2082–2083 (2012).
Li, W. T. et al. Meta-analysis of QTL associated with tolerance to abiotic stresses in barley. Euphytica 189(1), 31–49 (2013).
Wang, Y., Wang, Y., Wang, X. & Deng, D. Integrated meta-QTL and genome-wide association study analyses reveal candidate genes for maize yield. J. Plant Growth Regul. 39, 229–238 (2019).
Safdar, L. B. et al. Genome-wide association study and QTL meta-analysis identified novel genomic loci controlling potassium use efficiency and agronomic traits in bread wheat. Front Plant Sci 11, 70 (2020).
Yang, Y. et al. Large-scale integration of meta-QTL and genome-wide association study discovers the genomic regions and candidate genes for yield and yield-related traits in bread wheat. Theor. Appl. Genet. https://doi.org/10.1007/s00122-021-03881-4 (2021).
Jin, T. et al. The genetic architecture of zinc and iron content in maize grains as revealed by QTL mapping and meta-analysis. Breed Sci. 63(3), 317–324 (2013).
Chardon, F. et al. QTL meta-analysis in Arabidopsis reveals an interaction between leaf senescence and resource allocation to seeds. J. Exp. Bot. 65(14), 3949–3962 (2014).
Martinez, A. K. et al. Yield QTLome distribution correlates with gene density in maize. Plant Sci. 242, 300–309 (2016).
Zhang, Y. et al. QTL meta-analysis of root traits in Brassica napus under contrasting phosphorus supply in two growth systems. Sci. Rep. 6, 33113 (2016).
Abdelraheem, A., Liu, F., Song, M. & Zhang, J. F. A meta-analysis of quantitative trait loci for abiotic and biotic stress resistance in tetraploid cotton. Mol. Genet. Genom. 292(6), 1221–1235 (2017).
Zhang, X., Shabala, S., Koutoulis, A., Shabala, L. & Zhou, M. Meta-analysis of major QTL for abiotic stress tolerance in barley and implications for barley breeding. Planta 245(2), 283–295 (2017).
Avni, R. et al. Genome based meta-QTL analysis of grain weight in tetraploid wheat identifies rare alleles of GRF4 associated with larger grains. Genes 9(12), 636 (2018).
Izquierdo, P. et al. Meta-QTL analysis of seed iron and zinc concentration and content in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 131(8), 1645–1658 (2018).
Islam, M., Ontoy, J. & Subudhi, P. K. Meta-analysis of quantitative trait loci associated with seedling-stage salt tolerance in rice (Oryza sativa L.). Plants 8(2), 33 (2019).
Raza, Q., Riaz, A., Sabar, M., Atif, R. M. & Bashir, K. Meta-analysis of grain iron and zinc associated QTLs identified hotspot chromosomal regions and positional candidate genes for breeding biofortified rice. Plant Sci. 288, 110214 (2019).
Chen, X., Yuan, L. & Ludewig, U. Natural genetic variation of seed micronutrients of Arabidopsis thaliana grown in Zinc-deficient and Zinc-amended soil. Front. Plant. Sci. 7, 1070 (2016).
Buescher, E. et al. Natural genetic variation in selected populations of Arabidopsis thaliana is associated with ionomic differences. PLoS ONE 5(6), e11081 (2010).
Ghandilyan, A. et al. Genetic analysis identifies quantitative trait loci controlling rosette mineral concentrations in Arabidopsis thaliana under drought. New Phytol. 184(1), 180–192 (2009).
Ghandilyan, A. et al. A strong effect of growth medium and organ type on the identification of QTLs for phytate and mineral concentrations in three Arabidopsis thaliana RIL populations. J. Exp. Bot. 60(5), 1409–1425 (2009).
Waters, B. M. & Grusak, M. A. Quantitative trait locus mapping for seed mineral concentrations in two Arabidopsis thaliana recombinant inbred populations. New Phytol. 179(4), 1033–1047 (2008).
Hubert, S. & Hedgecock, D. Linkage maps of microsatellite DNA markers for the Pacific oyster Crassostrea gigas. Genetics 168(1), 351–362 (2004).
Fishman, L., Kelly, A. J., Morgan, E. & Willis, J. H. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 159(4), 1701–1716 (2001).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, e251 (2020).
Löffler, M., Schön, C. C. & Miedaner, T. Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol. Breed. 23(3), 473–488 (2009).
Xu, Y., Li, P., Yang, Z. & Xu, C. Genetic mapping of quantitative trait loci in crops. Crop J. 5(2), 175–184 (2017).
Gupta, P. K., Rustgi, S. & Kulwal, P. L. Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol. Biol. 57, 461–485 (2005).
Korte, A. & Farlow, A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9, 29 (2013).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11(7), 459–463 (2010).
Visscher, P. M., Brown, M. A., McCarthy, M. I. & Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 90(1), 7–24 (2012).
Han, B. & Eskin, E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 8(3), e1002555 (2012).
Asins, M. J., Bernet, G. P., Villalta, I. & Carbonell, E. A. QTL analysis in plant breeding. In Molecular Techniques in Crop Improvement (ed. Mohan Jain, S. & Brar, D.S.) 3–21 (Springer, Dordrecht, 2010).
Serin, E. A. et al. Construction of a high-density genetic map from RNA-Seq data for an Arabidopsis bay-0× Shahdara RIL population. Front. Genet. 8, 201 (2017).
Flint, J. & Mackay, T. F. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 19(5), 723–733 (2009).
Salvi, S. & Tuberosa, R. The crop QTLome comes of age. Curr. Opin. Biotechnol. 32, 179–185 (2015).
Gao, F., Robe, K., Gaymard, F., Izquierdo, E. & Dubos, C. The transcriptional control of iron homeostasis in plants: a tale of bHLH transcription factors?. Front. Plant. Sci. 10, 6 (2019).
Thomine, S., Lelièvre, F., Debarbieux, E., Schroeder, J. I. & Barbier-Brygoo, H. AtNRAMP3, a multispecific vacuolar metal transporter involved in plant responses to iron deficiency. Plant J. 34(5), 685–695 (2003).
Lanquar, V., Lelièvre, F., Barbier-Brygoo, H. & Thomine, S. Regulation and function of AtNRAMP4 metal transporter protein. Soil Sci. Plant Nutr. 50, 1141–1150 (2004).
Lanquar, V. et al. Mobilization of vacuolar iron by AtNRAMP3 and AtNRAMP4 is essential for seed germination on low iron. EMBO J. 24(23), 4041–4051 (2005).
Colangelo, E. P. & Guerinot, M. L. The essential basic helix-loop-helix protein FIT1 is required for the iron deficiency response. Plant Cell 16(12), 3400–3412 (2004).
Jakoby, M., Wang, H. Y., Reidt, W., Weisshaar, B. & Bauer, P. FRU (BHLH029) is required for induction of iron mobilization genes in Arabidopsis thaliana. FEBS Lett. 577(3), 528–534 (2004).
Yuan, Y. X., Zhang, J., Wang, D. W. & Ling, H. Q. AtbHLH29 of Arabidopsis thaliana is a functional ortholog of tomato FER involved in controlling iron acquisition in strategy I plants. Cell Res. 15(8), 613–621 (2005).
Bauer, P., Ling, H. Q. & Guerinot, M. L. FIT, the FER-like iron deficiency induced transcription factor in Arabidopsis. Plant. Physiol. Biochem. 45(5), 260–261 (2007).
Schwarz, B. & Bauer, P. FIT, a regulatory hub for iron deficiency and stress signaling in roots, and FIT-dependent and-independent gene signatures. J. Exp. Bot. 71(5), 1694–1705 (2020).
Guerinot, M. L. The ZIP family of metal transporters. Biochim. Biophys. Acta. Biomembr. 1465(1–2), 190–198 (2000).
Grotz, N. et al. Identification of a family of zinc transporter genes from Arabidopsis that respond to zinc deficiency. Proc. Natl. Acad. Sci. USA. 95(12), 7220–7224 (1998).
Colangelo, E. P. & Guerinot, M. L. Put the metal to the petal: metal uptake and transport throughout plants. Curr. Opin. Plant Biol. 9(3), 322–330 (2006).
van de Mortel, J. E. et al. Large expression differences in genes for iron and zinc homeostasis, stress response, and lignin biosynthesis distinguish roots of Arabidopsis thaliana and the related metal hyperaccumulator Thlaspi caerulescens. Plant Physiol. 142, 1127–1147 (2006).
Talke, I. N., Hanikenne, M. & Krämer, U. Zinc-dependent global transcriptional control, transcriptional deregulation, and higher gene copy number for genes in metal homeostasis of the hyperaccumulator Arabidopsis halleri. Plant Physiol. 142(1), 148–1467 (2006).
Lin, Y. F. et al. Arabidopsis IRT3 is a zinc-regulated and plasma membrane localized zinc/iron transporter. New Phytol. 182(2), 392–404 (2009).
Delhaize, E. et al. A role for the AtMTP11 gene of Arabidopsis in manganese transport and tolerance. Plant J. 51(2), 198–210 (2007).
Han, Y. et al. WRKY12 represses GSH1 expression to negatively regulate cadmium tolerance in Arabidopsis. Plant Mol. Biol. 99(1–2), 149–159 (2019).
Sheng, Y. et al. The WRKY transcription factor, WRKY13, activates PDR8 expression to positively regulate cadmium tolerance in Arabidopsis. Plant Cell Environ. 42(3), 891–903 (2019).
Shaul, O. et al. Cloning and characterization of a novel Mg2+/H+ exchanger. EMBO J. 18(14), 3973–3980 (1999).
Berezin, I. et al. Overexpression of AtMHX in tobacco causes increased sensitivity to Mg2+, Zn2+, and Cd2+ ions, induction of V-ATPase expression, and a reduction in plant size. Plant Cell. Rep. 27, 939–949 (2008).
Gaash, R. et al. Phylogeny and a structural model of plant MHX transporters. BMC Plant Biol. 13(1), 1–20 (2013).
Grusak, M. A. & DellaPenna, D. Improving the nutrient composition of plants to enhance human nutrition and health. Ann. Rev. Plant. Physiol. Plant. Mol. Biol. 50, 133–161 (1999).
Garcia-Oliveira, A. L., Tan, L., Fu, Y. & Sun, C. Genetic identification of quantitative trait loci for contents of mineral nutrients in rice grain. J. Integr. Plant. Biol. 51(1), 84–92 (2009).
Baxter, I. et al. Biodiversity of mineral nutrient and trace element accumulation in Arabidopsis thaliana. PLoS ONE 7(4), e35121 (2012).
Manickavelu, A. et al. Genetic nature of elemental contents in wheat grains and its genomic prediction: toward the effective use of wheat landraces from Afghanistan. PLoS ONE 12(1), e0169416 (2017).
Hill, W. G. & Zhang, X. S. On the pleiotropic structure of the genotype–phenotype map and the evolvability of complex organisms. Genetics 190(3), 1131–1137 (2012).
Descalsota-Empleo, G. I. et al. Genetic mapping of QTL for agronomic traits and grain mineral elements in rice. Crop J. 7(4), 560–572 (2019).
Getahun, B. B., Visser, R. G. & van der Linden, C. G. Identification of QTLs associated with nitrogen use efficiency and related traits in a diploid potato population. Am. J. Potato. Res. 97(2), 185–201 (2020).
Newton-Cheh, C. & Hirschhorn, J. N. Genetic association studies of complex traits: design and analysis issues. Mutat. Res.-Fund. Mol. M. 573(1–2), 54–69 (2005).
König, I. R. Validation in genetic association studies. Brief. Bioinform. 12(3), 253–258 (2011).
Raboy, V. Approaches and challenges to engineering seed phytate and total phosphorus. Plant Sci. 177(4), 281–296 (2009).
Acknowledgements
The authors gratefully acknowledge the supports from Shiraz University. No grant was available to support this research.
Author information
Authors and Affiliations
Contributions
N.S. contributed to methodology, software, data curation, data analysis, writing- original draft preparation; B.H. involved in conceptualization, data analysis, investigation, supervision, methodology, reviewing and editing; S.R. and P.S. contributed to conceptualization, reviewing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shariatipour, N., Heidari, B., Ravi, S. et al. Genomic analysis of ionome-related QTLs in Arabidopsis thaliana. Sci Rep 11, 19194 (2021). https://doi.org/10.1038/s41598-021-98592-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98592-7
This article is cited by
-
Meta-QTLs, ortho-MetaQTLs and candidate genes for grain Fe and Zn contents in wheat (Triticum aestivum L.)
Physiology and Molecular Biology of Plants (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
