
Abstract
Antler is a special bone tissue that has the ability to regenerate completely periodically. It is the fastest growing bone in the animal kingdom. Antler provides a valuable research model for bone growth and mineralization. Antler grows longitudinally by endochondral ossification with their growth center located in its tip. Many scholars have carried out detailed studies on morphology and gene expression of antler tip. However, few scholars have analyzed the protein expression patterns of antler tip at different development stages. This study used label-free proteomics approach to analyze the protein expression dynamics of the antler tip in six developmental periods (15, 25, 45, 65, 100 and 130 days after the previous antler cast) and costal cartilage. In result, 2052 proteins were confidently quantified, including 1937 antler proteins and 1044 costal cartilage proteins. Moreover, 913 antler core proteins and 132 antler-special proteins were obtained. Besides, the stages special proteins and differentially expressed proteins (DEPs) in different development stages were analyzed. A total of 875 DEPs were determined by one-way AVOVA. It is found that the growth period (15, 25, 45 and 65 days) showed more up-regulated protein including several chondrogenesis-associated proteins (collagen types II, collagen types XI, HAPLN1, PAPSS1 and PAPSS2). In ossification stages, the up-regulated proteins related with lysosome (CTSD, CTSB, MMP9, CAII) indicated that the antler has higher bone remodeling activity. Given the up-regulated expression of immune-related molecules (S100A7, CATHL7, LTF, AZU1, ELANE and MPO), we speculate that the local immune system may contribute to the ossification of antler tip. In conclusion, proteomics technology was used to deeply analyze the protein expression patterns of antler at different development stages. This provides a strong support for the research on the molecular regulation mechanism of rapid growth and ossification of velvet antler.
Similar content being viewed by others


Introduction
There are different types of bones with different characteristics1. John Currey strongly advocated studying bone tissues that were not familiar in order to broaden the understanding of the structure–function relationship of vertebrate bones2. He was interested on the study of antlers due to their special features. Antler is a head appendage of Cervidae different from other ruminants. Antler can regenerate completely periodically. Antler is the fastest growing bone in the animal kingdom. This makes antler a good model for studying bone growth. Currently, extensive research has been carried out on the regeneration and development mechanism of antler and substantial progress has been made. Morphology, histology and experiments have proved that the regenerated tissue that forms the antler blastema is derived from the pedicle periosteum (PP). PP is a derivative of the antler antlerogenic periosteum (AP) which is responsible for the initial pedicle and first antler formation3,4,5. Studies from different laboratories have shown that cells from AP and PP have stem cell properties. Therefore, velvet regeneration is considered to be a regeneration process based on stem cells6.
The growth and ossification of antler is different from that of long bones. Growth plates and secondary ossification centers are the characteristics of mammalian long bone growth but they are absent in growing antlers. The growth of antlers is a manifestation of cartilage ossification which involves endochondral ossification and intramembranous ossification. Each antler branch extends longitudinally through endochondral ossification. Moreover, the growth of bone around the antler shaft increases its diameter through intramembranous ossification7. Type II collagen is a marker of chondroprogenitor. Price et al. demonstrated the presence of type II collagen at the tip of antler by in situ nucleic acid hybridization and immunocytochemistry8.
The antler growth center is located at the tip. The antler growth is similar to epiphyseal growth plate in some aspects. The antler tip is a typical far and near zone and the transition is gradual. However, the boundary between different regions is complicated. Histologically, the antler tip was divided into five tissue layers from distal to proximal: reserve mesenchyme, pre-cartilage, transition zone, cartilage and mineralized cartilage. Ba et al. analyzed the gene expression patterns of these five layers and found the hub genes and signaling pathway related to chondrogenesis and osteogenesis9. Through RNA-seq methods, gene expression patterns of the skin, mesenchyme, pre-cartilage and cartilage also were analyzed and series of genes that contributed to chondrogenesis were identified10.
Antler usually takes 4–5 months with its longitudinal growth showing a typical S-shaped curve. Growth starts slowly in the spring and accelerates exponentially during the summer. During the growth phase, elongation of antlers in some large deer species (wapiti or moose) can exceed 2 cm/day. This represents the fastest tissue growth rate in the animal kingdom11. Elongation slows while testosterone rise as the days shorten in late summer. Moreover, the antlers become heavily mineralized while velvet is shed in preparation for the autumn mating season. The biological characteristics of rapid growth and ossification provide a valuable research model for bone growth and mineralization. To investigate the molecular mechanism, Zhao et al. performed gene expression analysis for 15-day, 60-day and 90-day antler based on the RNA-seq approach. These represented slow growth stage, rapid growth stage, and ossification stages respectively12,13.
Protein participates in various biological processes as the direct executor of gene function. Studies have shown that since transcripts fails to consider various post-translational modifications. Moreover, transcripts are only a moderate predictor of protein expression. Therefore, changes in RNA levels cannot completely determine changes in protein levels. Currently, label-free proteomics technology has been widely used in the study of biological development. This provides a more comprehensive and accurate understanding of biological development14,15. At present, the whole-genome sequences of sika deer (Cervus nippon) was assembled and deposited in the Genome Warehouse in the National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, under accession number GWHANOY00000000, which is publicly accessible at https://bigd.big.ac.cn/gwh. This provided strong data support for the mining of key genes and construction of network regulation for the rapid growth and ossification of antler.
In this study, based on the label-free proteomics technology, an in-depth analysis of the protein expression changes of sika deer antler was conducted in six different developmental periods across slow growth stages (15 and 25 days), rapid growth stages (45 and 65 days) and ossification stages (100 and 130 days). In total, 2052 proteins were confidently quantified, including 1937 sika deer antler proteins and 1044 sika deer costal cartilage proteins. A total of 132 antler-specific proteins were successfully obtained as compared with sika costal cartilage. 875 differentially expressed proteins were screened using one ANOVA method. By analyzing the expression trend of these proteins in different biological periods of antler, we found that the main characteristic of antler growth period may be chondrogenesis, while the ossification stage is mainly the reconstruction of bone matrix. Moreover, we speculate that the local immune system may contribute to the ossification of antler tip. In conclusion, the protein expression dynamics during the regeneration cycle of sika deer antler was described. This was beneficial in determining the rapid growth and ossification mechanisms of antler.
Result
Protein identification based on label-free
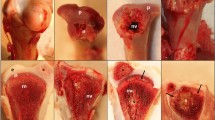
In this study, antlers from 6 representative time points (Fig. 1) and costal cartilage were collected and LC–MS/MS was performed three times per sample. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD024323 (https://www.ebi.ac.uk/pride/). Based on the sika deer protein sequences database, 3218 protein groups with 35,379 unique peptides at FDR < 0.01 were identified. Robust detection was filtered in at least two replicates of any sample. In result, we obtained 2052 proteins, including 1937 antler protein and 1044 costal cartilage proteins (Table S1). Pearson correlation analysis was performed to evaluate the MS/MS data (Fig. 2, Table S2). The Pearson correlation coefficients of 3 LC–MS/MS repetitions ranged from 0.989 to 0.999. This indicated high technical reproducibility of the mass spectrometer. In addition, the antler of 15 days, 25 days, 45 days, and 65 days had a high correlation (R = 0.836–0.967). However, the correlation with the antler of 100 days and 130 days was relatively low (R = 0.606–0.853). Sample cluster analysis yielded similar results. The first four periods were clustered into one category and then clustered within 100 days and 130 days. Moreover, the amounts of proteins in the first four periods were similar while the latter two periods were relatively small (Fig. 3A).

Flowchart of proteomic analysis of sika deer antlers at different development stages. 3 4-year-old male sika deer were selected. The two antlers from first deer were harvested at 15 and 25 days respectively, the two antlers of the second deer were harvested at 45 and 65 days respectively, and the two antlers of the last deer were harvested at 100 and 130 days respectively. LC–MS/MS was performed three times per sample.

Pearson correlation analysis of sika deer antlers in 6 different development stages.

Overview of proteins identified for antler in different development stages and costal cartilage. (A) The upset plot diagram showing overlapping proteins identified from antlers in six different development stages. (B) The top nine terms of GO enrichment result for 913 antler core proteins. (C) The Venn diagram showing overlapping between antler core proteins and costal cartilage proteins.
Core proteins for sika deer antler
The proteins detected in all samples can be considered as core proteins. In this experiment, 913 antler core proteins were detected accounting for 47.13% of the total identified proteins (Fig. 3A). To obtain a functional overview of these continuously expressed proteins, a gene ontology (GO) annotation enrichment analysis was performed. The core protein was mainly enriched in cell–cell adhesion, translation, Wnt signaling pathway and extracellular matrix organization (Fig. 3B, Table S3). Subsequently, the antler core proteins were compared with the costal cartilage proteins in order show the specificity of antler. A total of 132 antler core proteins was absent in the costal cartilage tissue (Fig. 3C). The network of predicted associations for 132 antler specific protein were constructed using STRING and Cytoscape (Fig. 4A). The top ten proteins ranked by degrees of the cytoHubba application in Cytoscape were recognized as hub proteins: CTNNB1, YBX1, ELAVL1, YARS, RPSA, EIF3B, HNRNPDL, UPF1, SF3B1, DDX5 (Fig. 4B).

The PPI network for antler-special proteins constructed by STRING and Cytoscape. (A) The whole network including 107 nodes and 289 edges. The disconnected proteins in the network did not be showed. (B) Sub-network of top ten hub proteins with their expanded network.
Analysis of stage-specific expression proteins
It is found that a total of 177 proteins were specifically expressed during the growth phase compared with the ossification phase (Fig. 3A). Among that, several proteins involved in chondrogenesis showed relatively higher LFQ intensity, such as collagen alpha-1(II) chain, hyaluronan and proteoglycan link protein 1, hyaluronan and proteoglycan link protein 3, bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase 1, bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase 2 (Table S1). The biological process enrichment results showed that these proteins were mainly involved in translational initiation, mRNA processing, Wnt signaling pathway, protein transport, osteoblast differentiation, endochondral ossification etc. Moreover, the cell component results showed that they were mainly localized in membrane, cytosol, nucleoplasm, endoplasmic reticulum etc. (Fig. 5A). 144 proteins were gone in the last time point of the ossification phase. Among that, aggrecan core protein is a major component of extracellular matrix of cartilagenous tissues and Collagen alpha-1(XI) chain may play an important role in fibrillogenesis by controlling lateral growth of collagen II fibrils. The enrichment results showed that these proteins were enriched into the biological process of translational initiation, chaperone-mediated protein folding and extracellular matrix organization (Fig. 5B). Although 100 days and 130 days were classified into ossification phase there were still significant differences. There were 33 proteins expressed specifically in 100 days. Among that, the LFQ intensity for keratin, type I cytoskeletal 19 is highest and it mainly is involved in the organization of myofibers. The second is Glutathione S-transferase A1, which is involved in the formation of glutathione conjugates of both prostaglandin A2 (PGA2) and prostaglandin J2 (PGJ2). Tartrate-resistant acid phosphatase type 5, a classical markers of bone resorption and osteoclast differentiation, is also concluded in the list of 100-day special protein. The enrichment results showed the proteins were mainly involved in negative regulation of superoxide anion generation and high-density lipoprotein particle remodeling and located in extracellular exosome (Fig. 5C). 53 proteins were screened as 130-day specifically expressed proteins. Among that, the immune response-related proteins were found with higher relative abundance, including azurocidin, neutrophil elastase, cathelicidin-7, myeloperoxidase and bactericidal permeability-increasing protein. Moreover, Matrix metalloproteinase-9, an important extracellular matrix degrading enzyme, is specifically expressed in 130-day antler. The enrichment results showed 130-day special proteins were mainly related with phagocytosis, response to yeast, collagen catabolic process, leukocyte migration and ossification and located in extracellular space, extracellular exosome and extracellular matrix (Fig. 5D).

The GO enrichment result for antler stages-special proteins. (A–D) respectively represent the results of specific expressed proteins for the first four stages, the first five stages, 100 days and 130 days.
Analysis for significant differential expression protein
A total of 875 differentially expressed proteins were screened with adjusted p-values < 0.05 using one-way ANOVA (Table S4). We divided 693 DEPs proteins into five clusters according to the trends in the expression changes in the six different developmental stages by GProX clustering analysis (Fig. 6). Proteins with similar expression patterns were grouped together. The proteins related to Cluster 1 showed a relatively stable expression before 65 days and were up-regulated after that. In this category, extracellular matrix organization, lysosome and glycosaminoglycan degradation was a focus of special attention (Table S5). Cluster 2 proteins were down-regulated at 130 days. The proteins were mainly composed of gene expression regulatory proteins such as translation initiation, protein folding, protein transport. Cluster 3 showed a peak expression at 100 days including regulation of endopeptidase activity, platelet degranulation, extracellular matrix organization, fatty acid degradation etc. Cluster 4 decreased at 100 days and then showed a dramatic increase at 130 days including platelet aggregation and regulation of actin cytoskeleton. Cluster 5 contained the most DEPs and had a dramatic decline at 100 days and 130 days mainly involving translation, gene expression, TCA cycle and spliceosome.

Global dynamics of proteins during antler development. The main function class was searched for GO (Biological process) and KEGG enrichment.
Discussion
Proteomics is a very useful method for studying biological development. In this study, we used label-free proteomics technology to identify the proteins of antler in the differential development stages and obtained a comprehensive proteomic data of sika deer antlers, containing 1937 proteins with high degree of credibility. Among these, 15 types of collagen were included, such as collagen I, II, III, IV, V, VI, VII, IX, X, XI, XII, XIV, XV, XVIII and XX. Collagen type II is a major matrix component of hyaline cartilage and collagen X is specifically expressed in hypertrophic zone in endochondral ossification. Besides, several bone proteins were also found in sika deer, such as osteopontin, bone sialoprotein (BSP), osteomodulin, SPARC. The data will provide valuable clues for understanding the molecular mechanisms of antler rapid growth and ossification.First, we analyzed the antler core proteins. Among that, 132 proteins were absent in costal cartilage and the to ten hub proteins were involved in gene expression regulation. CTNNB1 is a key downstream component of the classical Wnt signaling pathway, which plays a key role in promoting cartilage formation and bone formation during antler development9,16,17. Some studies have shown that CTNNB1 together with TCF proteins regulates osteoblast expression of osteoprotegerin, an important inhibitor of osteoclast differentiation18. The loss of CTNNB1 in osteoblasts leads to osteopenia and stabilization of CTNNB1 leads to high bone mass. The antler-special proteins indicated that antler had a unique developmental mechanism different from costal cartilage.
The hierarchical clustering results show that the antlers at 6 time points can be roughly divided into two stages, the growth phase (15, 25, 45 and 65 days) and the ossification phase (100 and 130 days). The antlers grow exponentially during the growth phase, but almost stagnates during the ossification phase19. Therefore, we screened the antler stage-specific proteins and significant differential expression protein. The results found that the expression pattern of these proteins has a distinct feature: in the growth phase, more protein expression is up-regulated. This unbalanced pattern has been revealed by previous quantitative proteomic analysis of cartilage formation20. Undifferentiated cells express mixed genes making the biological functions of these upregulated proteins more complicated compared with chondrogenic cells. Golan-Mashiach also reported that stem cells express promiscuous genes and have many differentiation options21. However, in the differentiated state, the expression of most genes decreases but expression of some specific genes increases. Therefore, the expression of cell protein may be related to its differentiation state.
In addition, collagen alpha-1(II) chain, collagen alpha-2(XI) chain and aggrecan, the main components of cartilage, were up-regulated in the growth phase of antler. Previous study showed that type II collagen was slightly expressed in young antlers (6 weeks) but absent in old antlers22. These proteins are required to maintain the normal physioligical function of cartilage. The deficiency will affect chondrocyte maturation, leading to the arrest of cartilage and long bone. Hyaluronan and proteoglycan link protein 1 (HAPLN1) with the highest relative abundance of special proteins for antler growth stages is also called cartilage-associated protein 1 (CRTL1). Hyaluronic acid is an important extracellular matrix in the process of cartilage formation. HAPLN1 stabilizes the aggregation of proteoglycan and hyaluronic acid by binding the hyaluronic acid chain in aggrecan. HAPLN1 may also be used as a growth factor to up-regulate the synthesis of aggrecan and type II collagen in cartilage23. Besides, two main enzymes were identified which are involved in the post-translational modification of chondrocyte ECM components, PAPSS1 and PAPSS2. In chondrocytes, the sulfation of proteoglycans is an important post-translational modification. In mammals, PAPSS plays an important role in the development of cartilage proteoglycan sulfation and adapts to the strong ECM synthesis during cartilage formation24. Therefore, it can be considered that chondrogenic differentiation may be the main characteristic of the rapid growth of antler.
The upregulated proteins during antler ossification were significantly enriched in lysosome, acidic membrane-bound organelle that contain proteins required for osteoclast bone resorptive function25. There is considerable evidence to suggest that Cathepsin B and cathepsin D are located in the lysosomes of osteoclasts26,27,28 and the inhibitors have marked inhibitory effect on bone resorption capacity29. Matrix metalloproteinases are a family of proteolytic enzymes that contribute to the degradation of the organic matrix of bones30. MMP9 is highly expressed in bone cells especially osteoclasts and regulates ECM degradation and bone remodeling. The MMP9 null mice showed improved connectivity density of the trabeculae31. The tartrate acid-resistant phosphatase (TRAP), which is commonly used as a specific marker of differentiated osteoclasts, is another lysosomal enzyme implicated in osteoclast resorptive function. It should be noted that the proteolytic enzymes generally require an acidic environment to be activated. Carbonic anhydrase II (CA II) is a zinc-containing metal enzyme that catalyzes the production of protons intracellularly from carbon dioxide32. These protons produced are transported by vacuolar H+-ATPases (V-ATPases) through the ruffled border cell membrane into the resorption lacuna33. ATP6V1A, a catalytic subunit of V-ATPases, is highly expressed in osteoclasts. Horng reported that knockdown of ATP6V1A can impairs acid secretion and ion balance in zebrafish34. The up-regulation of lysosomal-related proteins suggests that the bone remodeling activity of deer antler in the ossification stage is higher than that in the growth stage.
Bone growth and the immune system are believed to have a close interaction. Many immune factors affect the differentiation and bone resorption of osteoclasts by regulating the RANK/RANKL/OPG system such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IL-8. In this study, some immune-related molecules, such as Protein S100-A7 (S100A7), cathelicidin-7 (CATHL7), lactotransferrin (LTF), azurocidin (AZU1), neutrophil elastase (ELANE) and myeloperoxidase (MPO), were found to be up-regulated during ossification. These immune-related molecules play an important role in the regulation of osteoblast and osteoclast differentiation. S100A7 promotes osteoclast differentiation and activity by enhancing the effects of M-CSF and RANKL35,36,37. However, several other factors effectively promote the proliferation and differentiation of osteoblasts and bone formation while inhibiting bone resorption38,39,40,41,42,43,44,45. SHAO also found that immune-related factors are related to the process of intervertebral disc ossification when analyzing the differentially expressed genes in herniated discs with or without calcification46. Therefore, we speculate that the local immune system may contribute to the ossification of antler tip.
Although we applied proteomics approach to obtain more important information about the development of sika deer antler, our study still has some limitations. In this study, we only performed 3 LC–MS/MS replicates to monitor the performance of the equipment, which is necessary for label-free proteomics approach. Only one biological “replicate” was used per time point, which may produce inconclusive results47. To lower the influence of inter-individual variation, we pay more attention to the change of protein expression between the growth period (15, 25, 45, 65 days) and ossification period (100, 130 days), and not the change between two specific time points, i.e. 25 and 45 days. From some animal tissue developmental proteomics research, we have also obtained some information that the changes in protein expression at different development stage seem to be greater than the inter-individual variation48,49,50,51. Therefore, the results of this study are still meaningful for discovering the molecular mechanism of rapid growth and ossification of antler, although we still need some follow-up experiments to verify some of the results in this study. Another limitation is the choice of protein extraction protocol. In this study, we used SDT buffer to extract antler proteins, which was efficient to extract hydrophobic proteins. However, the extraction of protein from bone tissue usually requires a decalcification step with EDTA or HCl, which will help us extract more extracellular matrix proteins.
In summary, this study used label-free proteomics to analyze the protein expression profiles of sika deer antlers in different developmental stages. This will provide valuable information for studying the molecular mechanism of antler rapid growth and ossification.
Materials and methods
Samples selection and preparation
3 4-year-old male sika deer (Cervus nippon Temminck) with the same father from a local deer farm (Changchun, China) were selected. Antlers were harvested at 15, 25, 45, 65, 100 and 130 days after casting of the previous hard antlers, respectively. Before that, the deer needed to be anesthetized by 5% xylazine at 0.5 mg/kg body weight intramuscularly, which was performed in accordance with the ARRIVE guidelines (https://arriveguidelines.org). The antlers were washed with 75% ethanol to remove contaminants from the surface. The growing tips were removed and cut into small pieces (1 cm × 1 cm × 1 cm). These pieces were immediately ground into power in liquid nitrogen and stored at − 80 °C for protein extraction. All animal experimental protocols were approved and authorized by the Chinese Academy of Agricultural Sciences Animal Care and Use Committee.
Protein in-solution digestion
One gram of the raw powder was mixed with 5 mL SDT buffer (4% (w/v) SDS, 100 mM Tris/HCl pH 7.6, 0.1 M DTT). The sample was sonicated, then incubated in a periodic vortex ice bath for 2 h and centrifuged at 20,000 × g for 15 min at 4 °C to remove undissolved powder. The supernatant was collected and the protein solution was digested using Filter aided proteome preparation (FASP) method46. The peptides obtained by FASP were desalted with C18-SD Extraction Disk Cartridge. The lyophilized peptide was resuspended in 40 μL 0.1% formic acid solution.
LC–MS/MS analysis
5 µL of sample containing 1 µg peptide was loaded onto an EASY-nLC 1000 system (Thermo Science) equipped with trap column (2 cm × 100 μm, 5 μm C18) and analytical column (Thermo Science, 10 cm × 75 μm, 3 μm, C18). Each sample was conducted with three 3 LC–MS/MS runs. Buffer A was water with 0.1% formic acid; Buffer B was 84% acetonitrile with 0.1% formic acid. The flow rate was 300 nL/min. The mobile phase and gradient were as follows: the linear gradient of the B solution ranged from 0 to 45% in 0–100 min; at 100–108 min, the linear gradient of the B liquid ranged from 45 to 100%; at 108–120 min, solution B was maintained at 100%; at 120–130 min, solution B was maintained at 5%. The peptide fragments were analyzed using Q-Exactive mass spectrometer (Thermo Science). The mass spectrometer instrument was operated in positive mode with a 1.85 kV applied spray voltage and drying gas flow of 4 L/min at 325 °C was used. The scanning range was 300–1800 m/z. First-order mass spectrometry resolution was 70,000 at 200 m/z, AGC (Automatic gain control) target was 1e6, maximum IT was 50 ms and dynamic exclusion was 60.0 s. The m/z of polypeptide and polypeptide fragments were collected as follows: 20 fragments were collected after each full scan, MS2 activation type was HCD, isolation window was 2 m/z, MS2 resolution was 17,500 at 200 m/z, normalized collision energy was 30 eV and underfill was set for 0.1%.
Data analysis
The LC–MS/MS raw files were imported into MaxQuant software (version 1.5.3.17) for database retrieval. In this experiment, the protein database predicted using the combined methods of homology and de novo annotations with transcriptome and genome data of Cervus nippon (accession number GWHANOY00000000, https://bigd.big.ac.cn/gwh) were selected for retrieval and analysis. The database contains 21,449 protein sequences (Supplementary file 1). Relevant parameters settings are shown in Table 1. After the search was complete, the ‘proteingroups.txt’ output file was loaded in Perseus version 1.5.5.3. The proteins marked with reverse or potential contaminant were removed. The proteins identified with less than two unique peptides were also excluded from further analysis.
Bioinformatics analysis
Pearson correlation analysis were performed using the Perseus software. Upset plot and venn diagrams were drawn using TBtools (version 1.068). The network of protein–protein interaction was constructed by the online tool STRING (https://string-db.org/) with a filter condition (combined scores > 0.4) and the hub proteins were identified by degree using cytoHubba plugin in Cytoscape software (v3.8.0). The GO and KEGG pathway enrichment analyses were conducted based on DAVID Bioinformation Resources (version 6.8). The differentially expressed proteins for T15, T25, T45, T65, T100 and T130 were determined based on one-way ANOVA with adjusted p-values < 0.05. The GProX platform was used to cluster the differentially expression proteins with similar expression patterns. The number of clusters was set to 6 and a fixed regulation threshold (upper limit of 0.58 and lower limit of − 0.58) was used. The minimal membership for the plot was set as 0.5. Other parameters were set to default values.
Ethical approval
The deer used in this study were approved and authorized by the Chinese Academy of Agricultural Sciences Animal Care and Use Committee. All experimental procedures were carried out in accordance with the approved guidelines and regulations.
References
Landete-Castillejos, T. et al. Antlers—Evolution, development, structure, composition, and biomechanics of an outstanding type of bone. Bone 128, 115046 (2019).
Currey, J. D. Mechanical properties and adaptations of some less familiar bony tissues. J. Mech. Behav. Biomed. Mater. 3(5), 357–372 (2010).
Li, C. & Suttie, J. M. Electron microscopic studies of antlerogenic cells from five developmental stages during pedicle and early antler formation in red deer (Cervus elaphus). Anat. Rec. 252(4), 587–599 (1998).
Li, C. & Suttie, J. M. Deer antlerogenic periosteum: A piece of postnatally retained embryonic tissue?. Anat. Embryol. (Berl.) 204(5), 375–388 (2001).
Li, C., Suttie, J. M. & Clark, D. E. Histological examination of antler regeneration in red deer (Cervus elaphus). Anat. Rec. A Discov. Mol. Cell Evol. Biol. 282(2), 163–174 (2005).
Wang, D. et al. Deer antler stem cells are a novel type of cells that sustain full regeneration of a mammalian organ-deer antler. Cell Death Dis. 10(6), 443 (2019).
Price, J. S. et al. Deer antlers: A zoological curiosity or the key to understanding organ regeneration in mammals?. J. Anat. 207(5), 603–618 (2005).
Price, J. S. et al. Chondrogenesis in the regenerating antler tip in red deer: Expression of collagen types I, IIA, IIB, and X demonstrated by in situ nucleic acid hybridization and immunocytochemistry. Dev. Dyn. 205(3), 332–347 (1996).
Ba, H. et al. Transcriptomic analysis of different tissue layers in antler growth Center in Sika Deer (Cervus nippon). BMC Genom. 20(1), 173 (2019).
Yao, B. et al. Global analysis of tissue-differential gene expression patterns and functional regulation of rapid antler growth. Mammal Res. 64(2), 235–248 (2019).
Price, J. & Allen, S. Exploring the mechanisms regulating regeneration of deer antlers. Philos. Trans. R Soc. Lond. B Biol. Sci. 359(1445), 809–822 (2004).
Yao, B. et al. Transcriptomic characterization elucidates a signaling network that controls antler growth. Genome 61(12), 829–841 (2018).
Zhao, Y. et al. Comparative analysis of differentially expressed genes in Sika deer antler at different stages. Mol. Biol. Rep. 40(2), 1665–1676 (2013).
Casas-Vila, N. et al. The developmental proteome of Drosophila melanogaster. Genome Res. 27(7), 1273–1285 (2017).
Wang, J. et al. De novo transcriptome and proteome analysis of Dictyophora indusiata fruiting bodies provides insights into the changes during morphological development. Int. J. Biol. Macromol. 146, 875–886 (2020).
Zhang, H. L. et al. WNT4 acts downstream of BMP2 to mediate the regulation of ATRA signaling on RUNX1 expression: Implications for terminal differentiation of antler chondrocytes. J. Cell Physiol. 233(2), 1129–1145 (2018).
Mount, J. G. et al. Evidence that the canonical Wnt signalling pathway regulates deer antler regeneration. Dev. Dyn. 235(5), 1390–1399 (2006).
Glass, D. A. 2nd. et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8(5), 751–764 (2005).
Fennessy, P. F. et al. Antler Growth Patterns in Young Red Deer Stags (Springer, 1992).
Ji, Y. H. et al. Quantitative proteomics analysis of chondrogenic differentiation of C3H10T1/2 mesenchymal stem cells by iTRAQ labeling coupled with on-line two-dimensional LC/MS/MS. Mol. Cell Proteom. 9(3), 550–564 (2010).
Golan-Mashiach, M. et al. Design principle of gene expression used by human stem cells: implication for pluripotency. Faseb J. 19(1), 147–149 (2005).
Rucklidge, G. J. et al. Deer antler does not represent a typical endochondral growth system: Immunoidentification of collagen type X but little collagen type II in growing antler tissue. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 118(2), 303–308 (1997).
Urano, T. et al. Single-nucleotide polymorphism in the hyaluronan and proteoglycan link protein 1 (HAPLN1) gene is associated with spinal osteophyte formation and disc degeneration in Japanese women. Eur. Spine J. 20(4), 572–577 (2011).
Stelzer, C. et al. Expression profile of Papss2 (3′-phosphoadenosine 5′-phosphosulfate synthase 2) during cartilage formation and skeletal development in the mouse embryo. Dev. Dyn. 236(5), 1313–1318 (2007).
Lacombe, J., Karsenty, G. & Ferron, M. Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle 12(17), 2744–2752 (2013).
Gray, A. W., Davies, M. E. & Jeffcott, L. B. Localisation and activity of cathepsins K and B in equine osteoclasts. Res. Vet. Sci. 72(2), 95–103 (2002).
Goto, T. et al. Immunohistochemical localization of cathepsins B, D and L in the rat osteoclast. Histochemistry 99(5), 411–414 (1993).
Nakase, T. et al. Immunohistochemical detection of cathepsin D, K, and L in the process of endochondral ossification in the human. Histochem. Cell Biol. 114(1), 21–27 (2000).
McMichael, B. K., Wysolmerski, R. B. & Lee, B. S. Regulated proteolysis of nonmuscle myosin IIA stimulates osteoclast fusion. J. Biol. Chem. 284(18), 12266–12275 (2009).
Zhu, L. et al. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci. Transl. Med. 12(529), eaaw6143 (2020).
Nyman, J. S. et al. Differential effects between the loss of MMP-2 and MMP-9 on structural and tissue-level properties of bone. J. Bone Miner. Res. 26(6), 1252–1260 (2011).
Riihonen, R. et al. Membrane-bound carbonic anhydrases in osteoclasts. Bone 40(4), 1021–1031 (2007).
Duan, X. et al. V-ATPases and osteoclasts: Ambiguous future of V-ATPases inhibitors in osteoporosis. Theranostics 8(19), 5379–5399 (2018).
Horng, J. L. et al. Knockdown of V-ATPase subunit A (atp6v1a) impairs acid secretion and ion balance in zebrafish (Danio rerio). Am. J. Physiol. Regul. Integr. Comp. Physiol. 292(5), R2068–R2076 (2007).
Paruchuri, V. et al. S100A7-downregulation inhibits epidermal growth factor-induced signaling in breast cancer cells and blocks osteoclast formation. PLoS ONE 3(3), e1741 (2008).
Cubillos, S. & Norgauer, J. Low vitamin D-modulated calcium-regulating proteins in psoriasis vulgaris plaques: S100A7 overexpression depends on joint involvement. Int. J. Mol. Med. 38(4), 1083–1092 (2016).
Charoenpong, H. et al. Mechanical stress induced S100A7 expression in human dental pulp cells to augment osteoclast differentiation. Oral Dis. 25(3), 812–821 (2019).
Choi, Y. J. et al. Identification of azurocidin as a potential periodontitis biomarker by a proteomic analysis of gingival crevicular fluid. Proteome Sci. 9, 42 (2011).
Supanchart, C. et al. The antimicrobial peptide, LL-37, inhibits in vitro osteoclastogenesis. J. Dent. Res. 91(11), 1071–1077 (2012).
Zhang, Z. & Shively, J. E. Generation of novel bone forming cells (monoosteophils) from the cathelicidin-derived peptide LL-37 treated monocytes. PLoS ONE 5(11), e13985 (2010).
Horibe, K. et al. Roles of cathelicidin-related antimicrobial peptide in murine osteoclastogenesis. Immunology 140(3), 344–351 (2013).
Nakamichi, Y. et al. Roles of cathelicidins in inflammation and bone loss. Odontology 102(2), 137–146 (2014).
Yu, X. et al. LL-37 inhibits LPS-induced inflammation and stimulates the osteogenic differentiation of BMSCs via P2X7 receptor and MAPK signaling pathway. Exp. Cell Res. 372(2), 178–187 (2018).
Fan, F. et al. Lactoferrin preserves bone homeostasis by regulating the RANKL/RANK/OPG pathway of osteoimmunology. Food Funct. 9(5), 2653–2660 (2018).
Icriverzi, M. et al. Lactoferrin in bone tissue regeneration. Curr. Med. Chem. 27(6), 838–853 (2020).
Shao, J. et al. Sequencing and bioinformatics analysis of the differentially expressed genes in herniated discs with or without calcification. Int. J. Mol. Med. 39(1), 81–90 (2017).
du Sert, N. P. et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 18(7), e3000411 (2020).
Li, X. et al. A time-resolved multi-omic atlas of the developing mouse stomach. Nat. Commun. 9(1), 4910 (2018).
Sun, L. et al. Quantitative proteomics of Xenopus laevis embryos: Expression kinetics of nearly 4000 proteins during early development. Sci. Rep. 4, 4365 (2014).
Sun, L. et al. Proteomics of Xenopus development. Mol. Hum. Reprod. 22(3), 193–199 (2016).
Wang, X. et al. Proteomic analyses of sheep (Ovis aries) embryonic skeletal muscle. Sci. Rep. 10(1), 1750 (2020).
Acknowledgements
This study was supported by funds from the Agricultural Science and Technology Innovation Program of China (CAAS-ASTIP-201X-ISAPS) and the Special Economic Animals Sharing Platform in China (TZDW2018).
Author information
Authors and Affiliations
Contributions
R.Z. and X.X. conceived and designed the experiments. R.Z. performed the experiments and data analysis and prepared figures. R.Z. Y.L. and X.X. wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, R., Li, Y. & Xing, X. Comparative antler proteome of sika deer from different developmental stages. Sci Rep 11, 10484 (2021). https://doi.org/10.1038/s41598-021-89829-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89829-6
This article is cited by
-
De novo transcriptome assembly database for 100 tissues from each of seven species of domestic herbivore
Scientific Data (2024)
-
Comprehensive transcriptome analysis of sika deer antler using PacBio and Illumina sequencing
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
