
Abstract
Uveal melanoma is the most common primary intraocular malignancy. A vast majority of metastasizing tumors have mutations in the BAP1 gene. Here, we investigate the spatiotemporal timing of these mutations. The size of 177 uveal melanomas and 8.3 million individual tumor cells was measured. BAP1 sequencing results and BAP1 IHC were available and for 76 (43%) and 101 (57%) of these, respectively. Tumors with a BAP1 mutation had significantly larger volume (2109 vs. 1552 mm3, p = 0.025). Similarly, tumor cells with loss of BAP1 protein expression had significantly larger volume (2657 vs. 1593 μm3, p = 0.027). Using observations of the time elapsed between mitoses, the BAP1 mutation was calculated to occur when the primary tumor had a size of a few malignant cells to 6 mm3, 0.5 to 4.6 years after tumor initiation and at least 9 years before diagnosis. We conclude that BAP1 mutations occur early in the growth of uveal melanoma, well before the average tumor is diagnosed. Its timing coincides with the seeding of micrometastases.
Similar content being viewed by others

Introduction
Uveal melanoma (UM) represents 3–5% of all melanomas and arises from the uvea, a pigmented region of the eye composed of the iris, choroid, and ciliary body1. Most tumors are located in the choroid (90%)2. It is the most common primary intraocular malignancy in adults, affecting approximately 5 individuals per million per year worldwide2. More than one third of patients suffer from metastatic spread, after which the median survival is less than 1 year3,4. Gene expression profiles are of great value in stratifying patient prognosis. Further, mutations in the BRCA1 associated protein-1 gene (BAP1), a tumor suppressor located on chromosome 3p, is mutated in 47% of all UM and a vast majority of metastasizing UM5,6. This mutation leads to a dedifferentiated stem-like phenotype that correlates strongly with poor prognosis5,6.
BAP1 is one of many genes that undergo mutations to confer increased epithelial to mesenchymal transition of the tumor. In this mutation sequence, the BAP1 mutation has been assumed to occur relatively late in tumor progression, preceded by oncogenic mutations in G-protein subunits including GNA11 and GNAQ that are present in as high as 83–96% of UM7. It is thought that these subunit mutations are not sufficient for progression to metastatic disease. BAP1 mutations are thought to appear after the GNA11 or GNAQ mutations, correlating with a gratly increased risk for tumor progression5.
It is well-documented that larger UMs have a higher likelihood of harboring BAP1 mutations, supporting prognostic implications8,9. However, there is also evidence that smaller UMs may harbor BAP1 mutations and seed metastases10. The smallest UM of metastatic potential reported in the literature is 3.0 mm in diameter and 1.0 mm in thickness, corresponding to a volume of 4.7 mm3 in spherical cell models11. A study showed that of 61 choroidal melanomas with thickness ≤ 3 mm, 27% had monosomy 312,13. Another study found 6 of 59 choroidal melanomas with thickness ≤ 3 mm and diameter ≤ 9 mm developed metastases after treatment, highlighting the metastatic potential of small UMs14. Additionally, there are also mathematical models that show seeding of liver micrometastases when the primary UM is less than 10.0 mm3 in size12,13. These findings support the thought that BAP1 mutations can be present early in tumorigenesis and in small UMs.
Given the relationship between loss of BAP1 expression and poor prognosis, it is important to clarify the impact of tumor size and age on the likelihood of a BAP1 mutation. To the best of our knowledge, no study has investigated these spatial and temporal characteristics in clinical samples. We therefore use mathematical modeling and previously published calculations of the tumor size at which the seeding of micrometastases start as a reference and comparison for our own model. Thus, we aim to make estimations of the origin and dynamic evolution of the BAP1 mutated tumor cell clone in UM.
Results
Descriptive statistics
A total of 177 enucleated eyes with choroidal or ciliary body melanoma were included. BAP1 sequencing results and BAP1 IHC were available and for 76 (43%) and 101 (57%) of these, respectively. Forty of the tumors with BAP1 IHC available were used as a validation cohort. The mean patient age at enucleation was 61 years (SD 15). One hundred and twenty-three patients (69%) had AJCC stage IIb or IIIa disease. The mean tumor volume was 913 mm3 (SD 865) and the mean number of cells measured in terms of size and BAP1 expression in each tumor was 208 403 (SD 165 587). Eighty-six patients were deceased before the end of follow-up. Median follow-up for the 91 survivors was 46 months (SD 78, Table 1).
Of 76 sequenced tumors, 26 (34%) had a BAP1 mutation. Tumors with a BAP1 mutation had significantly larger mean volume than those with wild-type BAP1 (2109 vs. 1552 mm3, p = 0.025).
Similarly, tumor cells with loss of BAP1 expression had significantly larger mean volume (2657 μm3, SD 1283) than those with retained expression (1593 μm3, SD 602, p = 0.027, Fig. 1). The mean volume of all tumor cells regardless of BAP1 expression was 2105 μm3 (SD 936).

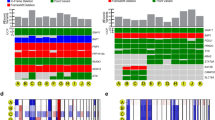
Results of whole exome sequencing and digital image analysis of BAP1 immunohistochemistry. (A) Distribution of BAP1 mutations and copy number variations (CNV) across the 76 included patients from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/projects/TCGA-UVM). Twenty-two patients had a BAP1 mutation, 1 patient had a BAP1 CNV gain, and 1 patient had a BAP1 CNV loss. (B) Distribution of the most frequently mutated genes as proportion of the cohort. (C) BAP1 immunohistochemistry. In this example, most tumor nuclei have retained their BAP1 expression (lilac) whereas some have lost their expression (blue). (D) Excerpt from digital image analysis of the same tumor section shown in B. Tumor cells with retained and lost BAP1 expression are indicated in red and blue, respectively. (E) Tumors with a BAP1 mutation had significantly larger mean volume than those with wild-type BAP1 (2109 vs. 1552 mm3, p = 0.025). (F) tumor cells with loss of BAP1 expression had significantly larger mean volume (2657 μm3, SD 1283) than those with retained expression (1593 μm3, SD 602, p = 0.027). Freq, Frequency. CNV, Copy number variation. * < 0.05.
Cell doubling time
The mean number of tumor cells per tumor was 433 941 547 (SD 410 738 729), calculated by dividing each tumor’s volume with the mean tumor cell volume of 2105 μm3. The mean number of tumor cells with loss of BAP1 expression was 251 520 892 (SD 368 488 894). The mean number of cell doublings required to reach the number of tumor cells and number of tumor cells with BAP1 loss was 27.9 (SD 1.7, smallest tumor 24.2, largest tumor 30.7) and 25.8 (SD 3.2, smallest tumor 19.5, largest tumor 30.4), respectively. The difference of 2.1 tumor doublings indicate that the mutation occurred when the lesion consisted of only 4 malignant cells (n = 22.1).
Median doubling times of 128 to 511 days in primary UM have been reported15,16. If each doubling took 128 days, the tumors in the present cohort consisted of 1 cell an average of 3571 days or 9.8 years before diagnosis (SD 0.6, min 8.5, max 10.8). Using the same doubling time for the cell population with loss of BAP1 expression, the first tumor cell with lost BAP1 expression appeared an average of 3302 days or 9.0 years before diagnosis (SD 1.1, min 6.8, max 10.7).
If the doubling time was 511 days instead, the first malignant cell appeared 39.1 years before diagnosis (SD 2.4, min 33.9, max 43.0), with the BAP1 mutation occurring 3 years later at 36.1 years before diagnosis (SD 4.5, min 27.3, max 42.6).
Curve fitting
We tested curves based on linear, logarithmic, inverse, quadratic, cubic, compound, power, S-shaped, logistic, growth and exponential functions. Goodness of fit to our data was tested in model summaries and ANOVA tables. In the first step, inverse, compound, power, power, S-shaped, growth, exponential and logistic functions were excluded as they could not be fitted to our data. All of the remaining functions (linear, logarithmic, quadratic and cubic) could be fitted (F-scores 3.2 to 9.8, R2 0.10 to 0.15, p = 0.03 to 0.003, Table 2).
Next, we used these functions to estimate the tumor volume at which the proportion of BAP1 mutant cells is zero by setting the y value (proportion of tumor cells with loss of BAP1 expression) to 0. We thereby arrived at 0 to 6 mm3, depending on which of the functions were used for calculation. The latter corresponds to a lesion with a diameter of 5 mm and a thickness of 1.5 mm.
Validation
We tested the linear, logarithmic, quadratic and cubic functions’ ability to solely based on tumor volume predict manual assessments of low versus high BAP1 expression (low < 33% BAP1 positive cells, high ≥ 33%) in a model validation cohort of 40 tumors. The included tumors had been independently graded by two ophthalmic pathologists’ ( H.E.G. and G.S.). True positive was defined as: Predicted to be positive (BAP1 ≥ 33%) by function and positive as graded by pathologist. True negative was defined as: Predicted to be negative (BAP1 < 33%) by function and negative as graded by pathologist.
The linear and quadratic functions predicted the classification of 65% of the cases, with a sensitivity and specificity of 31% and 88%, respectively and a Cohen’s Kappa of 0.21, indicating fair agreement.
The logarithmic function predicted the classification of 60% of the cases, with a sensitivity and specificity of 40% and 96%, respectively and a Cohen’s Kappa of 0.02, indicating slight agreement.
The cubic function predicted the classification of 65% of the cases, with a sensitivity and specificity of 69% and 88%, respectively and a Cohen’s Kappa of 0.57, indicating moderate agreement.
Implications of doubling time and cell size in a large external cohort
We applied our measurements of tumor cells with lost and retained BAP1 protein expression to the mean tumor from a previously published cohort of 8 033 consecutive patients4. In this cohort, the mean tumor diameter and thickness at diagnosis of UM was 11.1 and 5.5 mm, respectively. Using the formula for calculation of tumor and cell volume described by Char et al., a UM with these measurements would have a volume of 348 mm312,16. In a tumor with this volume, there would be approximately 165 320 665 tumor cells, if our mean tumor cell volume of 2105 μm3 μm3 was used.
As shown above, the volume of BAP1 mutant cells can be described as a function of tumor volume. According to these functions, a tumor with a volume of 348 mm3 would have loss of BAP1 expression in 35 to 41% of its cells, or about 57 862 233 to 67 781 473 cells.
To reach 165 320 665 tumor cells, 27.3 cell doublings would be required. To reach the estimated number of cells with loss of BAP1 expression, 25.8 to 26.0 doublings would be required.
Consequently, the BAP1 mutation occurred after 1.3 to 1.5 cell doublings, when the malignant clone consisted of only 3 malignant cells.
If each doubling took 128 days, a tumor with a volume of 348 mm3 consisted of 1 cell 3494 days, or approximately 9.5 years before diagnosis. Using the same doubling time for the cell population with loss of BAP1 expression, the first tumor cell with lost BAP1 expression appeared 3302 to 3328 days before diagnosis, when the tumor was 166 to 192 days or about 0.5 years old. If the doubling time was instead 511 days16, the tumor initiated 13 951 days or approximately 38.2 years before diagnosis and the first tumor cell with lost BAP1 expression appeared to 12 286 to 13 184 days or approximately 36.1 to 36.4 years before diagnosis. Consequently, with a doubling time of 511 days, the first tumor cell with lost BAP1 expression appeared when the tumor was 767 to 1665 days or 2.1 to 4.6 years old.
In summary, a tumor with a diameter of 11.1 mm and a thickness of 5.5 mm, which represents the average UM size at diagnosis, initiated 9.5 to 38.2 years before diagnosis, and its first cell with lost BAP1 expression appeared when the tumor was 0.5 to 4.6 years old, depending on the estimate of the proportion of tumor cells with loss of BAP1 expression in the average tumor and the mean time elapsed between mitoses.
Survival
Median patient metastasis-free survival after diagnosis of tumors with a BAP1 mutation was 2.4 years (standard error, SE 0.2, 95% confidence interval, CI 2.0 to 2.8), versus 16.0 years for tumors without a BAP1 mutation (SE 7.6, 95% CI 1.2 to 30.9, Log-Rank p < 0.0001, Fig. 2A). For tumors with IHC stains, we used retained BAP1 expression in 33% of tumor cells as a cutoff, in accordance with previous publications17,18,19. Of 177 patients, 79 had metastases before the end of follow-up. Of these 79 metastases, 79 (100%) occurred within 9 years from diagnosis.

Cumulative metastasis-free survival proportions. (A) Survival after diagnosis for patients with a BAP1 mutation or low immunohistochemical BAP1 expression (red) versus no BAP1 mutation or high immunohistochemical BAP1 expression (yellow, Log-Rank p < 0.0001). (B) Survival after tumor initiation, which was estimated to occur 22.3 years before diagnosis (SD 1.4). (C) Survival after BAP1 mutation, which was estimated to occur 20.6 years before diagnosis (SD 2.5). Green areas represent 95% confidence intervals.
Lastly, we examined patient survival with the concepts metastasis-free survival after tumor initiation and metastasis-free survival after BAP1 mutation. In contrast to using the day of diagnosis as starting point for survival analysis, we added the estimated time that had elapsed from tumor initiation and BAP1 mutation before diagnosis. We assumed a cell doubling time of 292 days, which is the median of previous observations16.
Metastasis-free survival after tumor initiation was defined as the proportion of patients not having suffered symptomatic and/or radiologically detectable metastases at a specific time after the estimated appearance of the first tumor cell. With a doubling time of 292 days, the mean duration from tumor initiation to diagnosis was 22.3 years (SD 1.4). Median Kaplan–Meier metastasis-free survival after tumor initiation was 32.7 years (SE 3.2, 95% CI 26.4 to 39.1, Fig. 2B).
Metastasis-free survival after BAP1 mutation was defined as the proportion of patients not having suffered symptomatic and/or radiologically detectable metastases at a specific time after the estimated appearance of the first tumor cell with loss of BAP1 expression. Two patients with BAP1 expression in 100% of their tumor cells were excluded from analysis, leaving 59 eligible patients. With a doubling time of 292 days, the mean duration from BAP1 mutation to diagnosis was 20.6 years (SD 2.5). Median Kaplan–Meier metastasis-free survival after BAP1 mutation was 31.0 years (SE 3.9, 95% CI 23.3 to 38.7, Fig. 2C).
Discussion
This study provides an estimate of the timing of the BAP1 mutation in the growth of UM. Adding to previous publications on the metastatic process20 and to doubling times of the primary tumor and metastases, we propose a model for the growth and dissemination of UM (Fig. 3)12,13,15,16. Our calculations showed that the first BAP1 mutant clone of an average UM appears when the tumor is 166 days to 1665 days old, within 2 cell doublings of the first UM clone. The timing of micrometastatic seeding has been estimated as 4.2 years after tumor initiation, which aligns with the calculated timing of the BAP1 mutation21. Thus, we provide a mathematical explanation of the occurrence of micrometastases during the tumor’s infancy and underscore the importance of BAP1 mutation in tumorigenesis.

A model for the growth and dissemination of uveal melanoma. (A) In a tumor’s infancy, mutations in G-protein subunits including GNA11 or GNAQ are accumulated. (B) In about half of uveal melanomas, subsequent mutations in BAP1 occur after 0.5 to 5 years, at a tumor volume of < 10 mm3. At this point, micrometastases are seeded to the liver. (C) As the primary tumor keeps growing, the BAP1 mutated clone has a survival advantage over BAP1 wild type tumor cells, gradually replacing the latter. The average primary tumor is diagnosed at a volume of 350 mm3, 9.5 to 38 years after tumor initiation. Micrometastases may remain dormant and undetectable for years. (D) At a later stage, the primary tumor is composed of BAP1 mutants only, with few exceptions. Eventually, liver micrometastases leave dormancy and start growing to radiologically detectable macrometastases. The entire process from primary tumor initiation to death from metastatic disease takes one to five decades. The minimum estimate is 9.5 years between tumor initiation and diagnosis, and 0 years from diagnosis to metastatic death for an overall course of 9.5 years. The maximum estimate is 38 years between tumor initiation and diagnosis, and 15 years from diagnosis to metastatic death for an overall course of 53 years. Later metastases are rare3,22. Note: The presence of micrometastases does not necessarily imply a worse prognosis. Subsequent events may be necessary for micrometastases to leave dormancy and start proliferating. Importantly, more than half of patients do not develop macrometastases at all3,4,6,22,23.
It is important to note that our model applies to melanoma cells, which are malignant by definition. UM can occur de novo or through malignant transformation of a nevus. Studies have shown that choroidal nevi, similar to UM, can harbor G-protein coupled receptor (GPCR) mutations in GNA11, GNAQ, and CYSLTR224,25. Our computational analyses assume that the initial cell is a malignant cell capable of losing the BAP1 gene, whether it formed de novo or growing in a nevus. To our knowledge, there are no BAP1-deficient choroidal nevi reported in the literature, so our model was able to focus solely on BAP1 mutagenesis in the evolution of UM.
The early onset of the BAP1 mutation is likely promoted by evolutionary pressure. Hypoxia and immune evasion may be two contributors. As the tumor grows, it will be at higher risk for hypoxia due to lower availability of nutrients. Studies of primary fibroblasts from individuals heterozygous for the germline BAP1 mutation showed that these cells have increased anaerobic glycolysis and decreased mitochondrial respiration compared to fibroblasts from wild-type BAP1 individuals26,27. This finding suggests that the BAP1 mutated clone can be favored under hypoxic conditions and gradually make up a larger proportion of the tumor. Furthermore, if BAP1 mutated cells are more stem-like, they also have a higher capability of immune evasion, thereby outliving the wild types. It has been documented that BAP1 mutations can downregulate the expression of genes responsible for immune checkpoints and increase inflammation in the tumor microenvironment28. A study has shown that a liver metastasis that arose 29 years after treatment of a class 1B UM harbored clonally expanded plasma cells, suggesting the role of antibody-mediated immunity in slow tumor growth and immune evasion28. More recently, another study has shown the effect of aneuploidy, commonly seen with BAP1 loss as monosomy 3, in immune suppression through activation of NF-kB and other pro-inflammatory signaling pathways29. Thus, both the hypoxic and stem-like properties would give BAP1 mutants an evolutionary advantage over wild type cells.
Our study found significant heterogeneity in BAP1 expression within tumors, which requires exploration in the context of UM tumorigenesis. BAP1 mutations tend to precede metastatic dissemination and they occur shortly after GPCR mutations in the UM evolutionary tree28. However, instances of monosomy 3 occurring after several UM mutations and cases in which metastases precede BAP1 loss have been reported30,31,32. A recent study identified 2 cases where primary UM homozygous for the BAP1 mutation gave rise to BAP1 heterozygous metastases30. However, metastases in both cases were negative for the BAP1 protein. This could be explained by BAP1 epigenetic silencing or the escape of the mutation from detection, as the authors suggest. That study utilized targeted resequencing with 423-fold coverage and without methylomic analysis, so it may have not detected smaller mutations in metastases. However, if this is replicated in additional studies, it would suggest that BAP1 loss can be clonal, supporting the observation of intratumor BAP1 heterogeneity in our study. Another investigation used digital PCR analysis to show that the order monosomy 3 shows heterogeneity with respect to other mutagenic events like gain in chromosome 8q31. Most recently, we found that there is significant intratumor heterogeneity of BAP1 expression, but that this has limited prognostic importance33. Robust applications of genomic analysis, such as digital PCR offering over 5000-fold coverage, are required to continue investigating the dynamic mutagenesis of BAP1 and tumor heterogeneity in UM.
It is important to note the role of tumor architecture and vascular framework, apart from the BAP1 mutation, in the promotion of metastasis in UM. Our study suggests that a BAP1 mutation occurs early in tumorigenesis, possibly when the tumor consists of a few malignant cells. However, this does not imply immediate initiation of metastases. Once a cell loses BAP1 expression, even after the GPCR mutation that drives proliferation, it needs to extravasate into vascular channels of the choroid. The necessary tumor architecture at this stage needs to be established and consists of small vascular channels in the primary tumor. It has been shown that tumors with high-grade vascularity patterns are significantly more likely to metastasize than tumors that do not, with mean vascular density positively correlating with the number of metastases, even in small UMs34. In another study, we showed that low BAP1 expression correlates with areas with areas of vasculogenic mimicry, consisting of de novo formations of tubules without endothelial lining that connect the UM to mature vasculature35. Thus, the requirements of the tumor microenvironment could explain the delay between the origination of the BAP1 mutant clone, the BAP1-GPCR double mutant clone, and micrometastatic spread.
The strengths of our study include its relatively large patient sample and use of our model in several different cohorts. By formulating this mathematical model, this study provides an early snapshot of BAP1 mutagenesis and suggests micrometastases can occur as early as after the first few mitoses of primary UM. One limitation of this study is its theoretical nature. We assumed cells were ellipsoid in shape, which is a frequently used assumption but not necessarily accurate for each individual tumor cell. Our model solely incorporated the BAP1 mutation. Several other mutations, such as EIF1AX and SF3B1, less often promote UM metastasis. Thus, a combined model may have increased the accuracy of our results. Since UM doubling times were calculated under separate postulations in the literature, we were only able to present our estimates in intervals and assume these values were constant. This is likely not the case, as we found BAP1 expression is a function of tumor volume. Our model did not take dynamic changes in the tumor environment during growth into account, including other mutations, chromosomal status, dormancy intervals and changes in tumor doubling time. Furthermore, from the observation that larger tumors harbor a greater proportion of BAP1 mutant cells follows that BAP1 mutated clone should have either a faster growth rate or a slower death rate. This assumption has not been used to modify calculations in the current study, and future studies may clarify this assumption based on observations rather than estimations. Some authors may question our findings that UMs contains a ratio of BAP1 mutant-to-wild type cells, arguing that UM is more often composed exclusively of mutants or wild-types. In our study, we have alternately used loss of immunohistochemical expression of BAP1 in tumor cell nuclei and BAP1 mutations detected by DNA sequencing as a marker for the same genetic event, using methods replicated in our previous studies17,18,19. It is also important to note that the presence of micrometastases is common regardless of BAP1 status23,36. Events that trigger micrometastases to leave dormancy and start proliferating in some patients may be even more important in terms of survival, considering that more than half of patients with UM do not develop metastases at all.
In conclusion, the BAP1 mutation occurs early in the growth of UM, well before the average tumor is diagnosed and the timing coincides with previous calculations of the tumor size at which seeding of micrometastases start. At the time of primary tumor diagnosis, the primary tumor, the BAP1 mutation and the liver micrometastases are one to several decades old. Considering an average patient age of about 60 years at diagnosis, these crucial tumor events occurred at an age of 20 to 50 years. Further studies of BAP1 mutagenesis and on factors that promote micrometastasis dormancy and reduce the risk for a switch to proliferation could improve the prospects for development of effective therapies and improved survival.
Methods
Patients and samples
The protocol for data collection and analysis of specimens from the L.F. Montgomery Ophthalmic Pathology Laboratory of Emory Eye Center, Atlanta, USA, was approved by the Emory University Institutional Review Board (reference 00,028,367) and from the Ophthalmic Pathology Laboratory of St. Erik Eye Hospital in Stockholm, Sweden by the Swedish Ethical Review Authority (reference 2020–02,835). The study adhered to the US Health Insurance Portability and Accountability Act and to the tenets of the Declaration of Helsinki. No protected health information was collected or handled in this project. All used tumor and patient data had been generated in previously published projects and was anonymized at the point of our analysis in this project.
The 177 included patients were collected from 3 different cohorts:
The first cohort (n = 61) consists of paraffin embedded and formalin-fixed eyes that were enucleated at St. Erik Eye Hospital and Emory Eye Center between years 1975 and 2017. They were included based on a set of inclusion criteria [histologically proven melanoma in choroid and/or ciliary body, patient deceased (requirement for waiver of informed consent by the Swedish Ethical Review Authority), availability of gene expression classification, sufficient formalin-fixed paraffin-embedded (FFPE) tissue for immunohistochemical staining and proper representation of tumor histopathology, availability of clinicopathological data including primary tumor largest basal diameter (LBD) and tumor thickness] and exclusion criteria (extensive tumor necrosis, hemorrhage or inflammation, abundant tumor pigmentation affecting visual examination of immunohistochemical protein expression and suboptimal staining results as determined by positive and negative internal and external controls). This cohort was published in 2019 and 202010,18,19,37.
The second cohort (n = 76) was collected from The Cancer Genome Atlas (TCGA), the National Cancer Institute, National Institutes of Health, USA. In supplemental information to the publication by Robertson et al. anonymized patient and tumor data including results from whole exome sequencing have been made available38. This data was downloaded to calculate the difference in tumor volume between tumors with and without a BAP1 mutation. The cohort originally consists of 80 patients, but 4 were excluded since they did not have complete tumor size data. This cohort was published in 201738.
The third cohort (n = 40) was collected from the Emory Eye Center for validation of findings. The included tumors had been independently graded for the level of BAP1 expression by two authors (E.S. and H.E.G.). This cohort was published in 201817.
Immunohistochemical staining and digital image analysis
Formalin fixed and paraffin-embedded (FFPE) tumor sections had been stained with monoclonal antibodies against BAP1 with a red chromogen (1:40, Santa Cruz Biotechnology, Dallas, Texas, USA). The Leica Bond-III automated system (Leica, Wetzlar, Germany) and digitally scanned to the .ndpi file format at × 400, using the Nano Zoomer 2.0 HT scan (Hamamatsu Photonics K.K., Hamamatsu, Japan). The QuPath Bioimage analysis software (v0.2.3) run on an Apple computer (Apple Inc., Cupertino, CA), had been used for digital image analysis39. Each full tumor section was then analyzed for the number of tumor cells, number of cells with loss of BAP1 expression and retained expression, respectively, and size measurements for each individual cell (maximum and minimum caliper). Cell measurements were obtained from the 61 tumors in the first cohort and were validated in the second and third cohorts. A workflow for morphometric analysis was established using the QuPath software. The staining, scanning, digital image analysis, and cell measurement steps used for the included cohorts have been described previously10,18,19,33.
Estimation of tumor and cell volume
The volume of tumors was estimated assuming a semi ellipsoid shape12,16:
where t is the tumor thickness and lbd is the largest basal diameter.
In calculations of the volume of tumor cells we assumed a prolate ellipsoid shape:
where a and b are the long and short calipers of the cell, respectively.
The number of cell doublings required to reach the number of cells in a tumor (x) was calculated as:
Model computation and statistical analysis
Differences with a p < 0.05 were considered significant, all p values being two-sided. Continuous variables including cell and tumor sizes did not deviate significantly from normal distribution, when evaluated by the Shapiro–Wilk test (p = 0.64). We therefore used the Student’s T-test for comparison of these variables. The ANOVA F-test and coefficient of determination (R2) was used to optimize the computational model and goodness of fit of S, compound, logistic, growth, exponential, linear, logarithmic, inverse, quadratic and cubic curves of the proportion of BAP1 mutated cells as a function of tumor size. Under the assumption that the cells are ellipsoid in shape, the volume formula for a rotating ellipsoid was used to calculate cell volumes. We used binary logistic regression without x-centering of the tumor volume variable to predict the probability of a BAP1 mutation as a function of the tumor volume. X-centering was defined as inclusion of the constant in the regression model. The range of doubling times of a given tumor was assumed to be 154–511 days12,13. Follow-up was defined as the time in months from uveal melanoma diagnosis to the last occasion metastasis-free patients were known to be alive. All statistical analyses were performed using IBM statistics version 26 (Armonk, NY, USA).
Change history
17 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-96113-0
References
Aronow, M. E., Topham, A. K. & Singh, A. D. Uveal melanoma: 5-year update on incidence, treatment, and survival (SEER 1973–2013). Ocul. Oncol. Pathol. 4, 145–151. https://doi.org/10.1159/000480640 (2018).
Kaliki, S. & Shields, C. L. Uveal melanoma: relatively rare but deadly cancer. Eye (London, England) 31, 241. https://doi.org/10.1038/eye.2016.275 (2017).
Stalhammar, G. Forty-year prognosis after plaque brachytherapy of uveal melanoma. Sci. Rep. 10, 11297. https://doi.org/10.1038/s41598-020-68232-7 (2020).
Shields, C. L. et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch. Ophthalmol. 127, 989–998. https://doi.org/10.1001/archophthalmol.2009.208 (2009).
Harbour, J. W. et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–1413. https://doi.org/10.1126/science.1194472 (2010).
Karlsson, J. et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 11, 1894. https://doi.org/10.1038/s41467-020-15606-0 (2020).
Decatur, C. L. et al. Driver mutations in uveal melanoma: associations with gene expression profile and patient outcomes. JAMA Ophthalmol. 134, 728–733. https://doi.org/10.1001/jamaophthalmol.2016.0903 (2016).
Gupta, M. P. et al. Clinical characteristics of uveal melanoma in patients with germline BAP1 mutations. JAMA Ophthalmol. 133, 881–887. https://doi.org/10.1001/jamaophthalmol.2015.1119 (2015).
Masoomian, B., Shields, J. A. & Shields, C. L. Overview of BAP1 cancer predisposition syndrome and the relationship to uveal melanoma. J. Curr. Ophthalmol. 30, 102–109. https://doi.org/10.1016/j.joco.2018.02.005 (2018).
Herrspiegel, C. et al. Nuclear expression of BAP-1 in transvitreal incisional biopsies and subsequent enucleation of eyes with posterior choroidal melanoma. Br. J. Ophthalmol. https://doi.org/10.1136/bjophthalmol-2020-316498 (2020).
Jouhi, S. et al. The Small Fatal Choroidal Melanoma Study. A survey by the European Ophthalmic Oncology Group. Am. J. Ophthalmol. 202, 100–108. https://doi.org/10.1016/j.ajo.2019.01.031 (2019).
Singh, A. D. Uveal melanoma: implications of tumor doubling time. Ophthalmology 108, 829–830. https://doi.org/10.1016/S0161-6420(00)00607-2 (2001).
Eskelin, S., Pyrhönen, S., Summanen, P., Hahka-Kemppinen, M. & Kivelä, T. Tumor doubling times in metastatic malignant melanoma of the uvea: tumor progression before and after treatment. Ophthalmology 107, 1443–1449. https://doi.org/10.1016/s0161-6420(00)00182-2 (2000).
Malcles, A. et al. Small metastasizing choroidal melanomas. Acta Ophthalmol. 93, e160-166. https://doi.org/10.1111/aos.12523 (2015).
Char, D. H., Kroll, S. & Phillips, T. L. Uveal melanoma. Growth rate and prognosis. Arch. Ophthalmol. 115, 1014–1018. https://doi.org/10.1001/archopht.1997.01100160184007 (1997).
Augsburger, J. J., Gonder, J. R., Amsel, J., Shields, J. A. & Donoso, L. A. Growth rates and doubling times of posterior uveal melanomas. Ophthalmology 91, 1709–1715. https://doi.org/10.1016/s0161-6420(84)34088-x (1984).
Szalai, E., Wells, J. R., Ward, L. & Grossniklaus, H. E. Uveal melanoma nuclear BRCA1-associated protein-1 immunoreactivity is an indicator of metastasis. Ophthalmology 125, 203–209. https://doi.org/10.1016/j.ophtha.2017.07.018 (2018).
Stålhammar, G., See, T. R. O., Phillips, S., Seregard, S. & Grossniklaus, H. E. Digital image analysis of BAP-1 accurately predicts uveal melanoma metastasis. Transl. Vis. Sci. Technol. 8, 11. https://doi.org/10.1167/tvst.8.3.11 (2019).
See, T. R. O., Stålhammar, G., Phillips, S. S. & Grossniklaus, H. E. BAP1 immunoreactivity correlates with gene expression class in uveal melanoma. Ocul. Oncol. Pathol. https://doi.org/10.1159/000502550 (2019).
Grossniklaus, H. E. Understanding uveal melanoma metastasis to the liver: the Zimmerman effect and the Zimmerman hypothesis. Ophthalmology 126, 483–487. https://doi.org/10.1016/j.ophtha.2018.09.031 (2019).
Bononi, A. et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 24, 1694–1704. https://doi.org/10.1038/cdd.2017.95 (2017).
Kujala, E., Mäkitie, T. & Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Invest. Ophthalmol. Vis. Sci. 44, 4651–4659 (2003).
Eide, N. et al. The results of stricter inclusion criteria in an immunomagnetic detection study of micrometastatic cells in bone marrow of uveal melanoma patients—relevance for dormancy. Pathol. Oncol. Res. 25, 255–262. https://doi.org/10.1007/s12253-017-0355-7 (2019).
Vader, M. J. C. et al. GNAQ and GNA11 mutations and downstream YAP activation in choroidal nevi. Br. J. Cancer 117, 884–887 (2017).
Nell, R. J. et al. Involvement of mutant and wild-type CYSLTR2 in the development and progression of uveal nevi and melanoma. BMC Cancer 21, 164 (2021).
Carbone, M. et al. BAP1 and cancer. Nat. Rev. Cancer 13, 153–159. https://doi.org/10.1038/nrc3459 (2013).
Wang, T. et al. An empirical approach leveraging tumorgrafts to dissect the tumor microenvironment in renal cell carcinoma identifies missing link to prognostic inflammatory factors. Cancer Discov. 8, 1142–1155. https://doi.org/10.1158/2159-8290.CD-17-1246 (2018).
Durante, M. A. et al. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 11, 496. https://doi.org/10.1038/s41467-019-14256-1 (2020).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. https://doi.org/10.1038/nature25432 (2018).
Shain, A. H. et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 51, 1123–1130 (2019).
de Lange, M. J. et al. Heterogeneity revealed by integrated genomic analysis uncovers a molecular switch in malignant uveal melanoma. Oncotarget 6, 37824–37835 (2015).
Singh, N., Singh, A. D. & Hide, W. Inferring an evolutionary tree of uveal melanoma from genomic copy number aberrations. Invest. Ophthalmol. Vis. Sci. 56, 6801–6809 (2015).
Stålhammar, G. & Grossniklaus, H. E. Intratumor heterogeneity in uveal melanoma BAP-1 expression. Cancers 13, 1143 (2021).
Grossniklaus, H. E. Tumor vascularity and hematogenous metastasis in experimental murine intraocular melanoma. Trans. Am. Ophthalmol. Soc. 96, 721–752 (1998).
Stålhammar, G., See, T. R. O., Phillips, S. S. & Grossniklaus, H. E. Density of PAS positive patterns in uveal melanoma: Correlation with vasculogenic mimicry, gene expression class, BAP-1 expression, macrophage infiltration, and risk for metastasis. Mol. Vis. 25, 502–516 (2019).
Eide, N. et al. Disseminated tumour cells in bone marrow of patients with uveal melanoma. Acta Ophthalmol. 91, 343–348. https://doi.org/10.1111/j.1755-3768.2012.02449.x (2013).
Herrspiegel, C., See, T. R. O., Mendoza, P. R., Grossniklaus, H. E. & Stålhammar, G. Digital morphometry of tumor nuclei correlates to BAP-1 status, monosomy 3, gene expression class and survival in uveal melanoma. Exp. Eye Res. 193, 107987. https://doi.org/10.1016/j.exer.2020.107987 (2020).
Robertson, A. G. et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 32, 204-220.e215. https://doi.org/10.1016/j.ccell.2017.07.003 (2017).
Bankhead, P. et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878–16878. https://doi.org/10.1038/s41598-017-17204-5 (2017).
Acknowledgements
The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
Open access funding provided by Karolinska Institute. Support for this study was provided to Mr. Uner from: The Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship (Boulder, CO). Emory Eye Center Trainee Pilot Grant (Atlanta, GA). To Dr. Grossniklaus from: An unrestricted departmental grant to the Emory Eye Center from Research to Prevent Blindness (New York, NY). National Institutes of Health, National Eye Institute, NIH NEI P30 06360. To Dr. Stålhammar from: The Royal Swedish Academy of Sciences (reference ME2019-0036). The Swedish Cancer Society (20 0798 Fk). The Swedish Society of Medicine (Cronqvists stiftelse, reference SLS 934014). The Swedish Eye Foundation (reference 2020–04-27). Karolinska Institutet (reference 2019–01900, 2020–013333 and 2020–02517). Region Stockholm (reference 20200356). The Crown Princess Margareta Foundation for the Visually Impaired (reference 157). Carmen and Bertil Regnér Foundation (reference 2020–00062).
Author information
Authors and Affiliations
Contributions
O.U. and G.S. conceived the project. T.S., E.S., H.G. and G.S. acquired the data. O.U., and G.S. performed the statistical analysis, prepared the figures and tables and wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the legend of Figure 1. “F) Tumor cells with loss of BAP1 expression had significantly larger mean volume (801 μm3, SD 223) than those with retained expression (524 μm3, SD 352, p = 0.042).” now reads: “(F) tumor cells with loss of BAP1 expression had significantly larger mean volume (2657 μm3, SD 1283) than those with retained expression (1593 μm3, SD 602, p = 0.027).”
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Uner, O.E., See, T.R.O., Szalai, E. et al. Estimation of the timing of BAP1 mutation in uveal melanoma progression. Sci Rep 11, 8923 (2021). https://doi.org/10.1038/s41598-021-88390-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88390-6
This article is cited by
-
A serum protein signature at the time of Uveal Melanoma diagnosis predicts long-term patient survival
BMC Cancer (2023)
-
Digital morphometry and cluster analysis identifies four types of melanocyte during uveal melanoma progression
Communications Medicine (2023)
-
The rationale for treating uveal melanoma with adjuvant melatonin: a review of the literature
BMC Cancer (2022)
-
Vasculogenic mimicry correlates to presenting symptoms and mortality in uveal melanoma
Journal of Cancer Research and Clinical Oncology (2022)
-
The prognostic implication of latitude in uveal melanoma: a nationwide observational cohort study of all patients born in Sweden between 1947 and 1989
Discover Oncology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
