
Abstract
The endoplasmic reticulum (ER) immunoglobulin binding proteins (BiPs) are molecular chaperones involved in normal protein maturation and refolding malformed proteins through the unfolded protein response (UPR). Plant BiPs belong to a multi-gene family contributing to development, immunity, and responses to environmental stresses. This study identified three BiP homologs in the Solanum tuberosum (potato) genome using phylogenetic, amino acid sequence, 3-D protein modeling, and gene structure analysis. These analyses revealed that StBiP1 and StBiP2 grouped with AtBiP2, whereas StBiP3 grouped with AtBiP3. While the protein sequences and folding structures are highly similar, these StBiPs are distinguishable by their expression patterns in different tissues and in response to environmental stressors such as treatment with heat, chemicals, or virus elicitors of UPR. Ab initio promoter analysis revealed that potato and Arabidopsis BiP1 and BiP2 promoters were highly enriched with cis-regulatory elements (CREs) linked to developmental processes, whereas BiP3 promoters were enriched with stress related CREs. The frequency and linear distribution of these CREs produced two phylogenetic branches that further resolve the groups identified through gene phylogeny and exon/intron phase analysis. These data reveal that the CRE architecture of BiP promoters potentially define their spatio-temporal expression patterns under developmental and stress related cues.
Similar content being viewed by others
Introduction
One of the best characterized molecular chaperones in the endoplasmic reticulum (ER) is the ER binding immunoglobulin protein (BiP), also known as the glucose receptor protein 78 (GRP78), is conserved across evolutionary kingdoms. BiP guides the co-translational translocation of nascent proteins into the ER, and chaperones protein folding and maturation. BiP contains N-terminal nucleotide-binding domain (NBD) and C-terminal substrate-binding domain (SBD). The NBD has two lobes surrounding the allosteric ATP-binding site to modulate substrate binding. The SBD has SBDβ and SBDα subdomains to bind the hydrophobic surfaces of newly translated proteins to prevent aggregation. The SBDβ is a pocket with two primary loops that surround the nascent polypeptide and, the SBDα lid is covering this pocket1. All BiP proteins have a C-terminal HDEL or KDEL signaling motif for ER retention2.
Across eukaryotes, BiPs contribute to the unfolded protein response (UPR). Under normal condition, the mammalian BiP binds to and inhibits three ER stress sensors, protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1α (IRE1α)3,4. BiP releases the sensors to activate ER-to-nucleus signaling cascades. BiP plays another key role in protein quality control by identifying and refolding misfolded proteins. The yeast Kar2p (BiP orthologue) binds to and inhibits the IRE1p, which is the master regulator of UPR. Dissociation of Kar2p/BiP releases the IRE1p/IRE1α to oligomerize and then splice the mRNA controlling production of the Hac1/XBP1 transcription factor5. In Arabidopsis, the ER stress sensors include IRE1a, IRE1b, bZIP28, and bZIP17. The IRE1a and IRE1b splice the mRNA controlling production of the bZIP60 (Hac1p/XBP1 orthologue) transcription factors. The bZIP28 and bZIP17 are transcription factors with transmembrane domains that are cleaved at the ER upon activation. In Arabidopsis, BiP regulates cleavage and removal of the bZIP28 transmembrane domain for nuclear translocation of the transcription factor6,7.
In contrast to yeast and mammals, plant BiP genes belong to a multi-gene family. The Arabidopsis, wheat (Triticum aestivum), citrus (Citrus sinensis), soybean (Glycine max), pepper (Capsicum annuum L.) and tobacco (Nicotiana tabacum) encode three or four BiPs which have been annotated and shown to help protect against many abiotic stresses including heat, cold, salt, heavy metal and osmotic stresses17,18,19,20,21. These BiPs also play crucial roles in biotic stress resistance and plant innate immunity1,2,3,4,5,16. Several studies link BiPs to the regulation of ER stress-mediated cell death6,7,8,9, hypersensitive cell death, and non-host HR programmed cell death induced by Xanthomonas oryzae pv. Oryzae in rice, Pseudomonas syringae pv. maculicola in Arabidopsis, and Pseudomonas syringae pv. tomato in soybean and tobacco, Phytophthora sojae in soybean10,11,12,13,14.
Arabidopsis thaliana has three well-studied BiP proteins. AtBiP1 and AtBiP2 are almost identical (99% protein identity) and ubiquitously expressed8,9. AtBiP3 is less conserved (80% protein identity with AtBiP1/2) and is only expressed in response to ER stress6,8. AtBiP1 and AtBiP2 expression correlates with the synthesis of seed storage proteins and is induced in flower organs and endosperm tissue9,10. These AtBiPs also mediate fusion of polar nuclei during male and female gametogenesis11,12,13. The three Arabidopsis BiP promoters have cis-regulatory elements (CREs) known as the unfolded protein response element (UPRE) and ER-stress response element (ERSE) that are transcription targets of bZIP60, bZIP28 and bZIP178,14. The AtBiP promoters have many other CREs for multiple factors having an additive effects on the levels or timing of gene induction15.
Although the Solanaceae family includes several agriculturally important crops, the BiP gene family in Solanaceae species remains largely unexplored, poorly annotated, and only partially characterized. This leaves a large gap in our knowledge about one of its major stress response pathways, the UPR. The first reported partial or complete cDNAs for tobacco BiPs identifying a multigene family was in 199115. This report was prior to complete genome sequencing an annotation of any Solanaceae species. Six identified genes were classified based on the uniqueness of their 3′ untranslated sequences and were named BiP-like protein (BLP)1, BLP2, BLP3, BLP4, BLP5, and BLP8. Certain BLP proteins were reported to show tissue-specific patterns of expression, stress-related expression, and responsiveness to ER stress induced by tunicamycin treatment2,15. Newer studies reported the cDNA sequences for three BiP homologs in C. annuum and S. lycopersicum and the annotated loci are publicly available in the genome databases. Recent studies reported the cDNA sequences for four BiP homologs in N. benthamiana and N. tabacum and named them based on homology with S. lycopersicum or Arabidopsis BiPs12,16. While the transcripts were identified, the loci sequences for these Nicotiana spp. are incomplete. Furthermore, the putative transcript or loci IDs provided in databases have significantly changed since the first reported tobacco BiPs making it difficult to cross-reference recently reported sequences with historic information.
The potato genome sequence was first published in 2011 and the genome sequence consortium has been actively updating the sequence accessions (solgenomics.net). Potato is the third most important global food crop in terms of human consumption (CIP; https://www.cipotato.org) and is threatened by many fungal, bacterial, and viral pathogens for which resistance likely requires activities of the BIP gene family. This study identifies and characterizes the BiP gene family in potato (Solanum tuberosum), that we now know encompasses three BiPs. We analyzed their evolution, structural features, promoter architecture, and their expression under both development and stress-induced conditions. We report new insights that suggest their promoter architecture is key to differentiate the various roles of potato BiPs in development and during ER stress.
Results
Identification and phylogeny of StBiP
Arabidopsis BiP proteins (AtBiP1-3) were used to query Ensemble Plants and Solanum Genome Network (SGN) and to find homologs in Solanaceae (Capsicum annuum, Nicotiana attenuata, N. benthamiana, N. tabacum, Solanum lycopersicum, S. tuberosum)17,18. We also retrieved a broader set of putative plant BiP homologs for Brachypodium distachyon, Glycine max, Oryza sativa, Sorghum bicolour, Triticum aestivum, Zea mays from Ensemble Plants. Proteins were selected that satisfied the e-value of e−25. Candidate proteins were categorized as functional BiPs based on three predefining criteria: (1) predicted localization to the ER; (2) five internal domains for ATP hydrolysis and substrate binding; (3) ER retention signal (XDEL). Forty-six BiPs were recovered from both searches. Most BiPs were in the range of 639 to 678 amino acids and their molecular weights were between 71 and 75 kDa. There was one soybean BiP that was 581 amino acids in length and 63.7 kDa and another N. benthamiana BiP that was 851 amino acids and 95.8 kDa. The protein PIs were generally between 4.71 and 5.03 (Table 1). We identified three putative S. tuberosum BiPs as PGSC0003DMG400012254, PGSC0003DMG400018544, and PGSC0003DMG400024707 (Table 1 and Supplementary Table S1 online). These were renamed StBiP1, StBiP2, and StBiP3 respectively. The amino acid sequence of StBiP1 and StBiP2 are highly similar (92%), whereas StBiP3 is more distantly related to StBiP1 and StBiP2 (75%).
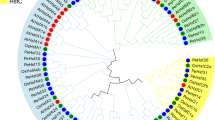
We constructed a phylogenetic tree using PhyML (v. 1.5) with 1,000 bootstraps (Fig. 1) which included the 41 plant BiPs as well as five vertebrate and Saccharomyces cerevisiae BiPs (Table 1 and Supplementary Table S1 online)19. The phylogenetic tree included three branches representing plants, animals, and fungi. The plant BiPs were aggregated in two groups. Group A has ten BiPs belonging to monocots and dicots and, includes the AtBiP3 and StBiP3. Surprisingly, there were no B. distachyon, Z. mays, or G. max genes represented in Group A. The monocot and Solanaceae BiP3-like proteins clustered in two sub-groups (MIII and SIII). These results indicate that there is an AtBiP3 specific lineage represented in Solanaceae.

Phylogenetic tree constructed using the PhyML method (v1.5) with Seaview (v4.7) (https://doua.prabi.fr/software/seaview) contains BiP proteins among fungi, plants, and animals. The output was visualized using iTOL (v4) and Adobe Photoshop CC (2017). Group A is highlighted in green and contains BiPs that cluster with the AtBiP3. Group B is highlighted in blue and represents a larger group. The identified subgroups include three monocot (MI, MII, and MIII), two Solanaceae (SI and SII) and soybean and Arabidopsis (GLYM and ATI). The AtBiPs and StBiPs are marked with red or black squares, respectively.
Group B was more diverse (Fig. 1) and the monocot and Solanaceae BiPs each clustered into two separate subgroups (MI, MII, SI and SII). The Arabidopsis and soybean BiPs clustered into distinct sub-groups (AT1 and GLYM). StBiP1 and StBiP2 were present in two Solanaceae sub-groups (SI and SII) closely related to the AT1 sub-group.
Intron/exon structure of BiP genes
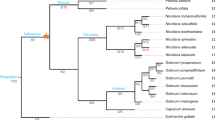
The intron positions and the frequency of intron phase combinations in related genes provide evidence of a common progenitor. Introns acquired in a progenitor and stabilized through evolution, maintain a non-random pattern. A random phase distribution of introns suggests that exons were shuffled through evolution, possibly creating new elements within gene products. The position of introns within a codon phase 0, 1, or 2, were mapped for the 46 BiPs (Fig. 2). The gene sizes of the 46 BiPs vary from 2,049 bps to 6,491 bps (Fig. 2). The yeast BiP (YJL034W) lacks introns. The representative animal BiPs possess seven introns with similar phase patterns suggesting a common origin. Notably, animal BiPs have intron phase 2 at the first intron while the majority of plant BIPs have phase 1 as the first intron. Near the 3′ end is a (1,1) symmetric exon. Within Group A genes, the SbBiP3 (SORBI_3001G118600) stands out because it contains one intron and two exons. All other Group A genes have a central (1,1) symmetric exon surrounded by asymmetric exons in (1,2), (2,1), (1,0) and (0,2) class. The AtBiP3 (AT1G09080.1) has a (1,2) asymmetric exon and both AtBiP3 and OsBiP3 (Os03g0710500) lacked the (0,2) asymmetric exon. The AtBiP3 had a (1,2) asymmetric exon not found in other Group A genes.

The structures of BiP genes, generated using GSDS 2.0 (https://gsds.cbi.pku.edu.cn/), starting from the transcription start site to the end of 3′ untranslated region in fungi, plants, and animals. Group A and Group B BiPs identified in green and blue boxes. The AtBiPs and StBiPs are marked with red or black squares, respectively, as in Fig. 1. The linear representation of each gene includes blue bars indicating 5′ and 3′ untranslated regions, yellow regions representing exons, and lines representing introns. The intron phase 0, 1, 2 are identified above each line. Red boxes surround the (1,1) symmetric exons. Most genes have one or two symmetric exons.
Group B includes an assortment of genes contain between five and eight exons and the MII subgroup of the intron-less rice and sorghum genes (Fig. 2). The AT1 subgroup contains the AtBiP1 and AtBiP2 genes which have the least number of exons (5) primarily comprised of (1,2), (2,1) (1,2) asymmetric exons and a single (1,1) symmetric exon. Most Group B genes have two (1,1,) symmetric exons in the central region, a (1,2) symmetric exon near the 5′ end, and (0,2) or (0,1) asymmetric exon near the 3′ end (Fig. 2). The common intron patterns and phases suggests a common ancestral origin for most of the Group A and Group B BiPs.
StBiP and AtBiP domain structures and amino acid sequences are highly conserved.
In all BiPs, the N-terminal nucleotide-binding domain (NBD) and C-terminal substrate-binding domain (SBD) are connected by a linker sequence that controls allosteric interactions20,21. The SBD has two subdomains SBDβ and SBDα (Fig. 3a). To compare the new plant BiPs with canonical structures, we used the I-TASSER server22 to generate three-dimensional structures of AtBiPs and StBiPs using the human and Chinese hamster BiPs as the threading templates (Fig. 3b; Supplementary Table S2 online). Supplementary Table S2 (see online) provides the confidence score (C-score), the TM-score and the RMSD values to explain the quality of the models for yeast, Arabidopsis and potato BiPs. The TM score and RMSD value correlate with the C-score22,23.

Domain structure of Arabidopsis and potato BiPs. (a) Schematic structures of the BiP domains. (b) The 3-D models are superimpositions of NBD, SBDα and SBDβ domains of AtBiPs and StBiPs and were generated using PyMol (v1.7.4) (https://pymol.org/). (c) Multiple sequence analysis of the AtBIP and StBiP proteins. The signal peptide, Domain1(β motif), Domain2 (γ motif), Domain3 (calmodulin-binding site), Domain4 (adenosine-binding motif), Domain5 (αβ motif), Domain6 (ER retention signal) are identified by bars above the alignment. The identical amino acids are identified by colors pointing to the high conservation among these proteins.
Next, we performed 3-D model superimposition of the well-characterized AtBiPs and candidate StBiPs (Fig. 3b) by overlaying the NBD, SBDα, and SBDβ to identify similarities. There are four structural subdomains oriented around one or more β-sheets (IA, IB, IIA, IIB) that divide the NBD into two lobes surrounding a central ATP binding pocket. This general folding structure is highly conserved among the AtBiPs and StBiPs with only minor variation (Fig. 3b). The SBDβ consists of eight β-strands forming a two-layered sandwich with five upper β-sheets and the three lower β-sheets. The SBDβ structure and amino acid sequences of the three AtBiPs and StBiPs are conserved. The SBDα consists of four helices. The 3-D models show that AtBiP2 has a shorter α-helical domain than AtBiP1 and AtBiP3. This corresponds to the gaps in the amino acid alignments that exist upstream of the domain 5 sequence (Fig. 3b, c). For all AtBiPs and StBiPs, except AtBiP2, the SBDα structure is conserved.
The I-Tasser predicted structures are consistent with amino acid conservation in sequence alignments of the candidate StBiP1, StBiP2 and StBiP3 proteins and other well-annotated plant BiPs. The first sequence alignment includes only Arabidopsis and S. tuberosum BiPs (Fig. 3c). The second alignment includes 46 BiPs used in the phylogenetic study (Supplementary Fig. S1 online). These alignments show the key conserved amino acid motifs that characterize members of the BiP gene family (Fig. 3c and Supplementary Fig. S1 online). Within the NBD and SBD are six amino acid sequence motifs that are remarkably well conserved among the 46 BiP sequences and are present in the StBiPs. The NBD contains the highly conserved β (Domain 1), γ (Domain 2), and adenosine binding (Domain 4) which provide the necessary ATPase binding and hydrolysis functions. The putative calmodulin-binding motif (Domain 3) located in the ATPase domain is also conserved among the StBiPs. The SBD contains the αβ domain (Domain 5), which includes a five- residues substrate-binding core that facilitates hydrogen-bonding with the peptide-substrate backbones. Finally, the StBiP1 and StBiP2 contain an ER retention HDEL signal sequence and StBiP3 contains a YDEL signal sequence.
Potato BiPs are differentially expressed under developmental and stress conditions
We analyzed the three StBiP expression profiles using the publicly available RNA-seq data that was generated from 15 different organs and tissues and presented the data as heatmaps (Fig. 4a)24. Under normal conditions, StBiP1 and StBiP2 are constitutively expressed in all tissues and generally show the same pattern of moderate to high expression in stolon, stem, shoot apex, petiole, young and mature tubers. StBiP1 is highly expressed in flowers while StBiP2 and StBiP3 are highly expressed in roots under normal growth and development.

Expression profiles of StBiPs under developmental and stress conditions. (a) A heat map of tissue-specific expression profiles generated from RNA-seq data using MeV (v4.9.0) based on ArrayExpress accession E-MTAB-55224 (https://mev.tm4.org/). The transcripts per million (TPM) values of representative transcripts were used to generate the heatmap. The color scale above heatmap shows the expression levels; red indicates high transcript abundance while green indicates low abundance. (b–d) The bar graphs show the average StBiPs transcript levels determined by RT-qPCR following: (b) treatment with 2 mM DTT and 150 mM NaCl; (c) heat (37 °C) and cold (4 °C) treatment; (d) confocal images showing transient expression of GFP fusions with PVY 6K2 and PVX TGB3. (e) transient expression of the potyvirus 6K2 and the potexvirus TGB3 proteins. *Significantly different from the control (H2O, 23 °C and GFP); student t test; p < 0.05; n = 6).
Since the UPR is an early and rapid response to environmental stress, we investigated whether these genes are upregulated to mitigate abiotic and biotic stress. Extensive prior studies of UPR related gene expression in Arabidopsis thaliana, Glycine max, Nicotiana species, and Solanum species treated leaves with DTT or NaCl. Time course experiments showed that the transcriptional response of several UPR related genes including BiP can be measured by RT-qPCR between 1 and 4 h after treatment25,26,27,28,29,30. We infiltrated potato leaves with 2 mM DTT or 150 mM NaCl to induce ER stress30,31. After four hours, the transcript levels of StBiP1 and StBiP2 remained unchanged while the StBiP3 transcripts increased 4.5-fold following salt treatment and 95-fold following DTT treatment above the mock control (H2O; Fig. 4b).
Prolonged heat and cold treatment are commonly used to measure gene responses to persistent ER stress28. Gene expression during persistent ER stress influences plant growth and pro-survival activities. BiP is typically considered to be a core component of the UPR during both early and prolonged responses to ER stress32. Here we exposed potato plants to cold (4 °C) for 16 h and gene expression was unaltered (Fig. 4c). Potato plants were exposed to heat (37 °C) stress for 16 h1 and there was a fourfold increase in StBiP2 and an approximately 200-fold increase in StBiP3 in the leaves (Fig. 4c). Biotic stress was applied by delivering viral proteins that are well known to be specific inducers of the UPR3,27. Binary vectors expressing the potato virus X (PVX) TGB3, potato virus Y (PVY) 6K2 proteins fused to GFP, or GFP alone (negative control) were delivered by agro-infiltration to potato leaves. We confirmed fluorescent protein expression using confocal microscopy (Fig. 4d). At 4 days, leaves expressing the PVY-6K2 displayed elevated levels of StBiP1, StBiP2, and StBiP3 transcripts that were between 2.1- and 4.7-fold higher than the GFP controls (Fig. 4e). Among the leaves expressing the PVX-TGB3, only StBiP2 was significantly induced. These combined data indicate that the expression of StBiP1 and StBiP2 are responsive to heat or viral factors while the expression of StBiP3 is responsive to DTT, NaCl, heat and viral factors.
StBiP promoters are enriched with developmental, hormone-response, and stress-related transcription factor binding sites
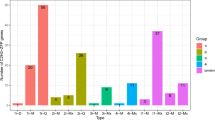
To identify the basis of differential gene expression, we selected 1,000 bp upstream of the predicted transcription start site for the StBiP and AtBiP promoters and derived the predicted cis-regulatory elements (CREs). CREs belonging to 30 families of transcription factors were identified. Across all the Arabidopsis and potato BiP promoters, there are between 22 and 24 combined basic leucine zipper (bZIP), heat shock transcription factor (HSF), myeloblastosis (MYB) and flowering time regulator SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) transcription factor binding sites. Notably, the StBiP1 lacks an HSF binding site, the StBiP3 lacks the SPL binding site.
Hierarchical clustering of StBiPs and AtBiPs identified two promoter groups based on the presence and the distribution of CRE in their promoters (Fig. 5a). One cluster consisted of the StBiP1, StBiP2, and AtBiP2 promoters. The second cluster consisted of the AtBiP1, AtBiP3 and StBiP3 promoters (Fig. 5a). The architectures of the AtBiP2 and StBiP2, and of the StBiP3 and AtBiP3 promoters were surprisingly conserved.

Distribution and frequency of CRE and ER-stress responsive elements on AtBiP and StBiP promoters. (a) The hierarchical clustering of CREs belongs to various transcription factor families identified using the TRANSFAC database release 2019.2. The average linkage distance was determined by hierarchical clustering and is presented on the right. The grid provides the number of sites that contain the CREs representing 25 transcription factor families. Shades of red were used to point to CREs that were highly represented (dark red), moderately represented (medium red), were represented once or twice (pale red), or not represented (white). The names of the promoters are identified on the right. (b) The distribution of ROOTMOTIFTAPOX1 (ATATT) elements (red arrows) in the 1,000 bp StBiP promoters are critical for root-preferential expression. These were promoters identified using the New PLACE database (https://www.dna.affrc.go.jp/PLACE). The promoter names are provided on the left. (c) The distribution of ER-stress responsive UPRE and ERSE in the 2000 bp AtBiP and StBiP promoters. The lines are drawn to scale with the nucleotide position relative to the transcription start site, represented above the lines. The colored arrows represent the positions of the CREs that are engaged by factors in the unfolded protein response. The names of these CRES are identified in the legend. The promoters are named on the right.
We identified key transcription factor families and CREs for differential gene expression during cell differentiation, organ development, and hormonal responses required for development (Fig. 5a–c and Supplementary Table S3 online)1,2. Regarding the MYB transcription factor family, the StBiPs and AtBiP1 and AtBiP2 the analysis identified: (a) MYB3R CREs that are components of the DP, Rb-like, E2F, and MuvB (DREAM) complex; (b) MYB24 CREs in StBiP1, AtBiP1, and AtBiP2 promoters for gibberellic acid and jasmonic acid-mediated stamen development; and (c) WEREWOLF CREs in the AtBiP2 promoter for epidermal cell patterning. The root specific ROOTMOTIFTAPOX1 motif is distributed among all three StBIPs with 13 copies in the StBiP3, eight copies in the StBiP2 and only one in the StBiP1 promoters (Fig. 5b). The copy number of root specific CREs correlates with the respective high, medium, and low levels of StBiP3, StBiP2 and StBiP1 in roots seen in Fig. 4a.
Several large families of transcription factors have known roles in key developmental and metabolic processes in response to adverse environments including temperature and water stress. These include the abscisic acid stress and ripening/water deficit stress (ABA/WDS), APETALA2/Ethylene-responsive factor (AP2/ERF), and basic helix-loop-helix (bHLH) transcription factor binding sites. Between 1 and 5 copies of the ABA/WDS and AP2/ERF binding sites are present in each AtBiP and StBiP promoters (Fig. 5a). Regarding the AP2/ERF transcription factor family binding sites, the BBM, RAP2.1, B3, and ESR1 are differently represented. The B3 CRE was found only in the AtBiP1 promoter while ESR1 CRE was only present in the StBiP1 promoter (Supplementary Table S3). Among the bHLH transcription factor binding sites, three CREs are differentially represented. The bHLH28(MYC5) CRE occurs in all Arabidopsis and potato promoters except for StBiP3. The bHLH64 is only present in the AtBiP3 promoter while BEE2 CREs occurs in StBiP1 and StBiP3 promoters.
There are 78 bZIP transcription factors in Arabidopsis which generally contribute to seed germination, sugar signaling, amino acid metabolism, salt stress, hypo-osmolarity responses, UPR, and pathogen defense responses. Regarding sucrose regulation and hormone signaling, the bZIP44/53/GBF6, AS1/OCS/TGA-like, and PEND CREs were prominent but differentially represented in the Arabidopsis and potato BiP promoters (see Supplementary Table S3 online). Only the AtBiP3 and StBiP3 promoters have PEND target CREs33,34,35.
While UPR-related elements were previously identified in the AtBiP promoters36,37, we compared the UPR element II (UPRE-II), UPRE-III, ERSE-I and ERSE-II in the promoter of AtBiPs and StBiPs (Fig. 5c, Supplementary Table S3). UPRE-II and ERSE-II elements were overlapping in StBiP1 and StBiP2 promoters. The UPRE-III elements were present on StBiP2 and all AtBiP promoters. Only AtBiP3 and StBiP3 promoters contain ERSE-I elements that are binding sites for the ER-stress induced AtbZIP28 factor (Fig. 5c)38,39.
The WRKY and CAMTA binding sites are well known for pathogen defense, abiotic stress, and the unfolded protein response. The WRKY binding sites were present in AtBiP1, StBIP1, and StBIP2, but lacking in the AtBiP3 and StBiP3 promoters (Fig. 5A and Supplementary Table S3). In particular, WRKY7, -11, and -17 transcription factors are known to control AtBIP1/2 expression by suppressing the AtbZIP28 arm of the UPR during plant immunity40. The StBIP3 has CAMTA binding sites, which are lacking in the StBiP1/2 promoters.
Discussion
The molecular chaperone BiP ensures protein homeostasis and is an essential regulator of ER stress transducers in eukaryotes. The characterization of BiPs is crucial to understanding cellular, tissue-specific, and developmentally related responses to ER stress. The BiP gene family has been investigated in several plant species, including Arabidopsis, rice, wheat, pepper and citrus36,37,41,42,43,44. Historic and recent studies using transgenic lines overexpressing various BiPs characterize their roles in plant tolerance to adverse environmental conditions or tissue specific gene expression2,15,32,36,43,45. Importantly, we had difficulties retrieving original data relevant to these publications, especially for Solanaceae family, because of the lack of proper gene nomenclature, annotation, presence of several genome assemblies, and unavailability of relevant nucleotide sequences in databases (GenBank, ENA, etc.)10,11,15,36. To resolve these issues, we retrieved relevant data directly from curated datasets and updated assemblies while cross comparing with the findings of the above-mentioned studies (Table 1 and Supplementary Table S1 online). This study reports a comprehensive phylogenetic analysis of already identified and some novel BiPs covering fungi, plant and animal kingdoms in order to identify and characterize BiPs in potato and dissect their evolutionary history.
We report three candidate BiPs (StBiP1, StBiP2, and StBiP3) in the S. tuberosum genome. Phylogenetic analysis, amino acid sequence analysis, 3-D protein modeling, and gene structure analysis confirmed that these potato gene candidates are true BiP genes. The phylogenetic tree divided the StBiPs into Group A and Group B, consistent with previous studies (Fig. 1)37,44,46. Both groups contained monocot and dicot BiPs. Group A contains AtBiP3 and StBiP3. Group B contains the StBiP1 and StBiP2 and shows higher diversification compared to Group A. Within Group B, StBiP1 and StBiP2 diverged more recently from AtBiP1 and AtBiP2. At the amino acid level, the StBiP1 and StBiP2, and the AtBiP1, AtBiP2 were highly similar (92% and 90% respectively) compared to StBiP3 and AtBiP3 (70–76%). The three StBiP and AtBiP proteins share the expected six protein functional domains that define NBDs and SBDs in eukaryotic BiPs. The 3-D model superimposition also revealed a higher degree of structural similarity between the BiP proteins in Arabidopsis and potato, indicating that their grouping is a result of the variations in non-conserved regions rather than functionally important conserved regions.
Gene structures are reliable indicators of the evolutionary history of gene families47. The exon/intron structures and intron numbers vary from zero to eight among the 46 BiP genes. Such diversity arises from insertions and deletions during evolution that alters the distance between the start codon and downstream intron peaks. The Group A gene structures and intron phase patterns are conserved except for SORBI_3001G118600. The Group B has 32 members and excluding the AtBiP1/2, SORBI_3004G0117700, and the four BiPs that lack introns, all other members have similar intron phase patterns throughout the genes. Although the gene structure and phylogenetic studies differentiated StBIP1, StBiP2, and StBiP3 into different groups, functional differences for plant development or the management of ER stress could not be assigned based on gene structure or protein phylogeny. Instead, these data support the functional redundancies often seen in plant experiments where individual BiP knockout mutations are partially or fully complemented by other BiP genes27.
In Arabidopsis, pepper, and rice, the BiP genes were primarily distinguishable by their expression profile42,43,45. This study showed that potato BiPs have distinguishable expression patterns. Under normal conditions, StBiP1 and StBiP2 are expressed in all tissues, whereas StBiP3 is only in roots. Treatments with high temperature and salt stress, induce only StBiP3 expression in leaves. StBiP1, StBiP2, and StBiP3 expression was elevated in response to expression of the PVX TGB3 and PVY 6K2 proteins, paralleling results in Arabidopsis for induction of AtBiP1, AtBiP2, and AtBiP348. These functional studies support the phylogenetic protein diversification of StBiP proteins, despite the highly conserved protein structures.
We propose that the promoter architecture, which controls the gene expression profiles, is key to the BiPs’ contributions to plant development. Understanding the promoter architecture is key to predicting gene functional specificity concerning tissue, timing, and pattern of expression. This study elucidates the various roles of the potato BiPs by combining gene expression profiles and analysis of promoter architecture. The frequency and the linear distribution of CREs in the StBiP promoter sequences produced two phylogenetic branches that coincide with the separation of Group A (BiP3) and Group B (BiP1/2). The identified CREs correspond to twenty-five transcriptional factor families and suggest fine regulation of BiPs under various developmental and stress-related stimuli. Profiling the distribution and frequency of CREs among StBiPs and AtBiPs revealed important similarities between the promoter architecture of Group B BiPs.
The StBiP1 and StBiP2 promoters are highly enriched with CREs related to cell differentiation, organ development, and hormone-regulated plant development. Considering only the potato BiPs, the presence of MYB3R-like target CREs in the BiP1/2 genes potentially coordinates the elevated levels of StBiP1 and StBiP2 as part of the gene network in shoots, stems, stolons, tubers, and stamens64,65. The AtBiP2 is the only promoter where we identified WEREWOLF CREs (MYB target) that play a major role in epidermal cell patterning and WRKY7/57 CREs that contribute to stress responses (Supplementary Table S3 online)49,50,51. The profile of CREs in AtBiP1 and AtBiP2 identified differences that could not be otherwise detected in gene expression assays using gene specific probes, because of their extensive sequence identity. These data suggest that AtBiP1 and AtBiP2 have overlapping and unique functions.
The StBiP3 promoter has fewer CREs involved in cell differentiation and organ development and is enriched with stress related CREs (Fig. 5a, c and Supplementary Table S3 online). The varied distribution of the root specific ROOTMOTIFTAPOX1 motif among all the StBiP promoters, likely contributes to different gene expression levels in root tissues. All StBiP promoters contain CREs representing the AP2, bHLH, ABA/EDS transcription factor families that link development, hormonal regulation, and stress response in plants. Some of the identified CREs recognized by AP2 transcription factors include B3 (mediation of auxin and brassinosteriod dependent development transitions), BBM and RAP2.6 (in cell proliferation, cell morphogenesis, pathogen infection, salt stress, and osmotic stress responses), and RAP2.1 (dehydration response)38,39,40,45,47,48,52.
All three StBiP promoters contain various bZIP transcription factor binding CREs important for biotic and abiotic stress-mediated signaling in plants. The presence of the AS1/OCS/TGA type elements, which are components of regulatory modules associated with hormonal regulation (auxin, salicylic acid, and jasmonic acid) under biotic stresses, wounding, and oxidative stress, might partially explain the activation of all BiP genes by biotic stressors. This study shows that StBIP1, StBiP2 and StBIP3 are consistently induced in response to PVY 6K2 which is an known activator of ER stress related gene expression48,53 The linear pattern of ER-stress related UPRE and ERSE elements in AtBiP and StBiP promoters potentially contribute to the differential expression of BiPs under normal and stressed conditions, and could explain the stress-responsive role of AtBiP3 and StBiP3.
Collectively, the promoter analysis results provide the ability to predict when, where, how, and how much each BiP can be expressed in potato. Overall, the identification of cis-regulatory architecture will extend our understanding of the complex developmental and stress related cis–trans relationships involved in the regulation of BiP genes. This analysis provides a foundation for future studies to dissect the components of transcriptional regulation of BiP genes using in vitro and in vivo strategies including Electrophoretic Mobility Shift Assay (EMSA), Chromatin Immunoprecipitation Sequencing (ChIP-Seq), promoter deletion, and other transgenic approaches for identifying condition-specific combinations of CRE modules. We anticipate further investigations will be useful for the functional characterization of BiPs, and for developing transgene-free crops that can withstand stress conditions using both classical and modern genome editing strategies.
In summary, we identified three BiPs in potato and named them based on their sequence similarity to Arabidopsis BiPs. Their gene structure and protein domain architectures were highly conserved during evolution, leading us to further investigate the basis for their distinguishable and sometimes overlapping roles in plant growth, development, and stress response. Figure 5a reveals that the differences between BiP promoter sequences produced phylogenetic branches that cluster StBiP1, StBiP2, and AtBiP2 as well as StBiP3 and AtBiP3 into separate groups. The pattern of CREs appears to be responsible for promoter differences which correlate with the different gene expression patterns observed between BiP1/2 and BiP3. Based on our findings, we propose a model in which the promoter architecture drives the spatiotemporal regulation of BiPs for plant growth, development, and stress responses. Accordingly, the distribution and frequency of various developmental, stress-responsive and tissue-specific CREs can be considered as the primary determinant of the fine regulation of BiPs both in Arabidopsis and potato.
Methods
Genome-wide identification of the BiPs in potato and phylogenetic analysis
We used BLASTP and the annotated Arabidopsis BiP genes (AtBiP1-3; AT5G28540.1, AT5G42020.2 and AT1G09080.2) which we retrieved from The Arabidopsis Information Resource 10 (TAIR10) (https://www.arabidopsis.org) to search for BiP homologs in the assemblies of Brachypodium distachyon (v3.0), Capsicum annuum (ASM51225v2), Glycine max (v2.1) Nicotiana attenuate (NIATTr2), Nicotiana benthamiana (v1.0.1), Nicotiana tabacum (v4.5), Oryza sativa (IRGSP-1.0), Sorghum bicolor (NCBIv3), Solanum lycopersicum (SL3.0), Solanum tuberosum (in the SolTub_3.0 Genome Assembly), Triticum aestivum (IWGSC), Zea mays (B73_RefGen_v4), assembly available at the Sol Genomics Network database and the Ensembl Plants database (release 43)17,18,19 within the publicly available Ensembl Genomes project54. An e-value threshold of 10–25 was used for the development of the first list of BiPs which included already annotated BiPs as well as candidate sequences of species in Solanaceae. Then subcellular localization of the protein sequences of identified potential candidates was assessed using DeepLoc-1.055. Only proteins with ER localization signals were identified as putative BiPs. The ENSEMBL Locus IDs were then used to retrieve the gene names and other identifiers from NCBI, UniPro, and TAIR databases (see Supplementary Table S1 online). Multiple sequence analysis was carried out using MUSCLE built into Seaview (v 4.7). The phylogenetic tree was generated using default settings of PhyML (v 1.5) build into Seaview (v 4.7) (https://doua.prabi.fr/software/seaview) with 1,000 bootstrap iterations56. The phylogenetic tree was visualized using iTOL (v4)57. Then Adobe Photoshop CC (2017) to compile diagrams into figures and provide labels and add color coding.
DNA extraction and sequencing
Rooted cuttings were grown in soil inside chambers with a 16 h photoperiod at 22 °C for 3 weeks. Leaf tissues were harvested in liquid nitrogen and ground using a motor and pestle. DNA extraction and purification were carried out using the Mini Genomic DNA Kit (IBI Scientific, Peosta, Iowa, USA). DNA was quantified using the Epoch spectrophotometer (Biotek, Winooski, VT) Powerwave XS2 with Gen 5.0 software. StBiP1, StBiP2, and StBiP3 genomic sequences were PCR amplified using gene-specific primers (Supplementary Table S4 online). Platinum SuperFi Green DNA Polymerase (Invitrogen) mix was used with PCR amplification conditions as follows; Initial denaturation 98 °C for 2 min, 98 °C 10 s, 60.9 °C 10 s, 72 °C for 2 min for 35 cycles, and final extension of 72 °C for 5 min. Amplified products were visualized using agarose gel (1%) electrophoresis. Sequencing was carried out using ABI 3,130 Genetic Analyzer (Applied Biosystems) using sequencing primers (Supplementary Table S4 online). The sequencing data was analyzed using Geneious Prime v. 2019.2.1. Gene sequences were deposited at NCBI Genbank under following accession numbers; MN982518 (StBiP1), MN982519 (StBiP2) and MN982520 (StBiP3).
Gene structures, domain analyses, and protein structure analyses
Intron–exon structures and intron phases of BiPs were visualized using GSDS 2.0 (https://gsds.cbi.pku.edu.cn/)58. Conserved domain analysis and visualization were carried out using Geneious Prime (v. 2019.2.1). Images were downloaded from GSDS 2.0. The I-Tasser structure and function prediction tool (Zhang lab) was used for structural modeling of the protein sequences. PyMol (v1.7.4) (https://pymol.org/2/) was used to visualize the protein domains of the chosen models and for the superimposition of the models. Selected mages were downloaded and then constructed into figures using Adobe Photoshop CC (2017).
Gene expression analysis based on the RNA-seq data
Transcriptomic analysis was carried out using RNA-seq data (accession number E-MTAB-552) generated by Potato Genome Sequencing Consortium (PGSC) available at the Expression Atlas Database (https://www.ebi.ac.uk/gxa/home)24. Transcripts per million (TPM) were calculated using raw counts by averaging technical replicates followed by quantile normalization of biological replicates using Limma59. TPMs were used to generate heatmap using MultiExpression Viewer (MeV) version 4.9.060. The diagrams and charts were compiled in Adobe Photoshop CC (2017).
Plant materials and stress induction
Solanum tuberosum cultivar ‘Russet Norkota’ were vegetatively multiplied in vitro on Murashige and Skoog (MS) medium (PhytoTech Labs, Lenexa, KS) or by cuttings placed into soil. All plants were grown inside chambers with a 16 h photoperiod at 22 °C for three weeks. The in vitro propagated plants were used for temperature stress experiments. These plants were subjected to an overnight 16 h at high (37 °C) or low (4 °C) temperature. Since the incubators for high and cold temperatures did not have lights, we subjected the control plants grown at regular (22 °C) temperature to an overnight 16 h of dark conditions to maintain consistent light conditions among the control and experimental plants. Leaves were harvested after the 16 h treatment for RNA extraction (below). Experiments requiring infiltrating leaves with solutions of DTT, NaCl or agrobacterium were carried out using rooted cuttings in soil according to Henriquez-Valencia et al.30. For abiotic stress, 2 mM dithiothreitol (DTT), 150 mM sodium chloride (NaCl) or H2O as mock treatment were infiltrated into leaves with a 1 mL needle-free syringe. Then leaf samples were harvest at 4 h post infiltration for RNA extraction and RT-qPCR (see below). For viral protein stress, PVX TGB3 and PVY 6K2 sequences were cloned by Gateway Technology (ThermoFisher) in pGWB505 binary vector for produce C-terminal GFP fusions. All plasmids were sequence verified and maintained in Agrobacterium tumefaciens sp. strain GV3101. A. tumefaciens carrying the GFP expression vector pXF7FNF2.0 was used as a mock control. A. tumefaciens harboring TGB3-GFP, 6K2-GFP or GFP were collected and suspended in a solution of 10 mM MES-KOH (pH 5.6), 10 mM MgCl2, 200 μM acetosyringone and adjusted to OD600 = 0.7. Then potato leaves (three weeks after rooting) were infiltrated with 1 mL needle-free syringe. These treated leaves were harvest at 4 days post infiltration for RNA extraction (below).
RNA extraction and RT-qPCR
Leaves were harvested, ground in liquid nitrogen, and RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Germantown, MD). Total RNA (2 µg) was reverse-transcribed using a High-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and random primers were employed for cDNA synthesis. The cDNAs were diluted 500 × and then qPCRs were performed using the SYBR Green Master Mix (Applied Biosystems). The qPCR amplification was performed for StBiP1, StBiP2, and StBiP3 using gene specific primers (Supplementary Table S5 online). The qPCR amplification of StACTIN58 (PGSC0003DMG400023429) was carried out as an internal control using gene specific primers (Supplementary Table S5 online). The ΔΔCt method was used for calculating relative gene expression. The data represent the mean of two technical replicates for three biological repetitions. Data was analyzed and charted using Microsoft Excel (2019). Charts were compiled using Adobe Photoshop CC (2017).
Confocal microscopy to confirm expression of viral proteins fused to GFP.
Leaf segments were placed on microscope slides and protected by cover slips before examination with an Olympus Fluoview FV1000 confocal laser scanning microscope and a 60 × objective (Olympus America Inc. Center Valley, PA). Laser excitation wavelength of 515 nm and Z-dimensions of 5 µm with 0.5 µm/slice. Image J 1.52p software61 was used for stacking Z-series images.
Ab initio promoter analysis
Promoter sequences representing 2000 bp from the transcription start site were retrieved from the Ensemble Plants database (https://plants.ensembl.org/; Ensembl Plants release 43, April 2019). CREs were identified using the New PLACE database (https://www.dna.affrc.go.jp/PLACE/), the TRANSFAC database (release 2019.2 https://genexplain.com/transfac/) and the Catalog of Inferred Sequence Binding Proteins (CIS-BP) Database (v 2.00) (https://cisbp.ccbr.utoronto.ca/) on 1,000 bp from the transcription start site62,63,64. Hierarchical clustering was carried out using the Manhattan correlation with the average linkage method in MultiExpression Viewer (MeV) (v4.9.0) https://mev.tm4.org/)60. The output of the hierarchical clustering was retrieved and the heatmap was recreated using Microsoft Excel (2019). UPRE and ERSE elements were identified in the 2000 bp promoter sequence (see Supplementary Table S3 online). Development and tissue-specific expression profiles were obtained from the Expression Atlas Database (https://www.ebi.ac.uk/gxa/home)24. Geneious Prime (v. 2019.2.1) was used to annotate and visualize the CREs and the diagrams were compiled using Adobe Photoshop CC (2017).
References
Williams, B., Verchot, J. & Dickman, M. B. M. B. When supply does not meet demand-ER stress and plant programmed cell death. Front. Plant Sci. 5, 1–9 (2014).
Leborgne-Castel, N., Jelitto-Van Dooren, E. P., Crofts, A. J. & Denecke, J. Overexpression of BiP in tobacco alleviates endoplasmic reticulum stress. Plant Cell 11, 459–470 (1999).
Ye, C., Dickman, M. B. M. B., Whitham, S. A. S. A., Payton, M. & Verchot, J. The unfolded protein response is triggered by a plant viral movement protein. Plant Physiol. 156, 741–755 (2011).
Ye, C.-M., Chen, S., Payton, M., Dickman, M. B. & Verchot, J. TGBp3 triggers the unfolded protein response and SKP1-dependent programmed cell death. Mol. Plant Pathol. 14, 241–255 (2013).
Lu, Y. et al. The unfolded protein response and programmed cell death are induced by expression of Garlic virus X p11 in Nicotiana benthamiana. J. Gen. Virol. 97, 1462–1468 (2016).
Reis, P. A. A. et al. The binding protein BiP attenuates stress-induced cell death in soybean via modulation of the N-rich protein-mediated signaling pathway. Plant Physiol. 157, 1853–1865 (2011).
Reis, P. A. B. & Fontes, E. P. B. N-rich protein (NRP)-mediated cell death signaling. Plant Signal. Behav. 7, 628–632 (2012).
Carvalho, H. H. et al. The molecular chaperone binding protein BiP prevents leaf dehydration-induced cellular homeostasis disruption. PLoS ONE 9, e86661 (2014).
Xu, H. et al. The ER luminal binding protein (BiP) alleviates Cd2+-induced programmed cell death through endoplasmic reticulum stress–cell death signaling pathway in tobacco cells. J. Plant Physiol. 170, 1434–1441 (2013).
Liebrand, T. W. H. et al. Chaperones of the endoplasmic reticulum are required for Ve1-mediated resistance to Verticillium. Mol. Plant Pathol. 15, 109–117 (2014).
Carvalho, H. H. et al. The endoplasmic reticulum binding protein BiP displays dual function in modulating cell death events. Plant Physiol. 164, 654–670 (2014).
Jing, M. et al. A Phytophthora sojae effector suppresses endoplasmic reticulum stress-mediated immunity by stabilizing plant Binding immunoglobulin Proteins. Nat. Commun. 7, 1 (2016).
Xu, G. et al. Plant ERD2-like proteins function as endoplasmic reticulum luminal protein receptors and participate in programmed cell death during innate immunity. Plant J. 72, 57–69 (2012).
Wang, D., Weaver, N. D., Kesarwani, M. & Dong, X. Induction of protein secretory pathway is required for systemic acquired resistance. Science 308, 1036–1040 (2005).
Denecke, J. The tobacco luminal binding protein is encoded by a multigene family. Plant Cell Online 3, 1025–1035 (1991).
Liebrand, T. W. H. et al. Endoplasmic reticulum-quality control chaperones facilitate the biogenesis of Cf receptor-like proteins involved in pathogen resistance of tomato. Plant Physiol. 159, 1819–1833 (2012).
Fernandez-Pozo, N. et al. The Sol Genomics Network (SGN)—from genotype to phenotype to breeding. Nucleic Acids Res. 43, D1036–D1041 (2015).
Kersey, P. J. et al. Ensembl Genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 46, D802–D808 (2018).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018).
Yan, M., Li, J. & Sha, B. Structural analysis of the Sil1-Bip complex reveals the mechanism for Sil1 to function as a nucleotide-exchange factor. Biochem. J. 438, 447–455 (2011).
Yang, J., Nune, M., Zong, Y., Zhou, L. & Liu, Q. Close and allosteric opening of the polypeptide-binding site in a human Hsp70 chaperone BiP. Structure 23, 2191–2203 (2015).
Yang, J. & Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Yang, J. & Zhang, Y. Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinforma. https://doi.org/10.1002/0471250953.bi0508s52 (2015).
Xu, X. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (2011).
Xu, Z., Song, N., Ma, L. & Wu, J. IRE1-bZIP60 pathway is required for Nicotiana attenuata resistance to fungal pathogen Alternaria alternata. Front. Plant Sci. 10, 1–10 (2019).
Moreno, A. A. et al. IRE1/bZIP60-mediated unfolded protein response plays distinct roles in plant immunity and abiotic stress responses. PLoS ONE 7, e31944 (2012).
Zhang, L., Chen, H., Brandizzi, F., Verchot, J. & Wang, A. The UPR branch IRE1-bZIP60 in plants plays an essential tole in viral infection and is complementary to the only UPR pathway in yeast. PLOS Genet. 11, e1005164 (2015).
Srivastava, R. et al. Response to persistent ER stress in plants: a multiphasic process that transitions cells from prosurvival activities to cell death. Plant Cell 30, 1220–1242 (2018).
Ruberti, C., Lai, Y. S. & Brandizzi, F. Recovery from temporary endoplasmic reticulum stress in plants relies on the tissue-specific and largely independent roles of bZIP28 and bZIP60, as well as an antagonizing function of BAX-Inhibitor 1 upon the pro-adaptive signaling mediated by bZIP28. Plant J. 93, 155–165 (2018).
Henriquez-Valencia, C. et al. bZIP17 and bZIP60 regulate the expression of BiP3 and other salt stress responsive genes in an UPR-independent manner in Arabidopsis thaliana. J. Cell. Biochem. 116, 1638–1645 (2015).
Iwata, Y. & Koizumi, N. An Arabidopsis transcription factor, AtbZIP60, regulates the endoplasmic reticulum stress response in a manner unique to plants. Proc. Natl. Acad. Sci. 102, 5280–5285 (2005).
Srivastava, R., Deng, Y., Shah, S., Rao, A. G. & Howell, S. H. BINDING PROTEIN is a master regulator of the endoplasmic reticulum stress sensor/transducer bZIP28 in Arabidopsis. Plant Cell 25, 1416–1429 (2013).
Gardner, B. M., Pincus, D., Gotthardt, K., Gallagher, C. M. & Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169–a013169 (2013).
Takayanagi, S., Fukuda, R., Takeuchi, Y., Tsukada, S. & Yoshida, K. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones 18, 11–23 (2013).
Mori, K. The unfolded protein response: the dawn of a new field. Proc. Japan Acad. Ser. B 91, 469–480 (2015).
Carolino, S. M. B. et al. Plant BiP gene family: differential expression, stress induction and protective role against physiological stresses. Braz. J. Plant Physiol. 15, 59–66 (2003).
Zhu, J. et al. Molecular cloning, phylogenetic analysis, and expression profiling of endoplasmic reticulum molecular chaperone BiP genes from bread wheat (Triticum aestivum L.). BMC Plant Biol. 14, 260 (2014).
Iwata, Y., Yoneda, M., Yanagawa, Y. & Koizumi, N. Characteristics of the nuclear form of the Arabidopsis transcription factor AtbZIP60 during the endoplasmic reticulum stress response. Biosci. Biotechnol. Biochem. 73, 865–869 (2009).
Liu, J. X. & Howell, S. H. bZIP28 and NF-Y transcription factors are activated by ER stress and assemble into a transcriptional complex to regulate stress response genes in Arabidopsis. Plant Cell 22, 782–796 (2010).
Arraño-Salinas, P. et al. WRKY7, -11 and -17 transcription factors are modulators of the bZIP28 branch of the unfolded protein response during PAMP-triggered immunity in Arabidopsis thaliana. Plant Sci. 277, 242–250 (2018).
Sarkar, N. K., Kundnani, P. & Grover, A. Functional analysis of Hsp70 superfamily proteins of rice (Oryza sativa). Cell Stress Chaperones 18, 427–437 (2013).
Noh, S.-J., Kwon, C. S., Oh, D.-H., Moon, J. S. & Chung, W.-I. Expression of an evolutionarily distinct novel BiP gene during the unfolded protein response in Arabidopsis thaliana. Gene 311, 81–91 (2003).
Wang, H., Niu, H., Zhai, Y. & Lu, M. Characterization of BiP genes from pepper (Capsicum annuum L.) and the role of CaBiP1 in response to endoplasmic reticulum and multiple abiotic stresses. Front. Plant Sci. 8, 1–15 (2017).
Guimarães, R. F. et al. Genome-wide identification and expression analysis of the molecular chaperone binding protein BiP genes in Citrus. Tree Genet. Genomes 14, 90 (2018).
Wakasa, Y., Hayashi, S. & Takaiwa, F. Expression of OsBiP4 and OsBiP5 is highly correlated with the endoplasmic reticulum stress response in rice. Planta 236, 1519–1527 (2012).
Sung, D.-Y., Kaplan, F. & Guy, C. L. Plant Hsp70 molecular chaperones: Protein structure, gene family, expression and function. Physiol. Plant. 113, 443–451 (2001).
Long, M., Rosenberg, C. & Gilbert, W. Intron phase correlations and the evolution of the intron/exon structure of genes. Proc. Natl. Acad. Sci. 92, 12495–12499 (1995).
Gayral, M. et al. Multiple ER-to-nucleus stress signaling pathways become active during Plantago asiatica mosaic virus and Turnip mosaic virus infection in Arabidopsis thaliana. bioRxiv 786137, 1. https://doi.org/10.1101/786137 (2019).
Huang, H. et al. Arabidopsis MYB24 regulates jasmonate-mediated stamen development. Front. Plant Sci. 8, 1 (2017).
Wang, P. et al. A key variant in the cis-regulatory element of flowering gene Ghd8 associated with cold tolerance in rice. Sci. Rep. 9, 9603 (2019).
Peng, J. Gibberellin and jasmonate crosstalk during stamen development. J. Integr. Plant Biol. 51, 1064–1070 (2009).
Sarkar, N. K., Thapar, U., Kundnani, P., Panwar, P. & Grover, A. Functional relevance of J-protein family of rice (Oryza sativa). Cell Stress Chaperones 18, 321–331 (2013).
Gaguancela, O. A. et al. The IRE1/bZIP60 pathway and bax inhibitor 1 suppress systemic accumulation of potyviruses and potexviruses in arabidopsis and nicotiana benthamiana plants. Mol. Plant-Microbe Interact. 29, 1 (2016).
Howe, K. L. et al. Ensembl Genomes 2020-enabling non-vertebrate genomic research. Nucleic Acids Res. 1, 1–7. https://doi.org/10.1093/nar/gkz890 (2019).
Almagro Armenteros, J. J., Sønderby, C. K., Sønderby, S. K., Nielsen, H. & Winther, O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 33, 3387–3395 (2017).
Gouy, M., Guindon, S. & Gascuel, O. SeaView Version 4: a multiplatform graphical user Interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
An upgraded gene feature visualization server. Hu, B. et al. GSDS 2.0. Bioinformatics 31, 1296–1297 (2015).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. https://doi.org/10.1093/nar/gkv007 (2015).
Saeed, A. I. et al. [9] TM4 microarray software suite. Methods Enzymol. https://doi.org/10.1016/S0076-6879(06)11009-5 (2006).
Rasband, W. S. ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA. https://imagej.nih.gov/ij/ (2014).
Higo, K., Ugawa, Y., Iwamoto, M. & Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27, 297–300 (1999).
Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 9, 326–332 (2008).
Matys, V. TRANSFAC(R) and its module TRANSCompel(R): transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110 (2006).
Acknowledgements
This work was supported by a grant from NSF (IOS #1759034). Additional support came from the Oklahoma Agriculture Experiment Station (OKL02961) to Rita K Miller and Texas A&M Agrilife to Jeanmarie Verchot.
Author information
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herath, V., Gayral, M., Adhikari, N. et al. Genome-wide identification and characterization of Solanum tuberosum BiP genes reveal the role of the promoter architecture in BiP gene diversity. Sci Rep 10, 11327 (2020). https://doi.org/10.1038/s41598-020-68407-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68407-2
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
