
Abstract
Tench (Tinca tinca L.) is one of the most valued species of the Cyprinidae. This species is commercially important and has been intensively domesticated in recent years. To avoid excessive production losses, the health of farm fish must be maintained. Characterization of the tench gut microbiome can help achieve this goal, as the gastrointestinal microbiome plays an important role in host health. As part of this characterization, investigating the influence of the environment and season will help to understand the interrelationship between host and gut microbiota. Therefore, our aim was to use high-throughput 16S rRNA gene amplicon sequencing to profile the gut microbiome of tench. We studied two populations in summer and autumn: wild tench living in a lake and tench living in a pond in a semi-intensive fish farm. We found that, in the gut microbiome of all fish, the most abundant phylum was Proteobacteria, followed by Firmicutes, Bacteroidetes and Actinobacteria. Together, these phyla constituted up to 90% of the microbial communities. The abundance of Candidatus Xiphinematobacter differed significantly between lake and pond fish in summer, but not in autumn. In pond tench, Methylobacterium abundance was significantly lower in summer than in autumn. Mean Shannon, Chao1 indices and observed OTU’s indicated that microbial biodiversity was greater in the gut of lake fish than in that of pond fish. Beta-diversity analysis showed significant divergence between groups with both weighted and unweighted UniFrac distance matrices. Principal coordinates analysis revealed that more of the variance in microbial diversity was attributable to environment than to season. Although some of the diversity in lake tench gut microbiota could be attributable to feeding preferences of individual fish, our results suggest that environment is the main factor in determining gut microbiome diversity in tench.
Similar content being viewed by others


Introduction
Tench (Tinca tinca L.) is one of the most valued species of Cyprinidae fish and has great commercial importance1. Tench is generally considered to be one of the original European cyprinid species, which most likely evolved from primitive Tertiary Paleoleuciscus in large lake systems of Central Europe2. This species plays important roles in the environment, local economies and research. Tench provide many benefits to their ecosystems, effectively preventing blooms of algae, and recirculating minerals and nutrients that are deposited on the bottom of lakes and streams when they stir up the muddy bottoms in search of food3. This behavior helps reduce eutrophication. In Europe, tench are utilized as food, as ornamental fish and for leisure purposes such as angling, and have been used as an indicator of water quality in the context of fish assemblage2. In several European countries, tench have been reared in farm ponds, either in monoculture or alongside common carp (Cyprinus carpio L.). In recent years, tench have undergone intensive domestication, similar to that of common carp centuries ago. Due to their flavor, the production of tench for consumers is increasing, which may, in the near future, lead to rearing of this species in recirculating aquaculture systems4.
Over the past decades, research has shown that the gut microbiota play a key role in the health and nutrition of the host5,6,7. Fish gut microbiota contribute to digestion and can affect growth, reproduction, overall population dynamics and the vulnerability of the host fish to disease8. Much research involving comparative analysis of the microbiome of fish of the same species has shown that factors like environment and diet are two of many factors that influence the structure of the fish gut microbiota9,10. These factors may affect the microbiome by changing the relative abundance of individual groups of microorganisms. These changes can have repercussions on physiological, hormonal or cellular functions, which can result in the development of diseases. Research on the microbiome is particularly valuable in the domestication of fish because it reveals differences between the structure of microorganisms colonizing the digestive tract of wild animals and that of domesticated animals. This information can be used to properly compose feed and enrich it with appropriate probiotics and other supplements. Because the fish-gut microbiome is important for host health, it is generally accepted that identification of the gastrointestinal microbiota is undoubtedly important for understanding the functional interactions between the microbes and the host11. Although there have been some studies on the gastrointestinal microbiome of various fish species10,12,13, to the best of our knowledge, there is a lack of studies on the gut microbiome of tench (T. tinca L.).
Recently, thanks to access to mass sequencing techniques, it has become possible to get detailed information about the structure of the gut microbiome and its changes. Therefore, in this work we characterized the gastrointestinal microbiome of tench (T. tinca L.) using a mass sequencing approach based on gene coding for 16S rRNA. We compared the microbiomes of the gastrointestinal tracts of tench from a semi-intensive farm (pond) and those from a natural body of water (lake). Moreover, we examined the influence of seasonality on the fish gut microbiome by analysis of fish caught in Autumn and Summer. Information concerning the influence of habitat and seasonality on the fish gut microbiome may help to understand how the microbiome is affected by these factors, which microorganisms are dominant and which are most beneficial for the host. Moreover, it can be used to enhance the economic benefits of aquaculture by supplementing the feed of farmed fish species with the necessary probiotics.
Methods
Fish and rearing conditions
This study was carried out on fish caught from a natural body of water – Kortowskie Lake and Pond. Kortowskie Lake is a flow lake with an area of 89.7 ha located in the south-western part of Olsztyn (N 53 °45′43″ E 20 °26′44″). The maximum depth of Kortowskie lake is 17.2 m with an average depth 5.2 m. The location of the pond from which fish were obtained is N 53 °57′46″ E 21 °5′47″. This pond is a mid-forest body of water with an area of 4 ha, a maximum depth of 2 m and an average depth of 1.1 m. In the pond, fry of the following species were co-cultured: tench (T. tinca L.), common crucian (Carassius carassius L.), carp (Cyprinus carpio L.) and grass carp (Ctenopharyngodon idella). In the growing season (May–September), the fish were fed twice a week with a mixture of grain (wheat, barley and maize) that weighed approx. 10% of the stocking weight.
Sampling
In autumn 2017 and summer 2018, 25 farm fish from the pond and 38 wild fish from the lake were randomly caught. Next, fish were weighed and measured. The final mean body weight and body length are shown in Table 1. After measurements, fish were stunned and decapitated quickly and correctly. The ventral body surface was wiped with a paper towel to remove excess mucus. All instruments, surfaces and the exterior of each fish were treated with 70% ethanol to sanitize the skin surface, and instruments were flame-sterilized prior to dissection. The ventral body surface was dried with a paper towel to remove any remaining ethanol. After opening the body cavity, the entire gastrointestinal tract and its contents were aseptically removed from each individual fish. The gut content was obtained by squeezing it into sterile tubes, after which it was stored at −20 °C until analysis.
An ethics statement is not required for this type of research. No specific permissions were required for the described studies. The habitats of the fish are not protected in any way and they do not contain endangered or protected species. Fish from Kortowskie Lake were caught with the permission of the local authorities (Faculty of Environmental Sciences, University of Warmia and Mazury in Olsztyn). Fish from the pond were captured with the permission of co-author KK, an owner of the pond.
DNA extraction
For isolation, we weighed around 100 mg gut content of each fish. Before isolation, samples of gastrointestinal contents were homogenized using plastic spatulas. Metagenomic DNA extractions were then performed with a QIAmp DNA Stool Mini Kit (Qiagen, Germany) following the manufacturer’s instructions. Qubit 2·0 Fluorometer (Invitrogen, Poland) was used to obtain accurate DNA quantification. The integrity of each DNA sample was assessed using 1% agarose gel electrophoresis. The purified DNA was suspended in 60 𝜇l of elution buffer and stored at −20 °C.
16S rRNA gene amplicon library preparation and sequencing
To check the quality and to be sure that the isolated DNA belongs to bacteria, we performed a PCR reaction using two universal 16S rRNA primers: 8 F and 534R14. Based on the results from PCR amplification and DNA yield we choose 32 samples from four groups (8 per group: Lake_Autumn_2017; Pond_Autumn_2017; Lake_Summer_2018; Pond_Summer_2018) for mass sequencing of 16S rRNA amplicons, which was done by an outside company (Genomed S.A, Poland). The Illumina protocol “16S Metagenomic Sequencing Library Preparation” was applied to prepare the 16S rRNA gene amplicons for the Illumina MiSeq system. The variable V3 and V4 regions of the 16S rRNA gene were amplified from bacterial DNA obtained from fish gut content samples. The PCR reactions were performed using 16S rRNA forward (5′ CCTACGGGNGGCWGCAG 3′) and reverse primers (5′ GACTACHVGGTATCTAATCC 3′) that were given by Klindworth et al.15. PCR amplification was performed accordingly to Illumina protocol. Amplicons were indexed using Nextera®XTIndex Kit accordingly to producer’s instructions. DNA was sequenced on an Illumina MiSeq instrument using 2 × 250 paired-end protocol. For sequencing, a Miseq Reagent Kit v3 (Illumina, San Diego, USA) was used for library sequencing.
Sequencing data analysis
Raw paired-end sequences (3,620,064 reads from 32 samples) were processed using the QIIME 216 software package (https://qiime2.org; version: 2018.8). Using Qiime 2 software, paired-end sequences were merged. This step reduced the 3,620,064 reads to 3,383,070 reads. Reads that software could not merge were excluded from further analyses. Next the data sequences underwent quality control using Deblur plugin in Qiime 217. Deblur uses sequence error profiles to associate erroneous sequence reads with the true biological sequence from which they are derived, resulting in high quality sequence variant data. Quality control was performed in two steps. First, an initial filtering process quality score (q = 20) was applied18. This step removed 539,889 reads (15.96% data). Second, Deblur workflow was applied. In this step based on the median quality score, all reads were trimmed to 435 bp length. Also chimeric sequences were detected and excluded from analyses. 16S rRNA OTUs were picked from the Illumina reads using a closed-reference OTU picking protocol against the Greengenes database (https://docs.qiime2.org/2018.8/data-resources; data files: 13_8) clustered at 97% identity and trimmed to span only the 16S rRNA V4 region flanked by sequencing primers 515F-806R. Taxonomy assignments were associated with OTUs based on the taxonomy associated with the Greengenes reference sequence defining each OTU. Out of the 283,854 Illumina reads from the V4 region of the bacterial 16S rRNA genes that passed the QIIME quality filters, 53.3% (151,382 reads) matched a reference sequence at 97% nucleotide sequence identity. Next, OTU counts were binned into genus-level taxonomic groups for plots preparation.
Sequencing data were exported as individual fastq files and have been deposited in Sequence Read Archive (SRA) NCBI (https://www.ncbi.nlm.nih.gov/) as Bioproject under the accession code: PRJNA542255.
Statistical analysis
Normality and homogeneity of variance of all weight and length data of fish were checked by Shapiro-Wilk’s and Levene’s test, respectively, using STATISTICA v.13.1 (StatSoft, Inc). To get a reliable statistical analysis, we rejected 4 samples with less than 1483 reads (3 from Lake_Autumn_2017 and 1 Pond_Autumn_2017) from the analysis due to the low number of reads assigned to taxon levels. The number of reads across samples was normalized by sample size and the relative abundance (%) of each taxon was calculated. All taxa present in the gut microbiome were considered for statistical analysis. Statistical analysis of intestinal microbial profiles was performed using the Statistical Analysis of Metagenomics Profiles (STAMP) program (http://kiwi.cs.dal.ca/Software/STAMP), retaining unclassified reads19. P-values were calculated by ANOVA followed by Tukey-Kramer post-hoc test and corrected for multiple comparisons using the Benjamini-Hochberg method for a False Discovery Rate (FDR) of 5%20. Furthermore, t-tests was used to check statistical differences between fish with extreme values of body weight and length in each group.
Alpha- and beta-diversity statistics were performed using QIIME 2 scripts diversity plugin, which supports metrics for calculating and exploring community alpha- and beta-diversity through statistics and visualizations in the context of sample metadata. In the calculation of alpha-diversity metrics, the normalization was performed using the “rarefaction” QIIME 2 process with standard parameters setting the max_rare_depth (upper limit of rarefaction depths) to mean sample size. Alpha-diversity metrics were calculated using ‘observed species’, ‘Chao1 index’ (species richness estimator), ‘Shannon’s diversity index’ and ‘Good’s coverage’. The alpha-diversity values at the same rarefaction level were calculated.
Differences in the beta-diversity of bacterial communities were verified using nonparametric Permutational Multivariate Analysis of Variance (PERMANOVA) test with 999 permutations. A pairwise significance test was also performed comparing groups from different sampling times and environments using the same distance matrix metrics (weighted and unweighted UniFrac distances). These tests were available in QIIME 2.
Beta-diversity metrics are an estimate of between-sample diversity of microbial profile and they were calculated by QIIME 2 “diversity beta-group-significance” script. We used both weighted (presence/absence/abundance matrix) and unweighted (presence/absence matrix) UniFrac distances21,22. The distance matrices were graphically visualized by three-dimensional PCoA representations.
Results
Qiime analysis of sequencing data
The sequence fastq files from the Illumina MiSeq were analyzed using QIIME 2 software. After filtering for quality, trimming length, and assigning taxonomies, the number of reads taxonomically classified according to the Greengenes database was 150,761 (Table 2). This value corresponded to an average number of 5384 ± 3298.33 per sample (range 1483–14,984). We identified 587 OTUs at 97% nucleotide sequence identity in tench gut content samples. After rarefaction, normalizing to the sample with the mean number of reads (4500), the observed species number per sample was between 7 and 89, corresponding to average number of counts per group between 31 and 49 (Table 2). Good’s coverage values for all groups were ≥ 0.99, indicating that sequencing coverage was attained and that the OTUs found in the samples were representative of the sampled population (Table 2). Although we observed that the mean values of observed species as well as Shannon diversity index (which reflects both the abundance and evenness of the species present) differed between environments (Table 2), these differences were not statistically significant. However, analyzing statistically the Chao1 index of the examined groups, we found that environment was a factor which affected species richness (P = 0.000036). Seasonality did not significantly affect the indices used in this study.
A Venn diagram (Fig. 1) was constructed to visualize OTUs common to each group of fish and present in the gut microbial community regardless of investigated factors and to find those unique to each specific group. The number of common OTUs presented in all groups was 26 and unique OTUs for each group varied from 14 to 55 (Fig. 1). List of the common and unique OTUs present in groups was reported in Supplementary Material (Table S1).

Numbers of common and unique OTUs present in the four groups of tench.
Gut microbiome of tench
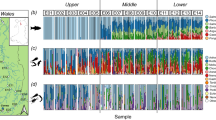
The gut microbiome of 32 fish representing four groups divided depending on environment and season of sampling were examined to characterize their structure and to reveal the differences between them. We successfully described the microbiome structure of each investigated group of fish at the phylum and genus level, and classified 13 phyla and 125 genera. The gut microbial communities of each group and of individual fish are presented at the phylum (Fig. 2) and genus (Fig. 3) levels.

Relative abundance (%) of the overall most prevalent phyla in the different tench groups (a) and in individual fish (b). In the figures, all bacteria with an overall abundance >0,5% were reported. Bacteria with an abundance less than 0,5% were pooled and indicated as “Others”.

Relative abundance (%) of the overall most prevalent genera in the different tench groups (a) and in individual fish (b). In the figures, all bacteria with an overall abundance >5% were reported. Bacteria with an abundance less than 5% were pooled and indicated as “Others”.
At the phylum level in each group, gut microbiota was dominated by the members of Proteobacteria, abundance on the mean level from 57.61% ±32.57 in Lake_Autumn_2017 to 83.49% ±11.90 in Lake_Summer_2018 (Fig. 2a). Firmicutes (5.65% ±7.95 to 21.72% ±22.89); Actinobacteria (0.86% ±0.72 to 9.71% ±11.22); Bacteroidetes 0.73% ±1.17 to 8.81% ±8.92) were less abundant (Fig. 2a). The bacterial community composition in individual fish was mostly similar to the mean phylum abundance in groups, except for fish 2LH17, 1LJ17, 4LJ17, 11LJ17, 8LH18, in which the gut microbiome was dominated by Firmicutes (45.10%, 44.06%, 60.55%, 61.40%, 40.52%, respectively) (Fig. 2b).
At the genus level, the gut microbiota of fish caught in summer 2018 was dominated by the members of Aeromonas, especially in Lake_Summer_2018 group (mean abundance of 62.61% ±20.25), where this genus dominated the gut microbiota of each fish (Fig. 3a,b). In contrast, in the fish catch in autumn, this genus was less abundant (from 10.05% ±16.05 in Pond_Autumn_2017 to 17.38% ±19.8 in Lake_Autumn_2017 (Fig. 3a). The gut microbiota of Pond_Autumn_2017 was dominated by the members of Methylobacterium, with a mean abundance of 38.88% ±20.6 of all bacteria at genus level, whereas in the other groups, we either did not observe any Methylobacterium, or in Pond_Summer_2018, we observed only a small amount (0.09% ±0.25) (Fig. 3a). In contrast, no genus clearly predominated in Lake_Autumn_2017. It is worth noting that, in fish 4LH18, 5LH18, 6LH18 and 9LJ18, we observed higher percent abundance of Eikenella (18.7%; 35.7%; 22;7%; 58.8% respectively), whereas in other fish they were less abundant, below 3%.
There were statistically significant differences between groups in terms of the abundance of some bacteria. Table 3 presents the relative abundance of phyla and genera that differed significantly between groups. The mean abundance of Methylobacterium differed significantly between groups (P = 0.00003), due to the high content of these bacteria in Pond_Autumn_2017. Also, the mean abundance of the genus Candidatus Xiphinematobacter differed significantly between groups (P = 0.00007), which made up a large portion of Verrucomicrobia, particularly in Lake_Summer_2018 (P = 0.02). The difference in mean abundance of unclassified Xanthomonadaceae was also significant (P = 0.04). Statistical analysis of all taxa were reported in Supplementary Table S2.
The results of post hoc multiple comparisons are shown in Table 4. Significant statistical differences of mean abundance of taxa were observed only between Pond_Summer_18 vs Lake_Summer_18 and Pond_Autumn_17 vs Pond_Summer_18 (Table 4).
QIIME 2 was used to compute microbial beta-diversity metrics. Analyses were performed using weighted and unweighted UniFrac distances. Data of UniFrac metrics was used to prepare three-dimensional plots using principal coordinates analysis (PCoA) (Fig. 4). Weighted and unweighted PCoA showed that all samples in Pond_Autumn_2017 were clustered together, and all of those from Lake_Summer_2018 were in a separate cluster. Most of samples in Lake_Autumn_2017 and Pond_Summer_2018 were scattered, nonetheless some of them were more clustered with Lake_Summer_2018 group (Fig. 4a,d). The PCoA plots suggest that both factors may have influenced the fish gut microbiome in all examined groups. However, environment seems to affected more on the fish gut microbiome than season. In Fig. 4(b,e) groups from the same environments clustered together more than dots on the season plots (Fig. 4c,f), where samples were less clustered and were scattered.

Beta-diversity metrics. Principal coordinate analysis (PCoA) of Unweighted (a–c) and Weighted (d–f) Unifrac distances of gut microbial communities associated to different environment and season. The figures show the plot of individual fish according to their microbial profile.
The statistical analysis (permutation multivariate analysis PERMANOVA) reflected the PCoA plot results, indicating a significant divergence between groups for both weighted (P = 0.001; Pseudo-F = 4.71) and unweighted (P = 0.001; Pseudo-F = 3.37) UniFrac distance matrices (Table 5). Result of pairwise test on unweighted and weighted UniFrac data revealed, that all group were significantly diverged (Table 5).
Discussion
Recent studies of the fish-gut microbiota of many species have focused on examining how environmental factors (e.g. diet, habitat) affect microbial communities and how changes in these factors influence the gut microbiome23,24. Since environmental factors are thought to play a role in shaping the intestinal microbiota of fish, we hypothesized that the gut microbiota of tench living in a semi-intensive pond differs from that of tench living in a lake, and that the gut microbiota will change across the seasons. Our results provide information on the gut microbiota of tench and highlight associations between environmental factors and gut microbiota. An understanding of these associations provides information that may be useful for addressing problems during the domestication of these valuable freshwater fish.
Our study indicated that, in the tench gut microbiome, the phyla Proteobacteria and Firmicutes predominate, followed by lesser percentages of Bacteroidetes and Actinobacteria (Fig. 2). This observation suggests that these phyla play valuable roles in the health and digestion of tench, based on three lines of reasoning. First, it is established that specific bacteria species in the gut microbiota are important for health and digestion25. For example, Ray et al.26 report numerous examples of amylase, protease-, lipase-, chitinase-, cellulose and phytase-producing bacteria isolated from the gastrointestinal tract of fish. Second, although it is difficult to estimate the contribution of specific bacteria to the function of the whole gut ecosystem, it is reasonable to expect that the overall gut microbiome will be strongly influenced by the predominant microorganisms27. Third, the presence of similar bacterial taxa in the gut microbiota of multiple fish species suggests that these bacteria are valuable for the host and could play important roles in digestion, nutrient absorption and immune response8. Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria have been found in the intestines of many marine and freshwater fish species9,10,28,29,30 and other Cyprinidae species31. Moreover, a number of studies of fish have found that these phyla are present in proportions similar to those in our study24,32,33. Thus, our results suggest that Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria may play important roles in the health and digestion of tench.
The structure of the gut microbiome differs between many species of fish. This mainly depends on external factors. However, some species might be unique to different hosts due to evolutionary factors and host genetics. Figure 1 presents 26 common OTUs that were always present in the tench gut microbiome regardless of changes in external conditions. Most of them belonged to Proteobacteria and Firmicutes (Table S1), which is consistent with other results of this study. One genus found in almost all examined fish was Aeromonas. A study conducted on another Cyprinidae species (grass carp) showed that Aeromonas dominated the gut microbiome34. Although their percent abundance was lower, Clostridium and Bacteroidetes were also present in all examined groups; these taxa have also been found in other herbivorous carps31. It is interesting to note that all these bacteria are widely known as cellulose-degrading bacteria, which are particularly important for food degradation in the gut of herbivores31. Thus, these findings suggest that these bacteria co-create the main stem of the Cyprinidae microbiome and are necessary to break down nutrients in digestive-tract contents.
The environment is one of the factors that affects the gut microbiota of fish35, and in our study, beta-diversity analysis based on the PERMANOVA test found statistically significant differences between groups from different environments (Table 5). This finding is consistent with that of a previous study on freshwater and marine fish7. In addition, differences in gut microbiota in various water habitats were observed in other Cyprinidae species related to tench, like grass carp, common carp36, silver carp (Hypophthalmichthys molitrix) and bighead carp (Hypophthalmichthys nobilis)9. In our study, there were statistically significant differences between the Pond_Summer_2018 and Lake_Summer_2018 groups at alpha-diversity level in terms of the percent abundance of Candidatus Xiphinematibacter, a representative of Verrucomicrobia (Table 4). Although the percent abundance of these bacteria was low, it was greater in the wild fish guts (Supplementary Table S2). It is possible that greater percent abundance of these bacteria in wild fish may be a result of the more diverse food which can be found in their habitat. The diet of the pond tench was supplemented with a mixture of grain, whereas the lake tench could eat their natural food, which might be insect larvae, small crustaceans or small snails37. Tench also eat dead plants and animals. Despite these differences in diet, alpha-diversity did not show statistically significant differences in percent abundance between Pond_Autumn_17 and Lake_Autumn_17. This observation can be explained by availability of food in lake while autumn, which was worse in contrast to summer and thus, diet was poorer and the biodiversity of gut microbiota was lower (See Shannon and Chao1 index Table 2). It is also worth adding that cause of these differences may be the result of the activity of the tench, which decreases in autumn, so that they need less food than in summer. On the other hand, as can be seen in Table 2, the number of OTUs and the biodiversity of the gut microbiota were usually higher in wild fish from the lake than in the farmed pond-fish. A previous study by Alaş et al.38 showed that the gut microbiome of tench varies depending on the season. All these findings suggest that the microbiome of wild fish is more complex due to the wider variety of food available in the lake.
Season is one of the factors that can shift the gut microbiota. Several reviews have indicated that seasonal variation and temperature changes influence fish-gut microbial composition39,40. Changes in total bacterial abundance of gut microbiota have been reported between summer and autumn periods41. Hagi et al.42 reported that the intestinal lactic acid bacteria composition of four Cyprinidae varied with seasons. It was revealed that abundance of lactic acid bacteria depended on the water temperature. Differences due to season were also apparent in largemouth bass (Micropterus salmoides) and spotted gar (Lepisosteus oculatus)43. However, in that study, seasonal changes were greater on the skin microbiome, suggesting that the gut is able to harbor a relatively stable community composition despite seasonal influences44. Our experiments are consistent with previous studies indicating that season is a factor which influences the gut microbiome. Although we did not observed any changes in abundance of lactic acid bacteria, it is interesting to note that beta-diversity analysis showed that season significantly affected on the composition of the gut microbiota (Table 5). This difference was mainly due to statistically significant changes in the percent abundant of Methylobacterium (P = 0.00003) belonging to Methylobacteriaceae (Table 4). Methylobacteriaceae members were previously detected in other studies45, which indicate that this family of bacteria is a part of the fish gut microbiome. Although Larsen et al.45 found that Methylobacteriaceae comprised below 1% of the entire gut microbiome in three species, our study showed that the abundance of Methylobacterium could be almost 40% (Pond_Autumn_17 group, Fig. 3, Supplementary Table S2). The reason for the unexpectedly high percent abundant of Methylobacteriaceae is not clear, but it might be related to temperature of water. According to Givens (2007)46, higher temperature could increase the abundance of Methylobacteriaceae in the gut microbiome of Lagodon rhomboids. These findings are in contrast to our results, which showed a higher amount of Methylobacteriaceae in Autumn, when the temperature of water was lower than in Summer. Thus, the influence of water temperature should be interpreted with caution. Zarkasi et al.47, in their study on the influence of season on gut microbiota composition, suggested that the gut microbiome was not affected directly by water temperature, but rather by diet.
It could be hypothesized that factors like body weight and length may also affect the fish gut microbiota and influence its composition and diversity, similarly to the factors described in this paper. To exclude the influence of these factors, we took the extreme values of body weight and length in each group and analyze them. We did not found statistically significant differences between the gut microbiome composition of these fish, which indicates that the observed differences are due to the analyzed factors. However, because we analyzed fry from a pond and wild fish at various stages of development, ontogeny might also have influenced the fish gut microbiota, as has been previously reported48.
Conclusions
To sum up, our research provides the first detailed description of the microbiome structure of tench (T. tinca L.) from different habitats. Alpha-diversity analysis based on the metrics used in our study found no statistically significant differences between the gut microbiome compositions of fish from different seasons. Only the environment had a significant effect on species richness. Analysis of percent abundance showed some significant differences between groups with respect to Methylobacterium and Candidatus Xiphinematibacter. Beta-diversity analysis on unweighted and weighted UniFrac showed statistically significant differences between each pair of groups that were compared. Both seasonality and environment had a significant influence on the microbiome structure. However, the environment had a stronger effect than seasonality, as is clearly shown in Fig. 4, where samples are clustered more tightly when classified by environment and less tightly when classified by season. However, the results of this study should be treated with caution. It cannot be excluded that ontogeny had an effect on gut microbiota structure. Nevertheless, future interspecific studies with large representative samples of individuals from different geographical areas and habitat types are needed to precisely define the role of these factors in shaping the gut microbial composition. Studies on fish gut microbiota may help to improve the welfare of fish and aquaculture practices. Future research on the specific functional role of these microorganisms within the tench gut microbiome is needed.
Data availability
All fastq files obtained from sequencing are available from the NCBI Sequence Read Archive (SRA) repository, (https://www.ncbi.nlm.nih.gov/sra; Bioproject accession number PRJNA542255). Rest relevant data are within the manuscript and its Supplementary Files.
References
Wang, J. et al. Tench farming in China: present status and future Prospects. Aquaculture international 14, 205–208 (2006).
Svobodova, Z. & Kolarova, J. A review of the diseases and contaminant related mortalities of tench (Tinca tinca L.). Veterinární Medicína 49, 19–34 (2004).
Benzer, S. S., Gul, A. & Yilmaz, M. Growth properties of tench (Tinca tinca L., 1758) living in Kapulukaya Dam Lake. Kastamonu Education Journal 18, 839–849 (2010).
Demska-Zakęś, K., Zakęś, Z., Ziomek, E. & Jarmołowicz, S. Impact of feeding juvenile tench (Tinca tinca (L.)) feeds supplemented with vegetable oils on hematological indexes and liver histology. Archives of Polish Fisheries 20, 67–75 (2012).
Brugman, S. & Nieuwenhuis, E. E. S. Mucosal control of the intestinal microbial community. Journal of Molecular Medicine 88, 881–8 (2010).
Viney, M. E. & Riley, E. M. From immunology to eco-immu-nology: more than a new name in Eco-immunology (eds. Malagoli, D. & Ottaviani, E.) 1–19 (Springer, 2014).
Wang, A. R., Ran, C., Ringø, E. & Zhou, Z. G. Progress in fish gastrointestinal microbiota research. Reviews in Aquaculture 10, 626–40 (2018).
Ghanbari, M., Kneifel, W. & Domig, K. J. A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 448, 464–75 (2015).
Eichmiller, J. J., Hamilton, M. J., Staley, C., Sadowsky, M. J. & Sorensen, P. W. Environment shapes the fecal microbiome of invasive carp species. Microbiome 4, 1 (2016).
Hennersdorf, P. et al. Microbial Diversity and Parasitic Load in Tropical Fish of Different Environmental Conditions. PLOS ONE 11, 3 (2016).
Dulski, T., Zakęś, Z. & Ciesielski, S. Characterization of the gut microbiota in early life stages of pikeperch Sander lucioperca. Journal of Fish Biology 92, 94–104 (2018).
Michl, S. C. et al. The malleable gut microbiome of juvenile rainbow trout (Oncorhynchus mykiss): Diet-dependent shifts of bacterial community structures. PLOS ONE 12, 5 (2017).
Rimoldi, S., Terova, G., Ascione, C., Giannico, R. & Brambilla, F. Next generation sequencing for gut microbiome characterization in rainbow trout (Oncorhynchus mykiss) fed animal by-product meals as an alternative to fishmeal protein sources. PLOS ONE 13, 3 (2018).
Nossa, C. W. et al. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J Gastroenterol. 16, 4135–44 (2010).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research 41, 1 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–6 (2010).
Amir, A. et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 25, 2 (2017).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature Methods 10, 57–9 (2013).
Parks, D. H. & Beiko, R. G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–21 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 57, 289–300 (1995).
Lozupone, C. & Knight, R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Applied and Environmental Microbiology 71, 8228–35 (2005).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and Qualitative Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Applied and Environmental Microbiology 73, 1576–85 (2007).
Roeselers, G. et al. Evidence for a core gut microbiota in the zebrafish. ISME Journal 5, 1595–608 (2011).
Sullam, K. E. et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Molecular Ecology 21, 3363–78 (2012).
Jandhyala, S. M. Role of the normal gut microbiota. World Journal of Gastroenterology 21, 8787 (2015).
Ray, A. K., Ghosh, K. & Ringø, E. Enzyme-producing bacteria isolated from fish gut: a review. Aquaculture Nutrition 18, 465–92 (2012).
Romero, J., Ringø, E. & Merrifield, D. L. The Gut Microbiota of Fish in Aquaculture Nutrition: Gut Health, Probiotics and Prebiotics (eds. Merrifield, D. & Ringø, E.) 75–100 (Wiley-Blackwell, 2014).
Li, J. et al. Comparative study on gastrointestinal microbiota of eight fish species with different feeding habits. Journal of Applied Microbiology 117, 1750–60 (2014).
Liu, H. et al. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Scientific Reports 6, 1 (2016).
Llewellyn, M. S., Boutin, S., Hoseinifar, S. H. & Derome, N. Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Frontiers in Microbiology 2, 5 (2014).
Li, T. et al. Multi-Omics analysis reveals a correlation between the host phylogeny, gut microbiota and metabolite profiles in cyprinid fishes. Frontiers in Microbiology 8 (2017).
Xia, J. H. et al. The intestinal microbiome of fish under starvation. BMC Genomics 15, 266 (2014).
Sevellec, M. et al. Microbiome investigation in the ecological speciation context of lake whitefish (Coregonus clupeaformis) using next-generation sequencing. Journal of Evolutionary Biology 27, 1029–46 (2014).
Li, H. et al. Isolation of cellulose—producing microbes from the intestine of grass carp (Ctenopharyngodon idellus). Environmental Biology of Fishes 86, 131–135 (2009).
Egerton, S., Culloty, S., Whooley, J., Stanton, C., Ross, R. P. The Gut Microbiota of Marine Fish. Frontiers in Microbiology 9 (2018).
Ni, J., Yan, Q., Yuhe, Y. & Zhang, T. Factors Influencing the Grass Carp Gut Microbiome and Its Effect on Metabolism. FEMS Microbiology Ecology 87, 704–14 (2014).
Kennedy, M. & Fitzmaurice, P. The biology of the tench Tinca tinca (L.) in Irish Waters. Proceedings of the Royal Irish Academy 69, 31–82 (1970).
Alaş, A., Altindağ, A., Yılmaz, M., Kirpik, M. A. & Ak, A. Feeding Habits of Tench (Tinca tinca L., 1758) in Beysehir Lake (Turkey). Turkish Journal of Fisheries and Aquatic Sciences 10, 187–194 (2010).
Nayak, S. K. Role of gastrointestinal microbiota in fish. Aquaculture Research 41, 1553–1573 (2010).
Ringø, E. et al. Effect of dietary components on the gut microbiota of aquatic animals. A never-ending story? Aquaculture Nutrition 22, 219–282 (2016).
Al-Harbi, A. H. & Uddin, M. N. Seasonal variation in the intestinal bacterial flora of hybrid tilapia (Oreochromis niloticus × Oreochromis aureus) cultured in earthen ponds in Saudi Arabia. Aquaculture 229, 37–44 (2004).
Hagi, T., Tanaka, D., Iwamura, Y. & Hoshino, T. Diversity and seasonal changes in lactic acid bacteria in the intestinal tract of cultured freshwater fish. Aquaculture 234, 335–346 (2004).
Ray, C. L. Characterization of the gut and skin microbiomes of wild-caught fishes from Lake Guntersville. Electronic Theses and Dissertations. M.Sc. Thesis, Preprint at, https://etd.auburn.edu/handle/10415/5314 (2016).
Tarnecki, A. M., Burgos, F. A., Ray, C. L. & Arias, C. R. Fish intestinal microbiome: diversity and symbiosis unravelled by metagenomics. Journal of Applied Microbiology 123, 2–17 (2017).
Larsen, A. M., Mohammed, H. H. & Arias, C. R. Characterization of the gut microbiota of three commercially valuable warmwater fish species. Journal of Applied Microbiology 116, 1396–404 (2014).
Givens, C. E. A fish tale: comparison of the gut microbiome of 15 fish species and the influence of diet and temperature on its composition. Preprint at, http://getd.libs.uga.edu/pdfs/givens_carrie_e_201212_phd.pdf (2012).
Zarkasi, K. Z. et al. Pyrosequencing-based characterization of gastrointestinal bacteria of Atlantic salmon (Salmo salar L.) within a commercial mariculture system. Journal of Applied Microbiology 117, 18–27 (2014).
Bledsoe, J. W. et al. Ontogenetic Characterization of the Intestinal Microbiota of Channel Catfish through 16S rRNA Gene Sequencing Reveals Insights on Temporal Shifts and the Influence of Environmental Microbes. PLOS ONE 11 (2016).
Acknowledgements
The study was supported by research grant from National Science Centre, Poland (https://www.ncn.gov.pl/) No. UMO-2017/25/N/NZ9/00064.
Author information
Authors and Affiliations
Contributions
T.D. did the formal analysis of the study, acquired funding and carried out the experiments and drafted the original manuscript. K.K. collected the samples and revised the manuscript. S.C. participated in the study’s design and supervised in study coordination. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dulski, T., Kozłowski, K. & Ciesielski, S. Habitat and seasonality shape the structure of tench (Tinca tinca L.) gut microbiome. Sci Rep 10, 4460 (2020). https://doi.org/10.1038/s41598-020-61351-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61351-1
This article is cited by
-
Gut Microbial Composition of Cyprinella lutrensis (Red Shiner) and Notropis stramineus (Sand Shiner): Insights from Wild Fish Populations
Microbial Ecology (2024)
-
Host-microbiota-parasite interactions in two wild sparid fish species, Diplodus annularis and Oblada melanura (Teleostei, Sparidae) over a year: a pilot study
BMC Microbiology (2023)
-
The impact of culture systems on the gut microbiota and gut metabolome of bighead carp (Hypophthalmichthys nobilis)
Animal Microbiome (2023)
-
Gut microbiome diversity and composition is associated with exploratory behavior in a wild-caught songbird
Animal Microbiome (2023)
-
Host individual and gut location are more important in gut microbiota community composition than temporal variation in the marine herbivorous fish Kyphosus sydneyanus
BMC Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
