
Abstract
Natural products isolation using protein based methods is an attractive for obtaining bioactive compounds. To discover neural stem cell (NSC) differentiation activators, we isolated eight inhibitors of Hes1 dimer formation from Psidium guajava using the Hes1-Hes1 interaction fluorescent plate assay and one inhibitor from Terminalia chebula using the Hes1-immobilized beads method. Of the isolated compounds, gallic acid (8) and 4-O-(4”-O-galloyl-α-L-rhamnopyranosyl)ellagic acid (11) showed potent Hes1 dimer formation inhibitory activity, with IC50 values of 10.3 and 2.53 μM, respectively. Compound 11 accelerated the differentiation activity of C17.2 NSC cells dose dependently, increasing the number of neurons with a 125% increase (5 μM) compared to the control.
Similar content being viewed by others

Introduction
Neurodegenerative diseases such as dementia and spinal cord injury severely compromise the quality of life. The discovery of neural stem cells (NSCs) in human adult brain1,2 led to the expectation of novel treatments involving NSC transplantation or accelerated differentiation of internal NSCs in the brain to overcome neurodegenerative diseases. However, the development of these approaches and small molecule NSC activators has been slow, possibly due to the complex mechanism underlying the differentiation of NSCs to neural cells.
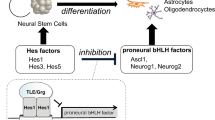
Basic helix-loop-helix (bHLH) factors play crucial roles in the differentiation of neural cells such as neurons, astrocytes and oligodendrocytes3. For example, proneural bHLH factors, such as Ascl1 (formerly Mash1), Neurog1 and Neurog2 regulate the generation of neurons. Olig1 and Olig2 are bHLH factors important for the maturation of oligodendrocytes. Hairy and enhancer of split (hes) bHLH factors promote the self-renewal of NSCs and the generation of astrocytes, as well as inhibiting the differentiation of NSCs by suppressing the expression of proneural genes. Recently, Kageyama et al. reported that oscillation in the concentration of bHLH factors directly controls the fate of NSCs4,5. Hes1 and Ascl1 proteins oscillate with 2- to 3-hour periods, whereas Olig2 oscillates with a 5- to 8-hour period. During cell fate choice, one bHLH factor is expressed in a sustained manner while the others are repressed. In neurons, Ascl1 is expressed sustainably while Hes1 and Olig2 expression is inhibited.
Hes1 dimerizes and binds to the promoter region of Ascl1 and other proneural genes (Fig. 1), making the inhibition of Hes1 an attractive approach to enhance neuronal differentiation. If the interested target protein was decided, it would be faster and effective to isolate the bioactive natural products using a “protein-based assay system”6. However, the number of reports of isolation using protein-based assay system was still limited (affinity chromatography7,8,9, protein-immobilized beads10,11,12,13,14,15,16,17,18, ultrafiltration19,20,21, and protein-protein interactions22,23). In our attempts to find Hes1 inhibitors effectively, we constructed a new Hes1-Hes1 interaction fluorescent plate assay which gave the first Hes1 dimer inhibitors24. Also, we have developed “target protein oriented natural products isolation (TPO-NAPI) methods” using protein-immobilized beads11,15,17,18. Lindbladione15, agalloside15, isomicromonolactam17 and inohanamine17 were isolated by using Hes1 protein beads and accelerated the differentiation of NSCs to neurons. Here, we report the isolation of eight Hes1 dimer inhibitors from Psidium guajava using the Hes1-Hes1 interaction fluorescent plate assay, which is the first example of natural products isolation using fluorescent plate assay of protein-protein interactions (PPIs). Moreover, the isolation using the TPO-NAPI method with Hes1 beads gave a Hes1 dimer inhibitor from Terminalia chebula.

Neural stem cell differentiation and fate control by bHLH factors.
Results and Discussion
It is essential to inhibit undesired PPIs in drug development, and bioactivity-guided isolation using the inhibitory activity of PPIs is an attractive method to obtain effective inhibitors. However, currently there are only a few examples of such approaches using PPI assay systems22,23. We previously developed a Hes1-Hes1 interaction fluorescent plate assay and reported the screening results of our natural products compound library (Fig. 2A)24. Glutathione-S-transferase (GST) fused rat Hes1(3–281) protein was expressed in Escherichia coli and purified rat Hes1 protein was immobilized on the bottom of 96 well plates. We prevented GST-GST interactions, which would result in false positives, by preparing GST-free Hes1 protein by GST cleavage with Turbo3C protease. After labeling Hes1 with Cy3, this florescent Hes1 protein was added to the wells of the above plate and incubated for 24 h at 4 °C. Hes1-Hes1 interaction was successfully detected as Cy3 fluorescence intensity. Using this assay system, we screened our 118 plant extract library and identified the MeOH extract of Psidium guajava leaves to contain naturally occurring compounds that inhibit Hes1 dimer formation. The MeOH extract (29.9 g) was fractionated using Diaion HP-20 with a MeOH-acetone solvent system to afford fractions 1A to 1C. Active fraction 1A (27.2 g) was suspended in 10% aq. MeOH and partitioned with hexane, EtOAc and BuOH to obtain hexane (1.1 g), EtOAc (5.7 g), BuOH (4.7 g) and aqueous (18.9 g) soluble fractions. Part of the active BuOH soluble fraction was subjected to ODS column chromatography and reverse-phase HPLC. Activity-guided separation yielded ten compounds (1–10; Fig. 3). The isolated compounds were identified as morin (1)25, isoquercitrin (2)26, methyl gallate (3)27, (+)-catechin (4)28,29, dihydrophaseic acid (5)30, quercetin (6)26,31, avicularin (7)32,33, gallic acid (8)34, protocatechuic acid (9)35 and 4-hydroxybenzoic acid (10)36 based on comparisons of their spectral data with spectra in the literature. The Hes1-Hes1 interaction inhibitory activities of the isolated compounds were examined (Fig. 4) and 3, 7, 8 and 9 produced moderate inhibition (IC50 12.7, 26.5, 10.3 and 23.8 μM). The most potent inhibitor was gallic acid (8). Commercially available gallic acid also exhibited comparable inhibition (IC50 8.9 μM). Inhibition by the gallic acid derivatives 3, 8, 9 and 10 showed that the number of phenolic hydroxyl groups affects inhibitory activity, with activity decreasing as the number of phenolic hydroxyl groups decrease.

Target protein-oriented isolation methods. (A) Hes1-Hes1 interaction fluorescent plate assay, (B) Hes1 immobilized beads method.

Structures of the isolated compounds.

Hes1 dimer formation inhibitory activities of the isolated compounds.
We recently developed another protein-based screening method, the target protein oriented natural products isolation method (TPO-NAPI) using protein beads (Fig. 2B). Agalloside, inohanamine, α-mangostine, BE-14106, isomicromonolactam, staurosporin and linarin were isolated as Hes1 binding compounds using the TPO-NAPI method15,17. Rat Hes1 (1–95) containing basic and helix-loop-helix domains was immobilized because the helix-loop-helix domain is known to be important for Hes1-Hes1 interaction; therefore, utilizing this domain in the beads method would likely be effective for screening Hes1 dimer inhibitors. GST-Hes1 immobilized beads were prepared by mixing freshly prepared GST-Hes1 protein with glutathione Sepharose 4B beads. GST-only beads were prepared as a control. After incubating the beads with plant MeOH extracts at 4 °C for 2 h, bound compounds were eluted by adding EtOH and heating at 100 °C for 3 min, then the eluted compounds were analyzed by HPLC. Of the 105 plant MeOH extracts screened using this method, the Bangladesh plant Terminalia chebula was found to contain a Hes1 binding compound. The MeOH extract (64.6 g) of Terminalia chebula bark was partitioned with hexane, EtOAc and BuOH to obtain hexane (1.5 g), EtOAc (3.6 g), BuOH (42.6 g), and aqueous (20.5 g) soluble fractions. The EtOAc fraction contained the target peak and was subjected to silica gel column chromatography to give eight fractions (1A-H). Fraction 1D contained the target peak and was separated by ODS column chromatography and reverse-phase HPLC to give compound 11 (0.4 mg). Compound 11 was identified as 4-O-(4″-O-galloyl-α-L-rhamnopyranosyl)ellagic acid by comparison of its NMR data with reported spectral data37. Next, we evaluated the Hes1 dimer inhibitory activity of compound 11 (Fig. 4) and found that it exhibited the most potent inhibition of Hes1 dimer formation of the compounds tested, with an IC50 value of 2.53 μM. This is the strongest Hes1 dimer inhibitor reported to date. The ability of 11 to accelerate the differentiation of C17.2 mouse neural stem cells was evaluated (Fig. 5). C17.2 cells (2 × 105 cells/mL) were seeded in a poly-L-lysine-coated 24 well plates and incubated for 24 h, then the cells were treated with DMSO (control), 100 μM valproic acid (VPA), 5 μM retinoic acid (RA) (positive controls) or 11 (1 or 5 μM) for 6 days. A confocal microscope was used to obtain images of differentiated neural cells after immunostaining class III β-tubulin (Tuj1) in neurons, and glial fibrillary acidic protein (GFAP) in astrocytes and nuclei (TO-PRO-3). The number of neurons increased following the addition of VPA, RA or compound 11, suggesting that these compounds enhanced the number of C17.2 neurons. In contrast, the control dish containing cells treated with DMSO contained small number of neurons. The differentiated neurons with 11 were 33.6% (1 µM) and 45.4% (5 µM), which are 67% and 125% increase compared to those of control. These results suggested that compound 11 accelerates the differentiation of NSCs to neurons by inhibiting Hes1 dimer formation.

Effect of 11 on NSC differentiation into neurons. Green: neurons, blue: nuclei, red: astrocytes. The number of neurons increased following the addition of compound 11 dose dependently. P values were analyzed by Student’s t test. P < 0.05 is considered statistically significant.
We also predicted the interaction between the HLH domain of Hes1 protein and compound 11 (4-O-(4″-O-galloyl-α-L-rhamnopyranosyl)ellagic acid) by performing in silico docking analysis of compound 11 with the HLH domain of Hes1. As shown in Fig. 6A,B, the galloyl site of compound 11 might interact with the loop region of Hes1, aiding the formation of Hes1(Arg46 of helix region)-Hes1(Glu76 of loop region) and preventing mutual recognition by Hes1 molecules. On the other hand, the ellagic acid site of compound 11 might bind with the helix region of Hes1, which consists of Ile50, Leu54 and Leu81, preventing hydrophobic core formation in the Hes1 dimer. Orange shows the hydrophobic region in Hes1 (Fig. 6C). Moreover, hydrogen bond formation between the ellagic acid site of compound 11 with Lys77 might obstruct Hes1(loop region)-Hes1(loop region) formation. Blue shows the hydrophilic region. As shown in Fig. 6C, the interaction of galloyl moiety with the hydrophilic region seems to be important. Therefore, the decrease of inhibition with decrease of number of phenolic hydroxyl groups in galloyl group would be reasonable. In addition, the α-L-rhamnopyranosyl unit appears to be an efficient linker, enabling tight interaction between compound 11 with the Hes1 monomer via its galloyl and ellagic acid sites.

Docking study of compound 11 to the Hes1 HLH domain. (A) NMR structure of the HLH domains of Hes1 dimers (PDB code: 2MH3). Green and pink show individual Hes1 HLH domains. (B–D) Docking study results of 11 with the HLH domain.
Conclusion
In conclusion, several Hes1 binding natural products were isolated using two target protein-oriented isolation methods: a Hes1-Hes1 interaction fluorescent plate assay and a Hes1-immobilized beads method. Ten compounds were isolated from Psidium guajava, of which eight showed Hes1 dimer inhibitory activity. 4-O-(4″-O-Galloyl-α-L-rhamnopyranosyl)ellagic acid (11) isolated using the Hes1 beads method showed potent Hes1 dimer formation inhibitory activity, with an IC50 of 2.53 μM. Compound 11 accelerated the differentiation of C17.2 NSC cells into neurons dose dependently, increasing the number of neurons with a 125% increase (5 μM) compared to the control. There are few reports of Hes1 dimer inhibitors and thus these results will be useful for evaluating Hes1-related mechanisms in cells. We believe that protein-based isolation is an effective approach for identifying bioactive compounds.
Methods
General experimental procedures
NMR spectra were recorded on JEOL ECZ600 spectrometers in a deuterated solvent whose chemical shift was used as an internal standard. Column chromatography was performed using Silica Gel 60N (Kanto Chemical Co., Tokyo, Japan), Chromatorex ODS (Fuji Silysia Chemical Ltd., Kasugai, Japan), Diaion HP-20 (Mitsubishi Chemical Co., Tokyo, Japan) and Sephadex LH-20 (GE Healthcare Japan, Tokyo, Japan). Preparative HPLC was performed using YMC-Pack SIL-06 (YMC Co. Ltd., Kyoto, Japan), COSMOSIL 5C18-AR-II, COSMOSIL Cholester, COSMOSIL πNAP (Nacalai Tesque, Inc. Kyoto, Japan) and CAPCELL PAK C18 MGII (OSAKA SODA Ltd., Osaka, Japan).
Plant materials
Psidium guajava (Myrtaceae) and Terminalia chebula (Combretaceae) were collected in Bangladesh in 2013 and were identified by one of the authors (S. K. Sadhu). Voucher specimens (KKB348 and KKB364) were deposited at the Department of Natural Products Chemistry, Graduate School of Pharmaceutical Sciences, Chiba University.
Plate assay for Hes1 dimer inhibitors
Hes1-bound microplate wells24 (Nunc ImmobilizerTM Amino Plate, Thermo) were incubated with 50 μL of Cy3-labeled-Hes1 in NET-N buffer (NET buffer (20 mm Tris-HCl, pH 7.5, 200 mm NaCl, 1 mm EDTA) containing 0.05% Nonidet® P-40, ca. 7 mg/L, dye/protein = 0.4) for 24 h at 4 °C. After removing the protein solution, the wells were washed twice with 200 μL of PBST, then each compound solution (in NET-N buffer, 50 μL) was added and the plates were incubated for 1 h at RT in the dark. The wells were washed twice with 200 μL of PBST, then dried under reduced pressure for 1 h in the dark. The Cy3 dye was excited at 544 nm and emission was monitored at 590 nm using a microplate reader (Fluoroskan Ascent, Thermo Labsystems, Vantaa, Finland). The assays were typically carried out in three individual wells, and the mean value and SD were calculated. The equilibrium between dimer formation between Cy3-Hes1/Cy3-Hes1 (in solution) and Cy3-Hes1/immobilized-Hes1 allowed the detection of Cy3-Hes1/immobilized-Hes1.
Typical screening procedure for the Hes1 beads method
To prepare GST-Hes1 beads, GST-ratHes11-95 (ca. 3.6 nmol) in PBS was added to pre-washed glutathione Sepharose 4B beads (bed volume 100 μL, GE Healthcare) and mixed at 4 °C for 1 h. The GST-Hes1 beads were washed five times with NET buffer, then suspended in 250 μL NET buffer. A MeOH or EtOAc plant extract (125 μg in EtOH, 25 μL) was added to the above GST-Hes1 freshly prepared beads (bed volume 100 μL) and gently mixed for 2 h at 4 °C. The beads were then washed by tapping the container with fingers three times with NET-N buffer. EtOH (70%, 150 μL) was added to the washed beads and the suspension was heated at 100 °C for 3 min in a heating block. The beads were collected by centrifugation (2000 rpm, 4 °C, 1 min) and the supernatant was centrifuged at 15000 rpm for 15 min. One-third of the supernatant was analyzed by HPLC. Control GST-beads were prepared in the same manner as GST-Hes1 beads by adding GST (ca. 3.8 nmol) in PBS to pre-washed glutathione Sepharose 4B beads (bed volume 100 μL, GE Healthcare). Lindobladione was used as a positive control in each experiment. Obvious differences in peak intensity for the results obtained using GST-Hes1-beads and GST-beads (control) were considered “hit” extracts containing a Hes1 binding natural product.
Extraction and isolation
The MeOH extract (29.9 g) of Psidium guajava (leaves; 139 g) was subjected to Diaion HP-20 (φ 70 × 210 mm) chromatography with a MeOH-acetone solvent system (1:0–0:1) to afford fractions 1A to 1 C. Active fraction 1A (27.2 g) was suspended in 10% aq. MeOH (150 mL) and partitioned with hexane, AcOEt and BuOH (400 mL × 3) to obtain hexane (1.1 g), AcOEt (5.7 g), BuOH (4.7 g) and aqueous (18.9 g) soluble fractions. An aliquot of the active BuOH soluble fraction was subjected to ODS column chromatography (φ 40 × 210 mm; MeOH-H2O = 3:7–1:0) to afford fractions 3A to 3E. Fraction 3B (134.6 mg) was subjected to ODS HPLC (COSMOSIL 5C18-AR-II; 40% MeOH + 0.1% HCOOH, flow rate 3.0 mL/min) to give compound 1 (4.6 mg). An aliquot of fraction 3D (124.5 mg) was subjected to ODS HPLC (COSMOSIL 5C18-AR-II; 40% MeOH + 0.1% HCOOH, flow rate 5.0 mL/min) to give compound 2 (7.3 mg). An aliquot of the active AcOEt soluble fraction was subjected to ODS column chromatography (φ 40 × 210 mm; MeOH-H2O = 3:7–1:0) to afford fractions 2A to 2I. Fraction 2B (236.5 mg) was subjected to ODS column chromatography (φ 20 × 460 mm; MeOH-H2O = 4:6–1:0) to give compound 3 (10.9 mg) and to afford fractions 4A to 4E. Fraction 4 C (32.9 mg) was subjected to silica gel column chromatography (φ 20 × 280 mm; CHCl3-MeOH = 30:1-0:1) to give compound 4 (10.3 mg). Fraction 4A (10.0 mg) was subjected to HPLC (YMC-Pack SIL-06, CHCl3-MeOH = 6:1, flow rate 3.0 mL/min) to give compound 5 (0.3 mg). Fraction 2 H (83.3 mg) was subjected to silica gel column chromatography (φ 15 × 310 mm; AcOEt-MeOH = 150:1–0:1) to give compound 6 (21.6 mg). An aliquot of fraction 2 F (68.7 mg) was subjected to HPLC (YMC-Pack SIL-06, AcOEt-MeOH = 7:1, flow rate 1.0 mL/min) to afford fractions 8 A to 8F. Fraction 8B (12.4 mg) was subjected to Sephadex LH-20 column chromatography (φ 15 × 400 mm; MeOH) to give compound 7 (0.8 mg). An aliquot of fraction 2A (192.6 mg) was subjected to Sephadex LH-20 column chromatography (φ 30 × 420 mm; MeOH) to afford fractions 15A to 15E. Fraction 15B (15.7 mg) was subjected to ODS HPLC (COSMOSIL 5C18-AR-II; 40% MeOH + 0.1% HCOOH, flow rate 3.0 mL/min) to give compound 8 (7.0 mg), compound 9 (2.0 mg) and compound 10 (0.8 mg).
Terminalia chebula (bark; 395 g) MeOH extract was partitioned with hexane, EtOAc and BuOH to obtain hexane (1.5 g), EtOAc (3.6 g), BuOH (42.6 g) and aqueous (20.5 g) soluble fractions. The EtOAc fraction containing the target peak was subjected to silica gel column chromatography (φ 50 × 250 mm; CHCl3-MeOH = 1:0–0:1) to give fractions 1A-H. Fraction 1D contained the target peak and was separated by ODS column chromatography (φ 25 × 200 mm; MeOH-H2O = 7:3–0:1) to give fractions 2A–F. Fraction 2A was subjected to ODS column chromatography (φ 20 × 200 mm; MeOH-H2O = 7:3-0:1) to give fractions 6A–G. Fraction 6E was separated by reverse-phase HPLC (CAPCELL PAK C18 MGII; 50% MeOH) to give compound 11 (0.4 mg). Compound 11 was identified as 4-O-(4″-O-galloyl-α-L-rhamnopyranosyl)ellagic acid by comparison of its NMR data with reported spectral data.
Cell culture
C17.2 cells were cultured in proliferation medium [DMEM (DS Pharma Biomedical Co., Ltd. Osaka, Japan) supplemented with 10% fetal bovine serum (Biowest, Nuaillé, France), 5% horse serum (Gibco, Life Technologies, Tokyo, Japan)]. Neural differentiation of C17.2 cells was initiated by transferring the cells to differentiation medium [Neurobasal Medium (Gibco) supplemented with 2% B-27 without vitamin A (Gibco), 1% antibiotic-antimycotic (Gibco)]. All cultures were maintained in a humidified incubator at 37 °C in 5% CO2/95% air.
Neural stem cell differentiation assay
C17.2 cells were dissociated using the proliferation medium 2 × 105 cells/mL on cover glass (MATSUNAMI GLASS) coated with poly-L-lysine and fibronectin/laminin. After incubation for 24 h, cells were washed with D-MEM and treated with individual compounds in differentiation medium for 6 days.
Immunofluorescence staining
C17.2 cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature (r.t.) and washed with 1% BSA in PBS three times. After blocking with 10% BSA in 0.3% Triton X-100 in PBS for 45 min at r.t., fixed cells were incubated with primary antibodies (anti-βIII-Tubulin; Neuronal Class III, Mouse-Mono, 1:400, R&D SystemsTM; anti-GFAP, 1:400, VERITAS) for 12 h at 4 °C. After washing three times with 1% BSA in PBS (500 μL/well), cells were incubated with secondary antibodies (Alexa Fluor® 488 goat anti-mouse IgG (H + L), 1:400, Life Technologies; Alexa Fluor® 555 goat ant-rabbit IgG (H + L), 1:200, Life Technologies) for 1 hr at r.t. in the dark. After washing three times with 1% BSA in PBS (500 μL/well), cells were treated with 200 μg/mL RNase (Nacalai Tesque) in PBS containing 1% BSA and 0.1% Triton X-100 for 1 hr at 37 °C in the dark. After washing three times with 500 μL/well PBS, cells were incubated in 30 μM TO-PRO®-3, 1% BSA, and 0.1% Triton X-100 in PBS for 10 min at r.t. in the dark. The cover glass on which the cells were attached was transferred onto slide glass and mounted with ProLong® Gold Antifade Reagent with DAPI (Invitrogen). Slide glasses were viewed and photographed with an LSM 780 (Carl Zeiss). Four pictures were taken per well, and the assays were carried out with three individual wells per condition.
In silico analysis
The initial 3D structures of 4-O-(4″-O-galloyl-α-L-rhamnopyranosyl)ellagic acid were constructed using Build in MOE (version 2016; Chemical Computing Group, Montreal, Canada) with standard geometric parameters. The ligand was then minimized using Energy Minimize in MOE with the Amber force field until the root-mean-square (rms) energy gradient was less than 0.001 kcal mol−1Å−1. The protein model was constructed based on the structure of HLH domain taken from the Protein Data Bank (PDB) (PDB code: 2MH3)38. Water molecules in the structure were removed. All hydrogen atoms were added and Amber all-atom charges were assigned for the whole protein. The molecular docking simulations were performed using the Induced Fit methods in MOE-Dock.
References
Eriksson, P. S. et al. Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317 (1998).
Goldman, S. A. Adult neurogenesis: from canaries to the clinic. J. Neurobiol. 36, 267–286 (1998).
Imayoshi, I. & Kageyama, R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 82, 9–23 (2014).
Imayoshi, I. & Kageyama, R. Oscillatory control of bHLH factors in neural progenitors. Trends Neurosci. 37, 531–538 (2014).
Kageyama, R., Shimojo, H. & Ohtsuka, T. Dynamic control of neural stem cells by bHLH factors. Neurosci. Res. 138, 12–18 (2019).
Arai, M. A. & Ishibashi, M. Target protein oriented natural products isolation. Comprehensive Natural Products III: Chemistry and Biology (ed. Saito, K.), Elsevier, in press.
Luo, H., Chen, L., Li, Z., Ding, Z. & Xu, X. Frontal immunoaffinity chromatography with mass spectrometric detection: A method for Finding active compounds from traditional Chinese herbs. Anal. Chem. 75, 3994–3998 (2003).
Rivera, G. S. M., Beamish, C. R. & Wencewicz, T. A. Immobilized FhuD2 siderophore-binding protein enables purification of salmycin sideromycins from Streptomyces violaceus. DSM 8286ACS Infect. Dis. 4, 845–859 (2018).
Wu, W. S., Cheng, W. C., Cheng, T. R. & Wong, C. H. Affinity-based screen for inhibitors of bacterial transglycosylase. J. Am. Chem. Soc. 140, 2752–2755 (2018).
Ohtsu, Y. et al. Selective ligand purification using high-performance affinity beads. Anal. Biochem. 338, 245–252 (2005).
Arai, M. A. et al. A method for the rapid discovery of naturally occurring products by proteins immobilized on magnetic beads and reverse affinity chromatography. Chem. Asian J. 4, 1802–1808 (2009).
Choi, Y. & van Breemen, R. B. Development of a screening assay for ligands to the estrogen receptor based on magnetic microparticles and LC-MS. Comb. Chem. High Throughput Screen 11, 1–6 (2008).
Kang, M. J. et al. Functional chromatography reveals three natural products that target the same protein with distinct mechanisms of action. ChemBioChem 15, 2125–2131 (2014).
Wubshet, S. G., Brighente, I. M. C., Moaddel, R. & Staerk, D. Magnetic ligand fishing as a targeting tool for HPLC-HRMS-SPENMR: α-Glucosidase inhibitory ligands and alkylresorcinol glycosides from Eugenia catharinae. J. Nat. Prod. 78, 2657–2665 (2015).
Arai, M. A. et al. Hes1 inhibitor isolated by target protein oriented natural products isolation (TPO-NAPI) of differentiation activators of neural stem cells. Chem. Sci. 7, 1514–1520 (2016).
Rush, M. D., Walker, E. M., Burton, T. & van Breemen, R. B. Magnetic microbead affinity selection screening (MagMASS) of botanical extracts for inhibitors of 15-lipoxygenase. J. Nat. Prod. 79, 2898–2902 (2016).
Arai, M. A. et al. Hes1 binding compounds isolated by target protein oriented natural products isolation (TPO-NAPI). J. Nat. Prod. 80, 538–543 (2017).
Arai, M. A. et al. GLI1 inhibitors isolated by target protein oriented natural products isolation (TPO-NAPI) with hedgehog inhibition. ACS Chem. Biol. 13, 2551–2559 (2018).
Chen, G. L., Tian, Y. Q., Wu, J. L., Li, N. & Guo, M. Q. Antiproliferative activities of Amaryllidaceae alkaloids from Lycoris radiata targeting DNA topoisomerase I. Sci. Rep. 6, 38284 (2016).
Ma, C. et al. Rapid screening of potential α-amylase inhibitors from Rhodiola rosea by UPLC-DAD-TOF-MS/MS-based metabolomic method. J. Funct. Foods 36, 144–149 (2017).
Wang, L., Liu, Y., Luo, Y., Huang, K. & Wu, Z. Quickly screening for potential α-glucosidase inhibitors from Guava leaves tea by bioaffinity ultrafiltration coupled with HPLC-ESI-TOF/MS method. J. Agric. Food Chem. 66, 1576–1582 (2018).
Hassig, C. A. et al. Ultra-high-throughput screening of natural product extracts to identify proapoptotic inhibitors of Bcl-2 family proteins. J. Biomol. Screen. 19, 1201–1211 (2014).
Arai, M. A. et al. Naturally occurring FANCF-Hes1 complex inhibitors from Wrightia religiosa. Med. Chem. Commun. 6, 455–460 (2015).
Arai, M. A., Masada, A., Ohtsuka, T., Kageyama, R. & Ishibashi, M. The first Hes1 dimer inhibitors from natural products. Bioorg. Med. Chem. Lett. 19, 5778–5781 (2009).
Tachakittirungrod, S., Ikegami, F. & Okonogi, S. Antioxidant active principles isolated from Psidium guajava grown in Thailand. Sci. Pharm. 75, 179–193 (2007).
Park, B. J. et al. Phenolic compounds from the leaves of Psidium guajava II. Quercetin and its glycosides. Chem. Nat. Comp. 48, 477–479 (2012).
Bailey, A. E., Asplund, R. O. & Ali, M. S. J. Nat. Prod. 49, 1149–1150 (1986).
Seto, R., Nakamura, H., Nanjo, F. & Hara, Y. Preparation of epimers of tea catechins by heat treatment. Biosci. Biotech. Biochem. 61, 1434–1439 (1997).
Takagaki, A. & Nanjo, F. Catabolism of (+)-catechin and (−)-epicatechin by rat intestinal microbiota. J. Agric. Food Chem. 61, 4927–4935 (2013).
Zhang, Z. et al. Gynostemosides A–E, megastigmane glycosides from Gynostemma pentaphyllum. Phytochemistry 71, 693–700 (2010).
Metwally, A. M., Omar, A. A., Harraz, F. M. & Sohafy, S. M. Phytochemical investigation and antimicrobial activity of Psidium guajava L. leaves. Pharmacogn. Mag. 6, 212–218 (2010).
Schieber, A., Hilt, P., Conrad, J., Beifuss, U. & Carle, R. Elution order of quercetin glycosides from apple pomace extracts on a new HPLC stationary phase with hydrophilic endcapping. J. Sep. Sci. 25, 361–364 (2002).
Ahmedu, A. A., Hassan, H. S. & Abudakar, M. U. Flavonoid glycosides from Byrsocarpus coccineus leaves. Schum and thonn (Connaraceae). Afr. J. Trad. CAM 4, 257–260 (2007).
Ban, J. Y. et al. Neuroprotective properties of gallic acid from Sanguisorbae Radix on amyloid β protein (25–35)-Induced toxicity in cultured rat cortical neurons. Biol. Pharm. Bull. 31, 149–153 (2008).
Chang, S. W. et al. Phytochemical Constituents of Bistorta manshuriensis. Nat. Prod. Sci. 15, 234–240 (2009).
Peungvicha, P. et al. 4-Hydroxybenzoic acid: a hypoglycemic constituent of aqueous extract of Pandanus odorus root. J. Ethnopharmacol. 62, 79–84 (1998).
Pfundstein, B. et al. Polyphenolic compounds in the fruits of Egyptian medicinal plants (Terminalia bellerica, Terminalia chebula and Terminalia horrida): Characterization, quantitation and determination of antioxidant capacities. Phytochemistry 7, 1132–1148 (2010).
Popovic, M. et al. The basic helix–loop–helix region of the transcriptional repressor hairy and enhancer of split 1 is preorganized to bind DNA. Proteins 82, 537–545 (2014).
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS), Strategic Priority Research Promotion Program, Chiba University, “Phytochemical Plant Molecular Sciences”, Takeda Science Foundation, the Naito Foundation, Astellas Foundation for Research on Metabolic Disorders, the Uehara Memorial Foundation, and a Workshop on Chirality at Chiba University (WCCU). This work was inspired by the international and interdisciplinary environments of the JSPS Core-to-Core Program “Asian Chemical Biology Initiative” and the JSPS A3 Foresight Program.
Author information
Authors and Affiliations
Contributions
Midori A. Arai designed the research. Midori A. Arai and Masami Ishibashi prepared the manuscript, Kaori Morita, Haruka Kawano, Yuna Makita, Manami Hashimoto, Akiko Suganami, and Yutaka Tamura did experiments. Samir K. Sadhu and Firoj Ahmed collected the plants.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arai, M.A., Morita, K., Kawano, H. et al. Target protein-oriented isolation of Hes1 dimer inhibitors using protein based methods. Sci Rep 10, 1381 (2020). https://doi.org/10.1038/s41598-020-58451-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58451-3
This article is cited by
-
Screening study of cancer-related cellular signals from microbial natural products
The Journal of Antibiotics (2021)
-
Total synthesis of lindbladione, a Hes1 dimerization inhibitor and neural stem cell activator isolated from Lindbladia tubulina
Scientific Reports (2020)
-
Isolation and evaluation of cardenolides from Lansium domesticum as Notch inhibitors
Journal of Natural Medicines (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
