
Abstract
Stripe rust fungus Puccinia striiformis f. sp. tritici (Pst) is a destructive pathogen of wheat worldwide. Pst has a macrocyclic-heteroecious lifecycle, in which one-celled urediniospores are dikaryotic, each nucleus containing one haploid genome. We successfully generated the first fully haplotype-resolved and nearly gap-free chromosome-scale genome assembly of Pst by combining PacBio HiFi sequencing and trio-binning strategy. The genome size of the two haploid assemblies was 75.59 Mb and 75.91 Mb with contig N50 of 4.17 Mb and 4.60 Mb, and both had 18 pseudochromosomes. The high consensus quality values of 55.57 and 59.02 for both haplotypes confirmed the correctness of the assembly. Of the total 18 chromosomes, 15 and 16 were gapless while there were only five and two gaps for the remaining chromosomes of the two haplotypes, respectively. In total, 15,046 and 15,050 protein-coding genes were predicted for the two haplotypes, and the complete BUSCO scores achieved 97.7% and 97.9%, respectively. The genome will lay the foundation for further research on genetic variations and the evolution of rust fungi.
Similar content being viewed by others


Background & Summary
The basidiomycete fungus Puccinia striiformis f. sp. tritici (Pst) is an obligate biotrophic pathogen that causes stripe (yellow) rust disease in wheat. Stripe rust has been reported in more than 60 countries, threatening 88% of wheat production worldwide and seriously affecting the global food supply1,2,3. The damage of this pathogen to agriculture is attributed to its massive genetic diversity because of sexual recombination mainly occurring in the Himalayan and neighboring regions (Nepal, Pakistan, and China), its long-distance dispersal across continents by means of nature and human transport, and its fast local adaptation through stepwise mutation and somatic hybridization, surmount the resistance of wheat cultivars and result in subsequent epidemics4,5,6,7,8. As a macrocyclic and heteroecious rust fungus, Pst has an extremely complex lifecycle, comprising five different types of spores (urediniospores, teliospores, basidiospores, pycniospores, and aeciospores) on two phylogenetically unrelated plant hosts: wheat is the primary host and barberry (Berberis spp.) is the alternate host9. The threat to wheat arises from urediniospores re-infecting and exponentially multiplying through the asexual cycle during the wheat growing season. The one-celled urediniospore is dikaryotic (N + N’), with a full set of haploid chromosomes in each separate nucleus (karyon), and is highly heterozygous10,11,12. Therefore, a high-quality haplotype-resolved genome assembly in nonhaploid rust fungi is important for in-depth research on genetic variation within and across species.
Although a haplotype-phased chromosome-scale genome of Pst has been reported, it has not been completely resolved and hundreds of gaps remain13. With the advancement of sequencing technologies and bioinformatics software, more and more complex genomes of animals and plants have achieved haplotype-resolved and telomere-to-telomere (T2T) construction14,15,16,17. Currently, PacBio High-Fidelity (HiFi) sequencing technology yields long reads averaging 10–25 kb and extremely low error rates (<0.5%), which are the main data types for high-quality genome assembly18,19. Furthermore, the trio-binning assembly strategy using short reads from two parental genomes provides a perfect approach for producing a completely haplotype-resolved diploid genome18,20. In this study, we combined PacBio HiFi sequencing technology and a trio-binning approach to obtain two primary haploid assemblies of the Pst isolate AZ2, which was derived from the Pst isolate A153 crossing with isolate XZ-2. Next, high-throughput chromosome conformation capture (Hi-C) sequencing technology was applied to scaffold the assembled data at the chromosome level. To reduce the influence of heterozygous genomic regions of the parents on haploid phasing, DNA data from haploid pycniospores from parental isolates A153 and XZ-2 were sequenced with single-cell genomic sequencing technology and used to partition HiFi reads into haplotypes.
Here, we successfully generated the first fully haplotype-resolved and nearly gap-free chromosome-scale genome for the dikaryotic wheat stripe rust fungus. The genome size of the two haploid assemblies was 75.59 Mb and 75.91 Mb, with both anchored onto 18 pseudochromosomes. In total, 15 and 16 gapless chromosomes were separately assembled for the two haplotypes, and the other chromosomes each contained only 1–2 gaps. A total of 15,046 and 15,050 protein-coding genes were predicted for the two haplotypes, and the complete BUSCO scores reached 97.7% and 97.9%, respectively. Meanwhile, a complete and circular mitochondrial genome (mitogenome) of Pst was also assembled, with a total size of 101,852 bp. Multiple assessment methods have confirmed the high continuity, correctness, and completeness of the haplotype-resolved assembly. This study will be a useful resource for community research on the pathogenicity, genetic variation, and evolution of the Pst genome.
Methods
Isolate selection and sexual hybridization
Sexual hybridization between Pst isolates A153 and XZ-2 was performed based on previously reported procedures21,22,23. When obvious nectars (or honeydews) formed, a partial nectar from one pycnium of A153 or XZ-2 was separately aspirated with a pipette gun for DNA extraction, and the remaining nectar from the same pycnium of A153 was transferred to the same pycnium of XZ-2 for mating and sexual hybridization. The aeciospores generated on the barberry host were collected to inoculate the susceptible wheat cultivar Mingxian 169 seedlings for the production of uredinium. Only a single urediniospore produced on Mingxian 169 was selected to inoculate the seedlings of Mingxian 169 and multiplied, forming the progeny isolate AZ2.
Genome and transcriptome sequencing
Genomic DNA of AZ2 was extracted from freshly harvested urediniospores using the previously described method24. For PacBio HiFi sequencing, an SMRT bell library was constructed and sequenced on the PacBio Sequel II system, and ~9.44 Gb consensus HiFi reads were generated using CCS software with default parameters, to achieve approximately 124 × coverage of the size of the haploid genome. Meanwhile, a DNA library with 350-bp fragment sizes was constructed and sequenced using the Illumina Novaseq PE150 platform, with ~77 × coverage of the haploid genome size. The Hi-C library was constructed using a 4-cutter restriction enzyme DpnII with fresh ungerminated AZ2 uredinospores, and ~18.22 Gb reads were generated on the Illumina Novaseq PE150 platform, with ~240 × coverage of the haploid genome size (Table 1).
AZ2 RNA was extracted separately from fresh urediniospores, 7 days and 9 days after inoculation on the susceptible wheat cultivar Mingxian169 using the Qiagen (Doncaster, Australia) Plant RNeasy kit as previously described25. Equal amounts of the three RNA samples were mixed for mRNA sequencing using Illumina Novaseq sequencing, and ~8.85 Gb reads were generated (Table 1). All sequencing studies were carried out at Novogene Corporation (Beijing, China).
Single-cell genomic sequencing of the pycniospore
The genomic DNA of A153 and XZ-2 from freshly harvested pycniospores was separately prepared and sequenced using single-cell genomic sequencing with multiple displacement amplification, both generating ~10 Gb reads on the Illumina Novaseq platform and achieving ~132 × coverage of the haploid genome size (Table 1). Sequencing was performed at Annoroad Gene Technology Corporation (Beijing, China).
Genome size and heterozygosity estimation
Before assembly, genome size and heterozygosity were estimated with Illumina short DNA reads. Jellyfish v2.3.026 was used to calculate the frequency distribution of the depth of clean data with 29-mer. The results were then imported to GenomeScope v1.027 to estimate the basic features of the genome with 29-mer. The haploid genome size of AZ2 was estimated to be 73.19 Mb, with a heterozygosity rate of 0.32% (Fig. 1).

The GenomeScope profle of Puccinia striiformis f. sp. tritici isolate AZ2 based on 29-mer.
Haplotype-resolved genome assembly
PacBio HiFi sequencing technology and a trio-binning strategy20 were combined using Hifiasm v0.16.128 with default parameters to generate a haplotype-resolved Pst assembly. In the first step, yak v0.1-r56 (https://github.com/lh3/yak) was used to count 19-mer with the Illumina short reads from pycniospores of the paternal isolate A153 and maternal isolate XZ-2. Next, HiFi reads from AZ2 were partitioned into haplotype-specific sets using parental sequencing data and subsequently assembled, respectively. Clean Hi-C paired-end reads were aligned with the assembly using Juicer v1.6.229 with the BWA algorithm to obtain the interaction matrix. The 3d-DNA v180922 pipeline30 was applied to reorder and scaffold the contigs. The position of the contigs was also manually adjusted based on the Hi-C heatmaps visualized using JuicerBox v1.9.831. Blastn searches against the NCBI nr/nt database were used to check potential contamination and none of the contigs had significant hits to noneukaryotic sequences, chloroplast sequences, mitochondrial sequences, or plant rRNA with E-value set as 1e-10. The obtained contigs were parsed by Purge Haplotigs v1.1.132 and Redundans33 to eliminate the redundancies.
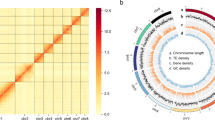
The final assembled genome contained two fully separated haplotypes, named AZ2A (75.59 Mb) and AZ2B (75.91 Mb), both with 18 pseudochromosomes (Table 2, Fig. 2). The genome size previously estimated using the k-mer frequency was similar to that of these assemblies. The contig N50 length of the two haplotypes was 4.17 Mb and 4.60 Mb, respectively. Remarkably, of the total 18 chromosomes, 15 and 16 were gapless while there were only five and two gaps for the remaining chromosomes of the two assembled haplotypes, respectively (Supplementary Table 1), suggesting good continuity of the genome assembly.

Overview of the haplotype-resolved genome assembly of Puccinia striiformis f. sp. tritici isolate AZ2. All 18 chromosomes of the AZ2 are drawn to scale and the ruler indicates chromosome length. Collinear regions between the two haplotypes are shown by gray lines. The cross-like shapes indicate the positions of the centromeres. The deep purple triangles indicate the presence of telomere sequence repeats.
Repeat and gene annotation
RepeatModeler v1.0.8 (https://www.repeatmasker.org/RepeatModeler/) constructed a de novo repeat library, which was then merged with Repbase library v23.09 (https://www.girinst.org/repbase/) and imported it into RepeatMasker v4.1.2-p134 for repeat prediction. A total of 27.88 and 28.38 Mb of repetitive sequences were identified, accounting for 36.89% of AZ2A and 37.39% of AZ2B, of which long terminal repeats (LTR) and DNA elements were the abundant repetitive elements despite unclassified repeats (Fig. 2, Supplementary Table 2).
The genome of repeats soft-masked was used for gene annotation using the funannotate pipeline (https://github.com/nextgenusfs/funannotate). Clean RNA-seq reads from AZ2 were aligned to the genome using Hisat2 v2.2.135 with ‘–max-intronlen 10000’, ‘–min-intronlen 20’ and default parameters for training gene models. The EST clusters of Pucciniamycotina were downloaded from the JGI MycoCosm website (http://genome.jgi.doe.gov/pucciniomycotina/pucciniomycotina.info.html) and used as transcript evidence. Proteins from previous Pst studies including Pst-104E36, Pst-DK091137, Pst93-21038, CYR3438 and Pst-134E13 were combined with the default UniProtKb/SwissProt curated protein database of funannotate as protein evidence. Genes were predicted using a suite of funannotate pipeline tools, including Augustus v3.3.339, GeneMark-ES v4.3240, CodingQuarry v2.041, SNAP v2006-07-2842 and GlimmerHMM v3.0.443. All the above gene models were combined using EvidenceModeler v.1.1.144 with default weight settings. A total of 15,046 and 15,050 protein-coding genes were predicted for AZ2A and AZ2B, respectively. The total lengths of the protein-coding genes were 23.93 Mb and 24.05 Mb, respectively (Table 3, Fig. 2). The mean lengths of the genes were 1.59 kb and 1.60 kb. There were 12,872 and 12,883 genes with an additional exon.
Mitochondrial genome assembly
Mitogenome of AZ2 was also assembled as in a previous study45. A multifasta file of Puccinia striiformis mitogenomes containing PST-7846, Pst-DK091137, Pst93-21047, Psh93TX-247 and CY3248 acted as the starting reference genome, and the mitogenome of AZ2 was assembled with PacBio HiFi reads using Canu v2.249. The assembled mitogenome was annotated with the GeSeq50 web browser (https://chlorobox.mpimp-golm.mpg.de/geseq.html) and the MITOS51 web server using genetic code 4 (http://mitos.bioinf.uni-leipzig.de/index.py). Next, the tRNA genes were then further evaluated using tRNAscan-SE v2.0.952. A graphical map of the mitogenome was drawn using mtviz (http://pacosy.informatik.uni-leipzig.de/mtviz). A complete circular mitogenome of AZ2 was assembled with a total size of 101,852 bp and a guanine-cytosine (GC) content of 31.44% (Fig. S1). In total, 14 protein-coding genes (atp6, atp8, atp9, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, cox1, cox2, cox3 and cob) and 24 tRNAs were detected in the AZ2 mitogenome located on the direct strand.
Chromosomal synteny analysis
To investigate differences between the two haplotypes, the command nucmer in MUMmer v4.053 with the parameters ‘–maxmatch -c 100 -b 500 -l 50’ was used for whole-genome alignments, and the alignment results were filtered using the command delta-filter with the parameters ‘-m -i 90 -l 100’. After format conversion with the command show-coords, SyRI v1.6.354 using the default parameters detected the syntenic regions and structural variations. Plotsr v1.1.155 was used to visualize the variations (Fig. 3). A total of 1128 syntenic regions with a cumulative size of 142.48 Mb (94.05%) were detected, indicating a high similarity between the two haplotypes. Furthermore, 227 translocations with a cumulative size of 1.70 Mb (~1.12%), 8 inversions with a cumulative size of 0.18 Mb (~0.12%), and 2778 duplications with a cumulative size of 5.02 Mb (~3.31%) were also detected.

The sequence collinearity and structural variants between AZ2A and AZ2B. The haplotype AZ2A is used as the reference sequence and the haplotype AZ2B is the query. Collinear regions between the two haplotypes are shown by gray lines.
Data Records
All raw sequencing data and genome assembly of Pst isolate AZ2 have been deposited in the National Center for Biotechnology Information (NCBI) under BioProject ID PRJNA1025922 and PRJNA1026770. The PacBio HiFi, Hi-C, Illumina sequencing reads and RNA sequencing reads of AZ2 have been deposited in the NCBI Sequence Read Archive database with accession group numbers SRP46553556. All raw sequencing data of A153 and XZ-2 have been submitted to the NCBI Sequence Read Archive database (SRR2634546057 and SRR2634546158). Genome assembly is available from GenBank in the NCBI with accession number GCA_039519205.159 and GCA_039519225.160. The genome assembly and gene annotation results were also deposited in the figshare database61.
Technical Validation
Evaluation of the assembled genome
The quality of genome assembly was evaluated using multiple methods. First, the accuracy of the Hi-C based chromosome construction was evaluated by chromatin contact matrix using HiC-Pro v3.0.062, and contact maps were plotted with hicPlotMatrix of HiCExplorer v3.7.263. The interactive Hi-C heatmap confirmed the good continuity of genome assembly (Fig. 4). Second, the BUSCO analysis using the basidiomycota odb9 database (genome mode) was performed to assess genome completeness using BUSCO v3.0.2b64 with Ustilago maydis as the reference species for Augustus gene prediction. The complete BUSCO scores (including single-copy and duplicated) of the two haplotypes accounted for 95.0% and 95.3%, respectively (Supplementary Table 3), suggesting good completeness of the genome assembly. Third, Illumina short reads and HiFi long reads from AZ2 were mapped to the assembly using BWA-MEM65 and minimap2 v2.2466, then QualiMap v2.267 was used to evaluate the mapping quality. Mapping rates were > 96%, and sequencing coverage reached 99.99%, indicating good consistency between the diploid genome with Illumina and HiFi sequencing reads (Supplementary Table 4). Fourth, the consensus quality value (QV) and completeness of the genome were evaluated using Merqury v1.368 with meryl v1.3 (under 19-mer) count. QVs for AZ2A and AZ2B, and shared AZ2A and AZ2B were 55.57, 59.02, and 56.96 (Genome accuracy > 99.999%), respectively (Table 4). The completeness scores for AZ2A and AZ2B were 92.15% and 92.23%, respectively. Finally, telomeres were annotated by searching for the CCCTAA or TTAGGG repeat sequences based on the method described previously69. In total, 34 of the 36 telomeres were detected on AZ2A, except for one telomere on chromosome 8 and one telomere on chromosome 16. Except for chromosome 9 on AZ2B containing one telomere, the other 17 chromosomes each contained telomere sequences at either end (Fig. 2, Supplementary Table 1). In general, this assembly can be described as a nearly telomere-to-telomere genome.

Heatmap of genomic interactions (with a resolution of 20 kb) of AZ2A (a) and AZ2B (b) chromosomes using Hi-C data. The strength of the interaction was represented by the color from yellow (low) to red (high).
Evaluation of the gene annotation
The annotated and integrated proteins were also evaluated using BUSCO v3.0.2b64 with the basidiomycota odb9 database (protein mode). The complete BUSCO scores of the two haplotypes accounted for 97.7% and 97.9%, respectively, indicating high quality of the gene annotation (Table 5).
Code availability
All sofware and pipelines used in this study were performed with the parameters described in the Methods section. If no detail parameters were mentioned for the sofware, default parameters were used as suggested by developer.
References
Wellings, C. R. Global status of stripe rust: a review of historical and current threats. Euphytica. 179, 129–141 (2011).
Beddow, J. M. et al. Research investment implications of shifts in the global geography of wheat stripe rust. Nat. Plants. 1, 15132 (2015).
Chen, X. Pathogens which threaten food security: Puccinia striiformis, the wheat stripe rust pathogen. Food Secur. 12, 239–251 (2020).
Brown, J. K. & Hovmoller, M. S. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science. 297, 537–541 (2002).
Hovmøller, M. S., Sørensen, C. K., Walter, S. & Justesen, A. F. Diversity of Puccinia striiformis on cereals and grasses. Annu. Rev. Phytopathol. 49, 197–217 (2011).
Park, R. F. & Wellings, C. R. Somatic hybridization in the Uredinales. Annu. Rev. Phytopathol. 50, 219–239 (2012).
Schwessinger, B. Fundamental wheat stripe rust research in the 21st century. New Phytol. 213, 1625–1631 (2016).
Ali, S. et al. Origin, migration routes and worldwide population genetic structure of the wheat yellow rust pathogen Puccinia striiformis f. sp. tritici. PLoS Pathog. 10, e1003903 (2014).
Zhao, J., Wang, M., Chen, X. & Kang, Z. Role of alternate hosts in epidemiology and pathogen variation of cereal rusts. Annu. Rev. Phytopathol. 54, 207–228 (2016).
Lorrain, C., Gonçalves Dos Santos, K. C., Germain, H., Hecker, A. & Duplessis, S. Advances in understanding obligate biotrophy in rust fungi. New Phytol. 222, 1190–1206 (2019).
Badet, T. & Croll, D. The rise and fall of genes: origins and functions of plant pathogen pangenomes. Curr. Opin. Plant Biol. 56, 65–73 (2020).
Zhao, J. & Kang, Z. Fighting wheat rusts in China: a look back and into the future. Phytopathol. Res. 5, 1–30 (2023).
Schwessinger, B. et al. A chromosome scale assembly of an Australian Puccinia striiformis f. sp. tritici isolate of the PstS1 lineage. Mol. Plant-Microbe Interact. 35, 293–296 (2022).
Barros, C. P. et al. A new haplotype-resolved turkey genome to enable turkey genetics and genomics research. GigaScience. 12, (2022).
Shen, F., Xu, S., Shen, Q., Bi, C. & Lysak, M. A. The allotetraploid horseradish genome provides insights into subgenome diversification and formation of critical traits. Nat. Commun. 14, (2023).
Chang, Y., Zhang, R., Ma, Y. & Sun, W. A haplotype-resolved genome assembly of Rhododendron vialii based on PacBio HiFi reads and Hi-C data. Sci. Data. 10, 451 (2023).
Huang, Z. et al. Evolutionary analysis of a complete chicken genome. Proceedings of the National Academy of Sciences - PNAS. 120, e2078326176 (2023).
Li, H. & Durbin, R. Genome assembly in the telomere-to-telomere era. Nat. Rev. Genet. (2024).
Hon, T. et al. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data. 7, (2020).
Koren, S. et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. 36, 1174–1182 (2018).
Zhao, J. et al. Identification of eighteen Berberis species as alternate hosts of Puccinia striiformis f. sp. tritici and virulence variation in the pathogen isolates from natural infection of barberry plants in China. Phytopathology. 103, 927–934 (2013).
Tian, Y. et al. Virulence and SSR marker segregation in a Puccinia striiformis f. sp. tritici population produced by selfing a Chinese isolate on Berberis shensiana. Phytopathology. 106, 185–191 (2015).
Wang, L. et al. Inheritance and linkage of virulence genes in Chinese predominant race CYR32 of the wheat stripe rust pathogen Puccinia striiformis f. sp. tritici. Front. Plant Sci. 9, 120 (2018).
Schwessinger, B. & Rathjen, J. P. Extraction of high molecular weight DNA from fungal rust spores for long read sequencing. Methods in Molecular Biology. 1659, 49–57 (2017).
Zhao, J. et al. Distinct transcriptomic reprogramming in the wheat stripe rust fungus during the initial infection of wheat and barberry. Mol. Plant-Microbe Interact. 34, 198–209 (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6, 256–258 (2018).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 19, 460 (2018).
Pryszcz, L. P. & Gabaldón, T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113 (2016).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics. 25, 1–14 (2009).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Schwessinger, B. et al. A near-complete haplotype-phased genome of the dikaryotic wheat stripe rust fungus Puccinia striiformis f. sp. tritici reveals high interhaplotype diversity. mBio. 9, e2217–e2275 (2018).
Schwessinger, B. et al. Distinct life histories impact dikaryotic genome evolution in the rust fungus Puccinia striiformis causing stripe rust in wheat. Genome Biol. Evol. 12, 597–617 (2020).
Xia, C. et al. Folding features and dynamics of 3D genome architecture in plant fungal pathogens. Microbiol. Spectr. 10, e260822 (2022).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Lomsadze, A. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Testa, A. C., Hane, J. K., Ellwood, S. R. & Oliver, R. P. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics. 16, 170 (2015).
Leskovec, J. & Sosič, R. SNAP: a general-purpose network analysis and graph-mining library. ACM Trans. Intell. Syst. Technol. 8, 1 (2016).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 20, 2878–2879 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Kovar, L. et al. PacBio-based mitochondrial genome assembly of Leucaena trichandra (Leguminosae) and an intrageneric assessment of mitochondrial RNA editing. Genome Biol. Evol. 10, 2501–2517 (2018).
Cuomo, C. A. et al. Comparative analysis highlights variable genome content of wheat rusts and divergence of the mating loci. G3. 7, 361–376 (2017).
Xia, C. et al. Genome sequence resources for the wheat stripe rust pathogen (Puccinia striiformis f. sp. tritici) and the barley stripe rust pathogen (Puccinia striiformis f. sp. hordei). Mol. Plant-Microbe Interact. 31, 1117–1120 (2018).
Li, C. et al. The complete mitochondrial genomes of Puccinia striiformis f. sp. tritici and Puccinia recondita f. sp. tritici. Mitochondrial DNA Part B-Resour. 5, 29–30 (2019).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Tillich, M. et al. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11 (2017).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Goel, M., Sun, H., Jiao, W. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, (2019).
Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics. 38, 2922–2926 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP465535 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26345460 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR26345461 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_039519205.1 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_039519225.1 (2023).
Wang, J., Xu, Y., Peng, Y., Kang, Z. & Zhao, J. The genome assembly and annotation of Puccinia striiformis f. sp. tritici isolate AZ2. Figshare. https://doi.org/10.6084/m9.figshare.24265198.v6 (2023).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Ramírez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32, 292–294 (2016).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21 (2020).
Li, F. et al. Emergence of the Ug99 lineage of the wheat stem rust pathogen through somatic hybridisation. Nat. Commun. 10, 5068 (2019).
Acknowledgements
We would like to acknowledge the High-Performance Computing (HPC) of Northwest A&F University and High-Performance Computing (HPC) of State Key Laboratory of Crop Stress Biology for Arid Areas of Northwest A&F University for providing computing resources. This work was supported by grants from the National Key Research and Development Program of China (2021YFD1401001), the Natural Science Foundation of Shaanxi Province (2024JC-YBQN-0170), Shaanxi Postdoctoral Science Foundation (2023BSHEDZZ121) and the National “111 plan” of China (BP0719026).
Author information
Authors and Affiliations
Contributions
Zhensheng Kang and Jing Zhao designed and supervised the study. Yiwen Xu, Yuxi Peng and Yiping Wang collected and created the experimental materials. Jierong Wang analyzed the data and wrote the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Xu, Y., Peng, Y. et al. A fully haplotype-resolved and nearly gap-free genome assembly of wheat stripe rust fungus. Sci Data 11, 508 (2024). https://doi.org/10.1038/s41597-024-03361-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03361-6
