
Abstract
Echeneis naucrates, as known as live sharksucker, is famous for the behavior of attaching to hosts using a highly modified dorsal fin with oval-shaped sucking disc. Here, we generated an improved high-quality chromosome-level genome assembly of E. naucrates using Illumina short reads, PacBio long reads and Hi-C data. Our assembled genome spans 572.85 Mb with a contig N50 of 23.19 Mb and is positioned to 24 pseudo-chromosomes. Additionally, at least one telomere was identified for 23 out of 24 chromosomes. Furthermore, we identified a total of 22,161 protein-coding genes, of which 21,402 genes (96.9%) were annotated successfully with functions. The combination of ab initio predictions and Repbase-based searches revealed that 15.57% of the assembled E. naucrates genome was identified as repetitive sequences. The completeness of the genome assembly and the gene annotation were estimated to be 97.5% and 95.4% with BUSCO analyses. This work enhances the utility of the live sharksucker genome and provides a valuable groundwork for the future study of genomics, biology and adaptive evolution in this species.
Similar content being viewed by others

Background & Summary
Live sharksucker (Echeneis naucrates), also known as the sluggard in the ocean, is in the Echeneidae family, order Carangiformes (Fig. 1). This sharksucker is widely found in tropical and warm temperate waters1, and ranging from coastal areas to those offshore2. The key distinctive characteristic to distinguish it from other fishes is the oval-shaped sucking disc, which is a highly modified dorsal fin and used to attach to hosts. The oval-shaped sucking disc comprises of 21–28 laminae and extends from the top of the head to the front part of the body3. The hosts of live sharksucker encompass whales, sharks, dolphins, sea turtles, divers and vessel hulls4,5,6,7. With a host, proposed benefits to live sharksucker comprise conveyance (via “hitchhiking”), shielding from predators, enhanced courtship and reproductive capacity, improved gill aeration and expanded feeding opportunities8. The unique suction cups and adsorption habits make the live sharksucker a good research subject for bionic study9,10, aid in fishing11 and adaptive evolution, such as the commensalism relation between remora fish and shark12. Nonetheless, our comprehension of the biological context of the live sharksucker remains constrained.

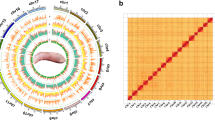
Morphological characteristics of E. naucrates.
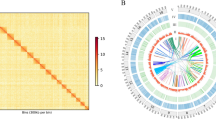
Genome sequencing has played a pivotal role in advancing various aspects of basic biology. High-quality reference genomes could profoundly enhance our understanding of the genetic foundation and the evolutionary process underlying unique biological characteristics in the live sharksucker. Although the chromosome-level live sharksucker genome has been released on NCBI with GenBank assembly accession GCA_900963305.113,14 and GCA_900963305.215, the completeness of genome assembly and annotations still require further refinement. For instance, the released chromosome-level genome assembly remained incomplete with many gaps (average 110.13 N’s per 100 kbp) (Fig. 3b). Not only that, a number of annotation details, including information related to repeats and non-coding RNAs, have not been made publicly available and remain inaccessible.

Hi-C interaction heat map for genome assembly of E. naucrates. The interaction density is quantified based on the number of supporting Hi-C reads and depicted using a color gradient ranging from white (low density) to dark red (high density).

Comparison of genome assembly of E. naucrates with the previous version. Contig distribution maps for chromosomes of E. naucrates between the assembly (a) in this study and (b) the previous version. The bars in grey represent entire lengths of chromosomes, in which the positions of telomeres are shown. The contig numbers and the sizes of chromosomes were shown behind the bars.
In this study, we generated 33.14 Gb of PacBio High fidelity (HiFi) long-reads with the N50 length of 18.11 kb, and 89.93 Gb of Illumina paired-end sequencing short-reads for genome assembly (Table 1). An additional 76.64 Gb of high-throughput chromatin capture (Hi-C) sequencing data were utilized to validate the genome assembly through a comparison with the scaffolding data. Leveraging these integrated sequencing data, we constructed a high-quality chromosome-level reference genome of E. naucrates. Specifically, a 572.85 Mb genome was assembled, comprised of 54 contigs with the contig N50 length of 23.19 Mb. A total of 570.71 Mb (99.63% of the contig-level genome) of the assembled sequences were positioned to 24 pseudo-chromosomes with low missing bases (average 0.40 N’s per 100 kbp). Moreover, telomeres were identified for at least one end of 23 out of 24 chromosomes, totaling 38 telomeres (Fig. 3a and Table 7). In this enhanced genome assembly, we have improved upon previous gene annotations by amalgamating ab initio predictions, protein homology searches and transcriptome-assisted methods, which identified a total of 22,161 protein-coding genes. Through a dual approach involving both homology searches and ab initio predictions, 15.57% of the assembled E. naucrates genome was identified as repetitive sequences. BUSCO alignment analysis of assembly based on the actinopterygii_odb10 database revealed that our ultimate assembly encompassed 3, 551 (97.5%) complete BUSCOs. The consensus QV of genome assembly was 52.01. In summary, this high-quality chromosome-level reference genome serves as a valuable foundation for the utilization of genetic resources, and the further investigation of the unique biological characteristics, such as the oval-shaped sucking disc, in the live sharksucker.
Methods
Sample collection and preparation
A single fish (~1500 g) was obtained in June 2022 from Northern South China Sea. The sampled fish in this study was permitted by the Animal Care and Use Committee of Fisheries College of Jimei University (Animal Ethics no. 1067) and performed by the regulations and guidelines established with this committee. Dorsal muscle, dorsal fin, skin, skull, and skull muscle tissues were collected and preserved in liquid nitrogen until the extraction of DNA and RNA. Dorsal muscle tissues were utilized for DNA sequencing to construct the genome assembly, while all tissues were utilized for RNA sequencing. The quality and quantity of genomic DNA samples were assessed through 1% agarose gel electrophoresis and the Pultton DNA/Protein Analyzer (Plextech).
WGS Illumina library construction, sequencing and assembly
To create the whole-genome sequencing (WGS) Illumina library, a paired-end library was constructed with an insert size of 300 bp adhering to the Illumina standard protocol. Then, DNA was purified, quantified, and sequenced from both ends using the Illumina NovaSeq 6000 sequencing platform. In total, a sum of 89.93 Gb raw reads was obtained (Table 1). After filtering process by using fastp v 0.23.216 with default parameters to remove low quality and short reads, as well as trim adapters and polyG sequences, a set of 87.77 Gb clean data were retained (Table 1). The estimation of the genome size and heterozygosity for live sharksucker was then performed using GCE v 1.0.017 by k-mer analysis with clean Illumina short data following the default settings.
PacBio library construction, sequencing and assembly
To obtain the PacBio long reads, a SMRTbell library was established with a fragment size of 20 kb using the SMRTBell template preparation kit 1.0 (PacBio) in accordance with the manufacturer’s instructions. The library was sequenced with the PacBio Sequel II system in Circular Consensus Sequence (CCS) mode. Upon the elimination of low-quality reads, a sum of 33.14 Gb reads with an average length of 17.90 kb were retained and then processed with the CCS v 6.0.0 algorithm with default parameters. With these PacBio long reads, the initial contigs were subsequently assembled using the Hifiasm v 0.16.1 algorithm18 with the default settings. After that, the purge_haplotigs v1.0.419 with the parameter of ‘-a 70 -j 80 -d 200’ was employed to eliminate redundant sequences. This procedure resulted in a contig-level assembly of about 588.30 Mb comprised of 54 contigs, with the N50 and maximum contig size of 23.19 Mb and 29.49 Mb, respectively.
Hi-C library preparation, sequencing and chromosome assembly
Hi-C data were used to anchor contigs onto chromosomes. Briefly, dorsal muscle tissue (~1 g) of E. naucrates was fixed with 1% formaldehyde for 10–30 min at room temperature (20–25 °C) to congeal proteins involved in chromatin interactions within the genome. DNA was digested with the 4-cutter restriction enzyme MboI. The overhangs of restriction fragments were filled and labeled with biotinylated nucleotides, followed by ligation in a compact volume. Following the cross-link reversal, the ligated DNA was purified and fragmented to a size range of 300–500 bp. Subsequently, ligation junctions were extracted by binding to streptavidin beads and prepared for Illumina NovaSeq 6000 sequencing. In total, 76.64 Gb of Hi-C reads were obtained (Table 1). After filtering reads with average quality scores less than 20 and removing adapters using fastp v 0.23.216 with the default settings, a total of 76.56 Gb clean data were retained (Table 1). We also utilized the HiCUP pipeline20, with the parameter of ‘--re1 ^GATC,MboI’ in hicup_digester step, to remove the erroneous mappings and duplicated contigs to yield the interaction matrix. This matrix served as the foundation for anchoring the contigs onto chromosomes through the utilization of approximately 169.29 Mb read pairs (~ 68.27%) via the 3D-DNA pipeline21 with the default settings. The scaffolds were subjected to a manual assessment and refinement process utilizing Juicebox Assembly Tools22 in order to rectify any instances of chromosome translocation and inversion. By integrating this Hi-C data, the contig-level assembled sequences were positioned onto 24 pseudo-chromosomes, encompassing a cumulative length of 570.71 Mb, covering ~99.63% of the contig-level genome (Fig. 2).
RNA library construction and transcriptome sequencing
Total RNA was extracted from five tissues of the live sharksucker, including dorsal muscle, dorsal fin, skin, skull, and skull muscle using TRIzol reagent (Invitrogen). To assess RNA quality, both a NanoDrop ND-1000 spectrophotometer (Labtech) and a 2100 Bioanalyzer (Agilent Technologies) were employed. The paired-end raw sequencing was performed using the Novaseq 6000 Platform. In sum, 33.01 Gb of clean data were generated from the RNA-seq library after filtering process using fastp v 0.23.216 with default parameters (Table 1).
Repetitive sequence annotation
Repeat elements within the live sharksucker genome were comprehensively identified through a dual approach involving both homology searches and ab initio predictions. The ab initio prediction of repeat elements was executed using both Tandem Repeat Finder v 4.0923 and LTR_FINDER_parallel v1.123 with default parameters. Subsequently, novel repeats were predicted utilizing RepeatMasker according to the de novo repetitive sequence library constructed with LTR_FINDER_parallel and RepeatModeler v 2.024 following default parameters. To identify known repeat elements for genome sequences, RepeatMasker v 4.0.925 and RepeatProteinMask v 4.1.0 (http://www.repeatmasker.org) with default parameters were employed, by querying the genome sequences against the Repbase database26. The integration of ab initio predictions and Repbase-based searches unveiled that 15.57% of the assembled E. naucrates genome was identified as repetitive sequences (Fig. 4). Among which, repetitive DNAs, LINEs, SINEs and LTRs covered 5.74%, 4.03%, 2.27% and 1.85% of the entire genome, respectively (Table 3).

Chromosome-level assembly genomic landscape of E. naucrates. Circos plot from the outer to the inner layers represents the following: (a) GC content (range: 31% - 63%); (b) gene density (range: 0% - 87%); (c) repeat density (range: 0% - 100%); (d) LTR retroelement density (range: 0% - 39%); (e) LINE density (range: 0% - 94%); and (f) DNA transposons density (range: 0% - 65%). a-f were drawn in 500-kb sliding windows, and y-axes represent the proportion of respective elements within the window.
Gene prediction and annotation
Using the repeat-masked genome, the prediction of protein-coding genes within the live sharksucker genome was approached through three strategies: ab initio predictions, homologous searches and RNA-sequencing methods. Ab initio prediction was conducted utilizing Augustus v 3.3.227 and Genscan28 tools with default parameters. In parallel, homologous gene prediction was based on the retrieval of protein sequences from various species, comprising Caranx melampygus (GenBank assembly accession: GCA_019059645.129,30), Echeneis naucrates (GenBank assembly accession: GCA_900963305.113,14), Danio rerio (GenBank assembly accession: GCA_000002035.431), Seriola dumerili (GenBank assembly accession: GCA_002260705.132,33), Takifugu rubripes (GenBank assembly accession: GCA_901000725.334), and Seriola lalandi (GenBank assembly accession: GCA_002814215.135,36). These protein sequences were downloaded from the NCBI database and subjected to alignment with our live sharksucker genome via tBLASTn (E-value ≤ 1e-5). Subsequently, the homologous genome sequences were aligned with the corresponding proteins through the utilization of Genewise v 2.4.037 to obtain precise gene annotation. A pooled RNA-seq dataset of five tissues, each sequenced separately, were aligned to the assembled genome utilizing HISAT2 v 2.1.038 with default parameters, and subsequently the putative transcript structures were predicted using StringTie v1.3.539 and TransDecoder v 5.1.0 (https://github.com/TransDecoder/TransDecoder) with default parameters. Three gene models underwent merging to eliminate redundancy using MAKER v 2.31.1040 and HiFAP (Wuhan OneMore Tech Co., Ltd., https://www.onemore-tech.com/) with default parameters, resulting in the identification of 22,161 and 22,086 genes, respectively (Fig. 4 and Table 4).
The predicted protein-coding gene sets were functionally annotated based on NCBI nonredundant protein (NR), Swiss-Prot41 (http://www.gpmaw.com/html/swiss-prot.html), TrEMBL (http://www.uniprot.org), eukaryotic orthologous groups of proteins (KOG)42, AnimalTFDB v4.0 (http://bioinfo.life.hust.edu.cn/AnimalTFDB4/?#/), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) using BLASTp43 (E-value ≤ 1e-5). The annotation of gene sets compared with InterPro and Pfam databases were performed via InterProScan v 5.6144 with parameters “--goterms–pathways--dp”. Finally, 21,402 genes (representing roughly 96.90% of total predicted genes) were effectively annotated by at least one of these databases (Table 5).
Non-coding RNA prediction and annotation
The ribosomal RNAs (rRNAs), microRNAs (mRNAs) and small nuclear RNAs (snRNAs) were predicted by using INFERNAL v.1.145 according to the rfam46 and miRBase47 databases. Transfer RNAs (tRNAs) were annotated with tRNAscan-SE v 1.3.148 following the default parameters. Taken together, non-coding RNAs, comprising 2,107 rRNAs, 1,786 miRNAs, 1,408 snRNAs and 12,200 tRNAs were predicted from the E. naucrates genome (Table 6).
Identification of telomeres
Based on the common characteristic sequences (CCCTAA/TTAGGG) of vertebrates, telomere sequences are identified through pattern searching at both ends of each chromosome, where the characteristic sequence repeats at least four times within a 50 kb region. All 38 telomeres were annotated within the 23 chromosomes, with no telomere sequence detected on chr7 (Fig. 3a and Table 7).
Data Records
The raw sequencing dataset of E. naucrates in this study can be achieved from Sequence Read Archive (SRA) under SRP45789349, including WGS Illumina sequencing data (SRR25859131), Pacbio HiFi sequencing data (SRR25859130) and Hi-C sequencing data (SRR25859129). The assembled genome of E. naucrates was deposited at GenBank under accession GCA_031770045.150. Furthermore, files of the assembled genome, protein-coding gene annotation, non-coding RNA prediction and repeat annotation of E. naucrates were deposited in Figshare database51.
Technical Validation
Assessing the quality of the genome assembly
We initially used QUAST v 5.2.052 to evaluate the integrity and quality of E. naucrates genome assembly. The contig N50 (the length at which half of the total sequence resides in contigs of this size) has shown a significant improvement, reaching 23.19 Mb, which significantly surpasses previous E. naucrates genome versions of 12.4 Mb (GenBank assembly accession: GCA_900963305.1, GCA_900963305.2). Furthermore, in this study, the genome exhibits an exceptionally low gap count (average 0.40 N’s per 100 kbp) (Table 2; Fig. 3a), marking a substantial reduction compared to the previous versions of average 110.13 N’s per 100 kbp (Fig. 3b). Next, we remapped Illumina paired-end clean reads and PacBio long reads to the final assembled genome using BWA53 and Minimap254, resulting in mapping rates of 99.62% and 99.98%, respectively. Homozygous SNP rate was 0.00% when aligned Illumina paired-end clean reads to the final assembly, underscoring the comprehensiveness of the complete genome (Table 8). Furthermore, the completeness of the assembled genome sequence was assessed with Benchmarking Universal Single-Copy Orthologs (BUSCO, v 5.1.0)55 based on the actinopterygii_odb10 database. The BUSCO analysis of assembly showed that 3,551 (97.5%) of the complete orthologs, including 3,514 (96.5%) single-copy orthologs and 37 (1.0%) duplicated orthologs, as well as 14 (0.4%) fragmented orthologs were identified (Table 9). The consensus quality value (QV) of the assembly, estimated using Merqury56 (kmer = 21), was 52.01.
Assessing the quality of the genome annotation
The BUSCO analysis of annotation based on the actinopterygii_odb10 database, which was used to assess the integrity of the annotated gene set, revealed that 95.4% (3,473) of the complete genes were identified, comprising 94.4% (3,437) single-copy genes, 1.0% (36) duplicated genes, and 1.3% (46) fragmented genes (Table 9).
Taken together, the comprehensive assessment of the E. naucrates genome surpassed that of other existing public E. naucrates genomes.
Code availability
No specifc code was used in this study. The data analyses adhered to the manuals and protocols offered by the creators of the corresponding bioinformatics tools, the parameter settings of which were outlined in the methods section.
References
Smith, J. L. B., Smith, M. M. & Heemstra, P. C. Smiths’ Sea fishes. 1st edn. (Macmillan South Africa, 1986).
Fao, R. Fichas FAO de identificación de especies para los fines de la pesca. Guía de campo de las especies comerciales marinas y de aquas salobres de la costa septentrional de Sur América. (Biología y Conservación de las Tortugas Continentales de Colombia., 1992).
Allen, G. R. & Erdmann, M. V. Reef fishes of the East Indies: Andaman Sea, Myanmar, Thailand, Indonesia, Christmas Island, Singapore, Malaysia, Brunei, Philippines, Papua New Guinea, Solomon Islands. (2012).
Strasburg, D. W. Some aspects of the feeding behavior of remora remora. UH Press (1962).
O’Toole, B. Phylogeny of the species of the superfamily Echeneoidea (Perciformes: Carangoidei: Echeneidae, Rachycentridae, and Coryphaenidae), with an interpretation of echeneid hitchhiking behaviour. Can J Zool 80, 596–623 (2002).
Ritter, E. K. Analysis of sharksucker, Echeneis naucrates, induced behavior patterns in the blacktip shark, Carcharhinus limbatus. Environ Biol Fish 65, 111–115 (2002).
Fertl, D. & Landry, A. M. Remoras. (Encyclopedia of Marine Mammals. Third Edition, 2018).
Fertl, D. & Landry, A. M. Sharksucker (Echeneis naucrates) on a bottlenose dolphin (Tursiops truncatus) and a review of other cetacean-remora associations. Mar Mammal Sci 15, 859–863 (1999).
Wang, S., Li, L., Zhao, W., Zhang, Y. & Wen, L. A biomimetic remora disc with tunable, reversible adhesion for surface sliding and skimming. Bioinspir Biomim 17, 036001 (2022).
Li, L. et al. Aerial-aquatic robots capable of crossing the air-water boundary and hitchhiking on surfaces. Sci Robot 7, eabm6695 (2022).
Carpenter, K. E. & Niem, V. H. FAO species identification guide for fishery purposes. (The living marine resources of the Western Central Pacific. Volume 3. Batoid fishes, chimaeras and bony fishes part 1 (Elopidae to Linophrynidae), 1999).
Das, S. & Bhattacharya, A. Symbiotic organisms search algorithm for short-term hydrothermal scheduling. AIin Shams Eng J 9, 499–516 (2016).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_900963305.1 (2019).
Miya, M. et al. Evolutionary origin of the Scombridae (tunas and mackerels): members of a paleogene adaptive radiation with 14 other pelagic fish families. PloS one 8, e73535 (2013).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_900963305.2 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant Biol 35, 62–67 (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, Unit 4 10 (2004).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268, 78–94 (1997).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_019059645.1 (2021).
Pickett, B. D., Glass, J. R., Ridge, P. G. & Kauwe, J. S. K. De novo genome assembly of the marine teleost, bluefin trevally (Caranx melampygus). G3 (Bethesda) 11, jkab229 (2021).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_000002035.4 (2017).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_002260705.1 (2017).
Araki, K. et al. Whole genome sequencing of greater amberjack (Seriola dumerili) for SNP identification on aligned scaffolds and genome structural variation analysis using parallel resequencing. Int J Genomics 2018, 7984292 (2018).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_901000725.3 (2021).
NCBI Assembly https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_002814215.1 (2017).
Purcell, C. M. et al. Insights into teleost sex determination from the Seriola dorsalis genome assembly. BMC Genomics 19, 31 (2018).
Birney, E., Clamp, M. & Durbin, R. Genewise and genomewise. Genome Res 14, 988–995 (2004).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650–1667 (2016).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18, 188–196 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res 12, 656–664 (2002).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–D124 (2005).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res 47, D155–D162 (2019).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP457893 (2023).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_031770045.1 (2023).
WangDanyang Chromosome-level genome assembly and annotation of live sharksucker, Echeneis naucrates. figshare https://doi.org/10.6084/m9.figshare.24079629.v1 (2023).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35, 543–548 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Acknowledgements
This work was supported by the Province Key Research and Development Program of Zhejiang (2021C02047).
Author information
Authors and Affiliations
Contributions
D.W. and T.G. conceived and designed the study. T.G. and K.L. performed animal work and prepared biological samples. T.G., Q.L. and D.W. conducted the genome assembly and analysis. D.W. and T.G. wrote the paper. D.W., T.G., K.L. and Q.L. revised the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, T., Liu, K., Liu, Q. et al. An improved chromosome-level genome assembly and annotation of Echeneis naucrates. Sci Data 11, 452 (2024). https://doi.org/10.1038/s41597-024-03309-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03309-w
