
Abstract
Background
Neonatal hypoxic-ischemic encephalopathy (HIE) is a kind of brain injury that causes severe neurological disorders in newborns. Metabotropic glutamate receptors (mGluRs) and ionotropic glutamate receptors (iGluRs) are significantly associated with HIE and are involved in ischemia-induced excitotoxicity. This study aimed to investigate the upstream mechanisms of mGluRs and the transcriptional regulation by nuclear respiratory factor 1 (NRF1).
Methods
The rat model of neonatal HIE was created using unilateral carotid artery ligation and in vitro oxygen-glucose deprivation paradigm. We used western blot, immunofluorescence, Nissl staining, and Morris water maze to investigate the impact of NRF1 on brain damage and learning memory deficit by HIE. We performed ChIP and luciferase activities to identify the transcriptional regulation of NRF1 on mGluRs.
Results
The neuronal NRF1 and some glutamatergic genes expression synchronously declined in infarcted tissues. The NRF1 overexpression effectively restored the expression of some glutamatergic genes and improved cognitive performance. NRF1 regulated some members of mGluRs and iGluRs in hypoxic-ischemic neurons. Finally, NRF1 is bound to the promoter regions of Grm1, Grm2, and Grm8 to activate their transcription.
Conclusions
NRF1 is involved in the pathology of the neonatal HIE rat model, suggesting a novel therapeutic approach to neonatal HIE.
Impact
-
NRF1 and some glutamatergic genes were synchronously downregulated in the infarcted brain of the neonatal HIE rat model.
-
NRF1 overexpression could rescue cognitive impairment caused by the neonatal HIE rat model.
-
NRF1 regulated the expressions of Grm1, Grm2, and Grm8, which activated their transcription by binding to the promoter regions.
Similar content being viewed by others


Introduction
Neonatal hypoxic-ischemic encephalopathy (HIE) is a severe neonatal brain injury caused mainly by perinatal asphyxia, restricted or suspended cerebral blood flow, and is a common cause of induced neonatal death.1 The incidence of neonatal HIE shows geographic inconsistency, ranging from approximately 1 to 8‰ worldwide and up to 26‰ in developing countries.2 Patients with severe HIE develop neurological sequelae such as cerebral palsy, mental retardation, and cognitive impairment and even suffer from the threat of death.3
Excitotoxicity triggered by the amino acid glutamate is now accepted as the primary pathogenic mechanism of HIE.4 Cerebral ischemia leads to excessive activation of glutamate receptors and ion channels, triggering malfunction of neuronal cells and producing excitotoxicity.5,6,7 Ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs) mediate excitatory neurotransmission in the brain, which is essential for memory formation learning, and several neurodegenerative diseases.8,9 The dysfunction of iGluRs and mGluRs exacerbates cognitive impairment in psychiatric disorders,10,11 Alzheimer’s disease,12 Parkinson’s disease,13 and epilepsy.14,15 The glutamate receptor-specific agents have been reported to achieve neuroprotective benefits on the hypoxic-ischemic (HI) brain by reducing neuron excitability.16,17,18 Unfortunately, the effects of NMDA receptor antagonists on perinatal HI injury are not very uniform. The observed side effects of these compounds preclude their clinical use.19 mGluRs in the pathophysiology of the HI brain have been a subject of attention for several years. The mGluR2/3 agonists and mGluR1/5 antagonists against ischemic brain injury have neuroprotective effects in the neonatal HI model.17,20,21,22 Therefore, studying the upstream mechanism of mGluRs downregulation in neonatal HIE has substantial therapeutic implications for neonatal HIE treatment.
Nuclear respiratory factor 1 (NRF1) was first identified to regulate cytochrome c transcriptionally.23 NRF1 participates in several biological processes, such as mitochondrial autophagy, DNA repair, and bio-metabolism, which affect neurological function, cardiac function, and tumor formation.24,25,26,27 Whole genome sequencing results predict that 5–6% of genes in neurons are regulated by NRF1 directly or indirectly.28 Our preliminary Chromatin immunoprecipitation (ChIP)-seq data identified multiple members of mGluRs and iGluRs as potential target genes of NRF1. Our previous studies have demonstrated that hypoxia caused a decrease in NRF1 expression in brain tissue.29 NRF1 was involved in hypoxia-induced cellular response through transcriptional regulation of hypoxia-inducible factor 1α, steroidogenic acute regulatory protein, and endothelin-1.29,30,31 Therefore, we hypothesized that NRF1 was involved in modulating glutamate receptors in the neonatal HIE rat model.
This study aimed to investigate whether the expression of mGluRs and iGluRs were related to NRF1 and to elucidate the transcriptional regulatory mechanisms of NRF1 on mGluRs in the neonatal HIE rat model.
Methods
Neonatal HIE rat model construction and lentivirus infection
Neonatal HIE rat model construction. The 7-day-old Sprague–Dawley rats were provided by the Experimental Animal Center of Nantong University. We ligated the right common carotid artery of the pups to block the blood supply to the right side of the brain. The pups were exposed for 2 h in a chamber filled with 8% O2 and 92% N2 at 37 °C. The pups were then returned to their mother and nourished for 24 h. The effectiveness of the model construction was evaluated using the Bederson Scale32 by an assessor who was blinded to the experimental groups. There are 5-point scales on Bederson Scale, including 0 (no deficits), 1 (lost forelimb flexion), 2 (lost forelimb flexion with lower resistance to lateral push), 3 (unidirectional circling), 4 (longitudinal spinning or seizure activity), and 5 (no movement).
Lentivirus infection: we used a brain stereotaxic apparatus to inject 5 μ l of lentivirus (1 × 107 pfu/μl) or 0.9% saline into the right ventricle of newborn 1-day-old Sprague–Dawley rats. After 7 days, we built the neonatal HIE model. When the mice were 4 weeks old, we then conducted behavioral tests.
The Ethics Committee on Animal Experimentation at Nantong University authorized all investigations. The Laboratory Animal Center at Nantong University approved all procedures (S20220219-009).
2, 3, 5-Triphenyltetrazolium chloride (TTC) staining
We used TTC staining to verify the success of the model histologically. Briefly, the brain tissues were quickly frozen for 10 min at − 20 °C. The brain was then cut into successive 2-mm-thick slices along the coronal plane, followed by incubation with 2% TTC solution (Sigma-Aldrich, MO) at 37 °C for 15 min, which was terminated by rinsing with PBS. Subsequently, the slice section was fixed with 4% paraformaldehyde for 8 h and photographed. The infarct ratio was calculated by FIJI ImageJ (National Institutes of Health).
Nissl staining
The 4 µm coronal sections were obtained from the frozen brain sample. We stained the sections with cresyl violet stain for 30 min and washed with deionized water. All stained sections were progressively dehydrated, cleared in xylol, and fixed with neutral balsam. The images were captured using Leica DM4000B.
3-[4,5-Dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay
Briefly, the cells were cultured in a 96-well culture plate and incubation with MTT reagent (5 mg/ml, Sigma-Aldrich, MO) for 4 h at 37 °C. DMSO solution dissolved the formazan crystal after removing the medium. The absorbance at 570 nm was detected using a microplate reader (Synergy 2, Biotech).
Morris water maze (MWM)
MWM tests of spatial learning-memory behavior were conducted as described by Zha et al.33 The testing occurred between 9:00 and 11:00 in the morning. The maze has four equal quadrants: the northeast (NE), southeast, southwest (SW), and northwest. Visual cues were posted on the laboratory walls around the maze to facilitate learning the location of the platforms. Rats were placed in the 150 cm circular pool with water temperature maintained at 22 ± 1 °C. In the center of NE, a circular escape platform (10 cm) was positioned, keeping it 2 cm below the water’s surface. Before experiments, we trained rats for 3 days. Then mice were placed in the middle of SE and allowed to find the platform. The time the mice first found the platform was recorded. On the sixth day, we removed the hidden platform from the destination quadrant. Mice were released in the SW quadrant and allowed 180 s free-swimming time. Then we recorded the time the mice found the escape platform and the frequency of crossing the destination platform.
Primary neuron and PC12 culture and treatments
Primary neuron was prepared from the cortex of neonatal Sprague–Dawley rats according to the method described before.34 Cortex tissue was isolated from fetal rats (16–17 dpc). After digestion using 0.05% trypsin, cells were dispersed in the presence of DNase I. Then, cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 humidified incubator. Two hours later, the culture medium was replaced with neurobasal media (Thermo Fisher Scientific, MA) containing B-27 supplement (Thermo Fisher Scientific, MA). Subsequently, 5 μM cytarabine was added on the third day to inhibit glial cell growth. The medium was semi-transformed every 2 days. Cells were used for experiments until 10 days later.
PC12 cells (gift from Xiaomei Wu, Nantong University) were cultured in DMEM supplemented with 10% FBS at 37 °C in a 95% humidified incubator with 5% CO2. To differentiate the cells, we replaced the medium with the culture medium containing 100 ng/ml nerve growth factor and cultured them for 10 days.35
For oxygen-glucose deprivation (OGD) treatments, neuron or PC12 cells were cultured in a glucose-free DMEM medium and exposed to 1% O2 for 0, 2, 4, and 6 h. To overexpress NRF1, we infected neurons or PC12 cells with an NRF1 expressing lentivirus at MOI = 50.
RNA isolation and real-time PCR
Total RNA was isolated by TRIzol reagent (Thermo Fisher Scientific, CA) and reverse-transcribed by HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, Nanjing, China) according to the instructions. The AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, China) performed real-time PCR. All primers used for real-time PCR are listed in Supplementary Table S1. We used the 2–ΔΔCt method to calculate relative expression.
RT2 Profiler PCR Array Test
Total RNA was extracted from the tissues with TRIzol reagent (Thermo Fisher Scientific, CA). Reverse transcription was performed using HiScript ® II 1st Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme, Nanjing, China). The cDNA was used on the real-time RT2 Profiler PCR Array in combination with RT2 SYBR®Green qPCR Mastermix (Qiagen, Hilden, Germany). Actb (Actin, beta), Hprt1 (Hypoxanthine phosphoribosyltransferase 1), B2m (Beta-2-microglobulin), and Rplp1 (Ribosomal protein, large, P1) were used as the assay reference gene. The RT2 Profiler PCR Array data analysis software calculates the fold change using the 2–ΔΔCt method. The QIAGEN web portal at GeneGlobe exported the data analysis report.
Western blot
RIPA lysed the brain tissue and neuron to obtain proteins. Proteins were separated by electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were incubated with primary antibodies, including anti-NRF1 (ab175932, Abcam, MA) or anti-β-actin (ab8227, Abcam, MA). The binding of primary antibodies was visualized with HRP-conjugated secondary antibodies (Jackson ImmunoResearch, PA). FIJI ImageJ software carried out the grayscale analysis (National Institutes of Health).
Immunofluorescence staining
Tissue sections (20 μm) were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. After being blocked by donkey serum, the brain sections were incubated with anti-NRF1 (ab175932, Abcam, MA) and anti-NeuN (MAB377, Millipore, CA). The primary antibodies were visualized by Alexa Fluor 555-conjugated donkey anti-rabbit IgG (A31572, Thermo Fisher Scientific, CA) and Alexa Fluor 488-conjugated donkey anti-mouse IgG (A21202, Thermo Fisher Scientific, CA). Then, the sections were counterstained with DAPI (Sigma-Aldrich, MO). A Leica SP8 confocal microscope captured the images.
ChIP assay
According to the manufacturer’s instructions, ChIP assays were performed using the SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads) (CST, MA). We fixed neurons in the culture medium containing 1% formaldehyde at room temperature for 10 min to crosslink proteins and DNA. Then cells were lysed and incubated with 0.5 μl of micrococcal nuclease for 20 min at 37 °C to digest DNA to 200–500 bp. The mixture was then immunoprecipitated with 2 μg of NRF1 antibody (ab34682, Abcam, MA) or a negative control IgG at 4 °C overnight. The purified DNA was amplified by real-time PCR with primers in Supplementary Table S1.
Dual-luciferase reporter assay
Fragments of rat Grms promoter region Grm1 (−1700/−1200), Grm1 (−400/+100), Grm2 (−1700/−1200), Grm8 (−1650/−1150), and Grm8 (−1250/−750) were cloned into the KpnI and HindIII restriction sites of the pGL3-basic vector (Promega, WI). All constructs were verified by sequencing. PC12 cells (80–90% confluence) were seeded and cultured in the 24-well plate the day before transfection. Cells were co-transfected with 250 ng reporter plasmids, 10 pmol NRF1 siRNA, and 10 ng of pRL-TK (Promega, WI) as an internal control by Lipofectamine 2000 Reagent. After 48 h, we treated cells with OGD for 2 h. Finally, the Dual-Luciferase Reporter Assay System detected the luciferase activities (Promega, WI). Firefly luminescence signal was normalized by Renilla luminescence signal.
Glutamate assay
The relative intracellular glutamate was analyzed using a Glutamate Assay Kit (ab83389, Abcam, MA). Briefly, cells were harvested and homogenized in assay buffer, and the insoluble fraction was removed by centrifugation. The absorbance at OD 450 nm was measured using a microplate reader (Synergy 2, BioTek).
Statistical analysis
GraphPad Prism v. 8 (GraphPad Software, CA) was used to analyze the data by Student’s t-test or One-way ANOVA. All the data were presented as mean ± SD. The level of significance was as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
NRF1 and some glutamatergic genes were synchronously downregulated in the neonatal HIE rats model
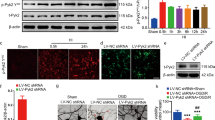
To construct the neonatal HIE rats, we used unilateral carotid artery ligation to block the blood supply to the brain of 7-day-old rats. Results in Fig. 1a, b demonstrated that infarction occurred in the right hemispheres. Nissl bodies reduced, disintegrated, and even disappeared on the infarct side (Fig. 1c). Besides, we evaluated the utility of the neonatal HIE rats using the Bederson Scale. We found that the motor ability of the neonatal HIE rats was significantly impaired (Fig. 1d). We also constructed HI neurons by OGD treatment. The cell viability declined time-dependent when neurons were subjected to OGD. NRF1-overexpressing cells exhibited increased cell viability compared with the control groups (Fig. 1e). We also found that NRF1 expression was significantly reduced in neurons on the injured side (Fig. 1f–i). The above results suggested that NRF1 levels were downregulated in the infarcted tissue of the neonatal HIE model.

Cerebral infarcts were determined using TTC staining (a) and Nissl staining (c). The percentage of infarct area and volume were displayed in relation to the whole brain (b, n = 6, ****p < 0.0001 by Student’s t-test). Bederson Scale evaluated the effectiveness of the model construction (d, n = 12). The NRF1 overexpression lentivirus infected primary neurons. The neurons were exposed to a glucose-free medium and 1% O2 for 2, 4, and 6 h (e). Brain sections were co-stained with anti-NRF1 and anti-NeuN antibodies to display NRF1 in neurons (f) and evaluated NRF1 level in NeuN+ cells (i, n = 5, ***p < 0.001 by Student’s t-test). Western blot detected the NRF1 level of IS and OS tissues (g, h, ***p < 0.001 by Student’s t-test). OS opposite side, IS infarct side.
Furthermore, we examined the gene expression in the infarct tissues. The glutamatergic candidate genes were remarkably downregulated (Fig. 2a). We verified the relevant gene expressions by real-time PCR (Fig. 2b). iGluRs, including Gria2, Grin1, Grin2b, Grin2c, Grik1, and Grik2 mRNA levels, were decreased on the infarct side. mGluRs, including Grm3, Grm5, and Grm8 mRNA levels, were significantly reduced on the infarct side. The results demonstrated that NRF1 was downregulated in parallel with iGluRs and mGluRs in infarcted tissues.

Total RNA of IS and OS brains were isolated and purified. Expressions of genes related to glutamatergic neurotransmitter were scanned by RT² Profiler PCR Array (a) and validated by real-time PCR (b, n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001 by Student’s t-test). OS opposite side, IS infarct side.
NRF1 overexpression rescued cognitive deficits and elevated glutamatergic candidate genes in the neonatal HIE rats model
We created the NRF1 overexpression in rat brain model to examine the role of NRF1 in HI-induced neurological damage (Fig. 3a). Next, we examined the effect of NRF1 overexpression on the integrity of brain tissue and cognitive function in the neonatal HIE model on the 28th day after birth. The brain tissue volume on the infarcted side was noticeably smaller than on the opposite side (Fig. 3b, c). There was no significant change in the avoidance latency from day 1 to day 3. From day 4 onwards, NRF1 OE rats started to shorten the escape latency with statistical significance (p < 0.01) (Fig. 3d). Neonatal HIE considerably decreased the frequency of platform passes (p < 0.001), whereas NRF1 overexpression significantly increased the frequency (p < 0.01) (Fig. 3e). The above results indicated that NRF1 improved the learning memory function.

The NRF1 overexpression lentivirus infected neonatal rats’ brains by stereotaxic injection (a). The infarct is the right cerebral hemisphere. The percentage (%) of brain volume loss was shown in relation to whole brain (b, c, n = 6, **p < 0.01 and ****p < 0.0001 by one-way ANOVA). MWM detected cognitive functions. The latency of reaching the platform during the training test (d, n = 12, *p < 0.05 and **p < 0.01 by Student’s t-test) and the frequency of crossing the target quadrant in the probe test (e, n = 12, *p < 0.05 and ***p < 0.001 by Student’s t-test, n.s. indicated no significance) were quantified. mRNA level of glutamate pathway-related factors Grin1, Grin2a, Grin2b, Grin2c, Gria1, Gria2, Grik1 and Grik2 (f) and metabotropic glutamate receptors Grm1, Grm2, Grm3, Grm4, Grm5, Grm7, and Grm8 (g) were measured by real-time PCR (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001 by Student’s t-test).
Furthermore, we observed altered expression of glutamatergic candidate genes in neonatal HIE rats’ infarcted brain tissue. iGluRs including Grin1, Grin2a, Grin2b, Gria2, Grik1, and Grik2 dramatically decreased on the obstructed side, and NRF1 overexpression restored the levels of Grin1, Grin2a, Grin2b, Gria2. However, neither infarcted nor NRF1 overexpression tissues significantly affected Grin2c or Gria1 (Fig. 3f). In the infarcted brain, Grm1, Grm2, Grm3, Grm7, and Grm8 levels also considerably decreased. Additionally, NRF1 overexpression reversed the decline in these mGluRs expression on the infarct side. However, Grm4 and Grm5 did not considerably change (Fig. 3g). The above results suggested that NRF1 effectively improved neonatal HIE rats’ cognitive function, accompanied by upregulation of iGluRs and mGluRs.
NRF1 effectively attenuated the reduction of mGluRs in OGD neurons
As shown in Fig. 4a, b, OGD treatment time-dependently decreased NRF1 levels. Similar to the results in vivo, Grm1, Grm2, and Grm8 expression levels drastically decreased after OGD treatment (Fig. 4c). To determine whether NRF1 impacted mGluRs expression, we make neurons overexpress NRF1 in OGD treatment (Fig. 4d, e). As shown in Fig. 4f, NRF1 overexpression significantly increased the expressions of Grm1, Grm2, and Grm8. The above results indicated that NRF1 successfully lessened mGluRs decrease in OGD neurons.

Primary neurons were treated with OGD by exposure to a glucose-free medium and 1% O2 for 0, 2, and 4 h. Then, cells were lysed to determine NRF1 level by western blot (a, b, **p < 0.01, and ***p < 0.001 by one-way ANOVA). After OGD treatment for 2 h, real-time PCR detected mRNA levels of Grm1, Grm2, Grm3, Grm4, Grm5, Grm7, and Grm8 in primary neurons (c, *p < 0.05 vs Con by Student’s t-test). Primary neurons were infected with lentivirus expressing NRF1 and followed with OGD treatment for 2 h. NRF1 mRNA expressions were determined by RT-PCR (d, e, *p < 0.05, and **p < 0.01 vs Vector group by Student’s t-test), and mRNA levels of Grm1, Grm2, Grm3, Grm4, Grm5, Grm7, and Grm8 were measured by real-time PCR (f, *p < 0.05, **p < 0.01, and ***p < 0.001 by Student’s t-test).
NRF1 transcriptionally activated metabotropic glutamate receptors in OGD neurons
We performed ChIP-PCR experiments to confirm whether NRF1 was bound to the Grm1, Grm2, and Grm8 promoter regions. As shown in Fig. 5a, NRF1 significantly enriched the promoter regions of mGluRs, including Grm1 (–1684/–1422 and –233/+28), Grm2 (–1617/–1517 and –1436/–1196), and Grm8 (–1662/–1518, –1432/–1216 and –1012/–832). Under OGD treatment, we discovered that the luciferase activity of the Grm1, Grm2, and Grm8 reporters drastically decreased. The outcome showed that OGD prevented Grm1, Grm2, and Grm8 from being transcribed (Fig. 5b). We transfected the NRF1 siRNA and evaluated the interference efficiency to demonstrate that Grm1, Grm2, and Grm8 were downregulated by NRF1 transcription (Fig. 5c). The results showed that NRF1 positively regulated the transcription of Grm1, Grm2, and Grm8 and that interference with NRF1 was linked to a decrease in the luciferase activities of Grm1, Grm2, and Grm8 reporters (Fig. 5e). The results above suggest that OGD therapy may inhibit NRF1 from restricting mGluR transcription. NRF1 knockdown under the OGD condition dramatically reduced intracellular glutamate (Fig. 5d), indicating enhanced glutamate release from neurons.

After lysis of primary neurons, ChIP enriched NRF1-bound DNA. Enrichment of the promoter region of mGluRs were determined by real-time PCR (a, *p < 0.05, **p < 0.01, and ***p < 0.001 vs IgG by Student’s t-test). PC12 cells were transfected with mGluRs reporter gene plasmids. The pRL-TK vector was co-transfected to normalize transfection efficiencies. Then cells were treated with OGD for 2 h. A dual-luciferase assay determined the luciferase activity. Results are presented as a luciferase/Renilla ratio (b, ***p < 0.001 vs Con by Student’s t-test). PC12 cells were transfected with siNRF1 or siCon. The interference efficiency of NRF1 was examined using western blot (c). The concentration of intracellular glutamate was measured in PC12 by OGD for 2 h (d, **p < 0.01 by Student’s t-test). PC12 cells were co-transfected with mGluRs reporter gene plasmids and siNRF1. The pRL-TK vector was co-transfected to normalize transfection efficiencies. Results are presented as a luciferase/Renilla ratio (e, **p < 0.01 and ***p < 0.001 vs siCon by Student’s t-test).
Discussion
In this study, we performed the common carotid artery ligation combined with hypoxia to investigate the mechanism of brain injury caused by perinatal hypoxia and ischemia.36 According to the infarct area and behavioral testing, the developed model is appropriate for studying non-mild neonatal HIE.
In an intrauterine HI model, mGluR1 expression in the dentate gyrus of ischemic animals was delayed and markedly reduced at postnatal day 7.37 We found that almost all types of mGluRs and numerous glutamate receptors were dramatically diminished in the newborn HIE model, which was consistent with previous studies. mGluR1 is widely distributed in the central nervous system.38 mGlu1α in the hippocampus steadily increases during postnatal development, suggesting a potential role in learning memory.39 mGluR1-deficient mice exhibit learning dysfunction and severe motor incoordination.40,41 mGlu2 has mainly been found in the presynaptic structures of mossy fibers and perforant paths in the hippocampus.42 mGluR2 expression increases rapidly during the first two weeks of life.43 Synaptic plasticity and learning complicated motor tasks are impaired in mGluR2-deficient mice.44,45 mGluR8 is broadly distributed in the central nervous system and is mainly localized in the active regions of presynaptic neurotransmitter release.12,46 mGlu8 transcript expression declines progressively during postnatal development47 and has been linked to anxiety and pain.48,49 Given the extensive significance of mGluRs in motor and cognitive activities outlined above, upregulating NRF1 could enhance cognitive skills in neonatal HIE rats by stimulating the transcription of mGluR1/2/8. The selective agents of mGluR1/2/5 showed the neuroprotective effects of ischemic brain injury in neonatal HIE rats.17,20,21,22 The neuroprotection mechanisms of mGluRs remain not fully understood and appear complex. Hilton et al. demonstrated that glutamate-induced cell death in immature hippocampal neurons was mediated primarily by mGluRs and subsequent metabotropic Ca2+ release from intracellular stores.50 Bratek et al. investigated that mGluR2/3 activation in a short time after neonatal HI triggered neuroprotective mechanisms by inhibiting oxidative stress and ROS production.17 We identified for the first time that NRF1 was severely reduced in the infarcted region and partially reversed the decrease in mGluR1, mGluR2, and mGluR8 in the neonatal HIE model. The functional NRF1 binding sites were present in the promoter regions of Grm1, Grm2, and Grm8. It has been reported that Grin1, Grin2b, and Gria2 are downstream target genes of NRF1.51,52 We also detected that NRF1 could rescue cognitive impairment caused by neonatal HIE. Therefore, we hypothesized that NRF1 transcriptionally regulated numerous glutamate receptors, which might impact brain function.
Numerous studies have demonstrated that hypoxia-induced glutamate over-release can activate iGluRs, leading to impaired neurological functions.53,54,55 Because of the restricted blood flow brought on by ligation and hypoxia, the brain receives less oxygen and glucose. The immediate fall in energy triggers a series of additional mechanisms, beginning with a failure of the ATP-dependent Na+-K+ pump, followed by membrane depolarization and excessive glutamate release.56,57 In the present study, interference with NRF1 expression on the infarct side increased glutamate release. We speculated that NRF1 affected glutamate release to some extent through transcriptional regulation of Na+/K+-ATPase subunits α1 and β1.58 The ameliorative effect of NRF1 on neurological function might associate with a reduction in glutamate release, which lessened neurotoxicity.
Furthermore, NRF1 might impact neonatal HIE through the following mechanisms. The energy failure results in energy metabolism disorder in mitochondria. NRF1 regulates the mitochondrial respiratory chain and mitochondrial DNA replication.59 For example, NRF1 regulates cytochrome c oxidase subunit genes in neurons, which is an essential enzyme of the energy-generating machinery.60 The evidence suggested a mechanism for coordinating regulation of NRF1 and energy consumption in the HI model. Mitophagy in neurons is another potential mechanism. Our previous study found that NRF1 was involved in the regulation of mitophagy by regulating the PINK1/Parkin pathway.25 PINK1/Parkin pathway is involved in the pathophysiological reoxygenation process after ischemia-reperfusion and neuronal OGD model.61 The reduction of NRF1 in the infarct zone might inhibit PINK1/Parkin-associated mitochondrial autophagy, resulting in neuronal cell loss.
NRF1 efficiently synchronized the expression of its target genes to create a constant and harmonic connection among energy metabolism, synaptic transmission, and autophagy. Therefore, NRF1 might be able to initiate and/or maintain the intricate processes of HIE. In the present study, we discovered that NRF1 regulated the transcriptional level of mGluRs in the neonatal HIE model and could effectively reverse the neurological impairment. The novel findings of the effect of NRF1 on HIE may create fresh concepts for future in-depth investigation.
References
Qin, X. et al. Mechanism and treatment related to oxidative stress in neonatal hypoxic-ischemic encephalopathy. Front. Mol. Neurosci. 12, 88 (2019).
Dixon, B. J., Reis, C., Ho, W. M., Tang, J. & Zhang, J. H. Neuroprotective strategies after neonatal hypoxic ischemic encephalopathy. Int. J. Mol. Sci. 16, 22368–22401 (2015).
Eunson, P. The long-term health, social, and financial burden of hypoxic-ischaemic encephalopathy. Dev. Med. Child Neurol. 57(Suppl 3), 48–50 (2015).
Shibasaki, J. et al. Changes in brain metabolite concentrations after neonatal hypoxic-ischemic encephalopathy. Radiology 288, 840–848 (2018).
Hernandez, D. E. et al. Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J. Cell Sci. 131, jcs214684 (2018).
Northington, F. J., Chavez-Valdez, R. & Martin, L. J. Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 69, 743–758 (2011).
Taniguchi, H., Anacker, C., Wang, Q. & Andreasson, K. Protection by vascular prostaglandin E2 signaling in hypoxic-ischemic encephalopathy. Exp. Neurol. 255, 30–37 (2014).
Nakanishi, S. Molecular diversity of glutamate receptors and implications for brain function. Science 258, 597–603 (1992).
Willard, S. S. & Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 9, 948–959 (2013).
Peyrovian, B. et al. The glycine site of NMDA receptors: a target for cognitive enhancement in psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 92, 387–404 (2019).
Stansley, B. J. & Conn, P. J. The therapeutic potential of metabotropic glutamate receptor modulation for schizophrenia. Curr. Opin. Pharm. 38, 31–36 (2018).
Caraci, F., Nicoletti, F. & Copani, A. Metabotropic glutamate receptors: the potential for therapeutic applications in Alzheimer’s disease. Curr. Opin. Pharm. 38, 1–7 (2018).
Zhang, Z. et al. Roles of glutamate receptors in Parkinson’s disease. Int. J. Mol. Sci. 20, 4391 (2019).
Hanada, T. Ionotropic glutamate receptors in epilepsy: a review focusing on AMPA and NMDA receptors. Biomolecules 10, 464 (2020).
Celli, R. et al. Targeting metabotropic glutamate receptors in the treatment of epilepsy: rationale and current status. Expert Opin. Ther. Targets 23, 341–351 (2019).
Mayor, D. & Tymianski, M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 134, 178–188 (2018).
Bratek, E., Ziembowicz, A., Bronisz, A. & Salinska, E. The activation of group II metabotropic glutamate receptors protects neonatal rat brains from oxidative stress injury after hypoxia-ischemia. PLoS One 13, e0200933 (2018).
Bratek, E., Ziembowicz, A. & Salinska, E. Pretreatment with group II metabotropic glutamate receptor agonist Ly379268 protects neonatal rat brains from oxidative stress in an experimental model of birth asphyxia. Brain Sci. 8, 48 (2018).
Zhang, X. & Peng, K. The function of the NMDA receptor in hypoxic-ischemic encephalopathy. Front. Neurosci. 14, 567665 (2020).
Cai, Z., Lin, S. & Rhodes, P. G. Neuroprotective effects of N-acetylaspartylglutamate in a neonatal rat model of hypoxia-ischemia. Eur. J. Pharm. 437, 139–145 (2002).
Bratek-Gerej, E., Bronisz, A., Ziembowicz, A. & Salinska, E. Pretreatment with Mglur2 or Mglur3 agonists reduces apoptosis induced by hypoxia-ischemia in neonatal rat brains. Oxid. Med. Cell Longev. 2021, 8848015 (2021).
Makarewicz, D., Duszczyk, M., Gadamski, R., Danysz, W. & Lazarewicz, J. W. Neuroprotective potential of group I metabotropic glutamate receptor antagonists in two ischemic models. Neurochem. Int. 48, 485–490 (2006).
Evans, M. J. & Scarpulla, R. C. Interaction of nuclear factors with multiple sites in the somatic cytochrome C promoter. characterization of upstream Nrf-1, Atf, and intron Sp1 recognition sequences. J. Biol. Chem. 264, 14361–14368 (1989).
Zhou, Y. et al. Nuclear respiratory factor 1 promotes spheroid survival and mesenchymal transition in mammary epithelial cells. Oncogene 37, 6152–6165 (2018).
Lu, Y. et al. Positive regulation of human Pink1 and Parkin gene expression by nuclear respiratory factor 1. Mitochondrion 51, 22–29 (2020).
Pepin, M. E. et al. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. Am. J. Physiol. Heart Circ. Physiol. 317, H674–H684 (2019).
Ramos, J. et al. NRF1 motif sequence-enriched genes involved in ER/PR -ve HER2 +ve breast cancer signaling pathways. Breast Cancer Res. Treat. 172, 469–485 (2018).
Satoh, J., Kawana, N. & Yamamoto, Y. Pathway analysis of ChiP-Seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul. Syst. Bio 7, 139–152 (2013).
Wang, X. et al. Transcription regulation of NRF1 on star reduces testosterone synthesis in hypoxemic murine. J. Steroid Biochem. Mol. Biol. 191, 105370 (2019).
Jiang, S. et al. Testosterone attenuates hypoxia-induced hypertension by affecting NRF1-mediated transcriptional regulation of ET-1 and ACE. Hypertens. Res. 44, 1395–1405 (2021).
Wang, D., Zhang, J., Lu, Y., Luo, Q. & Zhu, L. Nuclear respiratory factor-1 (NRF-1) regulated hypoxia-inducible factor-1alpha (HIF-1alpha) under hypoxia in Hek293t. IUBMB Life 68, 748–755 (2016).
Bederson, J. B. et al. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17, 472–476 (1986).
Zha, L. et al. NLRC3 delays the progression of Ad in APP/PS1 mice via inhibiting Pi3k activation. Oxid. Med. Cell Longev. 2020, 5328031 (2020).
Wu, X. M. et al. Bi-directionally protective communication between neurons and astrocytes under ischemia. Redox Biol. 13, 20–31 (2017).
Li, Z. et al. Ginkgolides protect Pc12 cells against hypoxia-induced injury by P42/P44 Mapk pathway-dependent upregulation of HIF-1alpha expression and Hif-1dna-binding activity. J. Cell Biochem. 103, 564–575 (2008).
Vannucci, R. C. & Vannucci, S. J. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev. Neurosci. 27, 81–86 (2005).
Simonyi, A., Zhang, J. P. & Sun, G. Y. Changes in mRNA levels for group I metabotropic glutamate receptors following in utero hypoxia-ischemia. Brain Res. Dev. Brain Res. 112, 31–37 (1999).
Martin, L. J., Blackstone, C. D., Huganir, R. L. & Price, D. L. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron 9, 259–270 (1992).
Shigemoto, R., Nakanishi, S. & Mizuno, N. Distribution of the mRNA for a metabotropic glutamate receptor (Mglur1) in the central nervous system: an in situ hybridization study in adult and developing rat. J. Comp. Neurol. 322, 121–135 (1992).
Aiba, A. et al. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in Mglur1 mutant mice. Cell 79, 365–375 (1994).
Aiba, A. et al. Deficient cerebellar long-term depression and impaired motor learning in Mglur1 mutant mice. Cell 79, 377–388 (1994).
Neki, A. et al. Metabotropic glutamate receptors Mglur2 and Mglur5 are expressed in two non-overlapping populations of Golgi cells in the rat cerebellum. Neuroscience 75, 815–826 (1996).
Catania, M. V. et al. Metabotropic glutamate receptors are differentially regulated during development. Neuroscience 61, 481–495 (1994).
Pekhletski, R. et al. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the Mglur4 subtype of metabotropic glutamate receptor. J. Neurosci. 16, 6364–6373 (1996).
Morishima, Y. et al. Enhanced cocaine responsiveness and impaired motor coordination in metabotropic glutamate receptor subtype 2 knockout mice. Proc. Natl. Acad. Sci. USA 102, 4170–4175 (2005).
Ferraguti, F. et al. Metabotropic glutamate receptor 8-expressing nerve terminals target subsets of gabaergic neurons in the hippocampus. J. Neurosci. 25, 10520–10536 (2005).
Duvoisin, R. M., Zhang, C. & Ramonell, K. A novel metabotropic glutamate receptor expressed in the retina and olfactory bulb. J. Neurosci. 15, 3075–3083 (1995).
Marabese, I. et al. Periaqueductal gray metabotropic glutamate receptor subtype 7 and 8 mediate opposite effects on amino acid release, rostral ventromedial medulla cell activities, and thermal nociception. J. Neurophysiol. 98, 43–53 (2007).
Duvoisin, R. M. et al. Increased measures of anxiety and weight gain in mice lacking the group iii metabotropic glutamate receptor Mglur8. Eur. J. Neurosci. 22, 425–436 (2005).
Hilton, G. D., Nunez, J. L., Bambrick, L., Thompson, S. M. & McCarthy, M. M. Glutamate-mediated excitotoxicity in neonatal hippocampal neurons is mediated by Mglur-induced release of Ca++ from intracellular stores and is prevented by estradiol. Eur. J. Neurosci. 24, 3008–3016 (2006).
Dhar, S. S. & Wong-Riley, M. T. Coupling of energy metabolism and synaptic transmission at the transcriptional level: role of nuclear respiratory factor 1 in regulating both cytochrome C oxidase and NMDA glutamate receptor subunit genes. J. Neurosci. 29, 483–492 (2009).
Dhar, S. S., Liang, H. L. & Wong-Riley, M. T. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome C oxidase: tight coupling of glutamatergic transmission and energy metabolism in neurons. J. Neurochem. 108, 1595–1606 (2009).
Chen, M. et al. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39, 3042–3048 (2008).
Bordi, F., Pietra, C., Ziviani, L. & Reggiani, A. The glycine antagonist Gv150526 protects somatosensory evoked potentials and reduces the infarct area in the Mcao model of focal ischemia in the rat. Exp. Neurol. 145, 425–433 (1997).
Wang, H. et al. The Glun3a subunit exerts a neuroprotective effect in brain ischemia and the hypoxia process. ASN Neuro. 5, 231–242 (2013).
Brekke, E., Berger, H. R., Wideroe, M., Sonnewald, U. & Morken, T. S. Glucose and intermediary metabolism and astrocyte-neuron interactions following neonatal hypoxia-ischemia in rat. Neurochem. Res. 42, 115–132 (2017).
Lai, M. C. & Yang, S. N. Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011, 609813 (2011).
Johar, K., Priya, A. & Wong-Riley, M. T. Regulation of Na(+)/K(+)-Atpase by nuclear respiratory factor 1: implication in the tight coupling of neuronal activity, energy generation, and energy consumption. J. Biol. Chem. 287, 40381–40390 (2012).
Scarpulla, R. C. Nuclear control of respiratory chain expression by nuclear respiratory factors and Pgc-1-related coactivator. Ann. N. Y. Acad. Sci. 1147, 321–334 (2008).
Dhar, S. S., Ongwijitwat, S. & Wong-Riley, M. T. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome C oxidase in neurons. J. Biol. Chem. 283, 3120–3129 (2008).
Lan, R. et al. Mitophagy is activated in brain damage induced by cerebral ischemia and reperfusion via the Pink1/Parkin/P62 signalling pathway. Brain Res. Bull. 142, 63–77 (2018).
Acknowledgements
We thank Xiaomei Wu from Nantong University, who kindly provided PC12 cells as gifts. We also thank Qianqian Luo and Yapeng Lu at Nantong University for their suggestions for experimental design.
Funding
The National Natural Science Foundation of China (31671206) and the Special Foundation for Excellent Young Teachers and Principals Program of Jiangsu Province supported the study.
Author information
Authors and Affiliations
Contributions
L.Z. and D.W. conceived the study. D.W., M.Q.X., and L.L.H. performed the experiments, analyses, and interpreted the data. D.W. and X.T.W. drafted the article, and all authors contributed to the manuscript. L.Z. reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, D., Xu, M., Huang, L. et al. Transcriptional regulation of NRF1 on metabotropic glutamate receptors in a neonatal hypoxic‑ischemic encephalopathy rat model. Pediatr Res 93, 1865–1872 (2023). https://doi.org/10.1038/s41390-022-02353-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-022-02353-9
