
Abstract
Glucocorticoid induction of pulmonary surfactant involves a mesenchyme-derived protein first characterized in 1978 by Smith and termed fibroblast-pneumocyte factor (FPF). Despite a number of agents having been postulated as being FPF, its identity has remained obscure. In the past decade, three strong candidates for FPF have arisen. This review examines the evidence that keratinocyte growth factor (KGF), leptin or neuregulin-1β (NRG-1β) act as FPF or components of it. As with FPF production, glucocorticoids enhance the concentration of each of these agents in fibroblast-conditioned media. Moreover, each stimulates the synthesis of surfactant-associated phospholipids and proteins in type II pneumocytes. Further, some have unique activities, for example, KGF also minimizes lung injury through enhanced epithelial cell proliferation and NRG-1β enhances surfactant phospholipid secretion and β-adrenergic receptor activity in type II cells. However, even though these agents have attributes in common with FPF, it is inappropriate to specify any one of these agents as FPF. Rather, it appears that each contributes to separate mesenchymal-epithelial signaling mechanisms involved in different aspects of lung development. Given that the production of pulmonary surfactant is essential for postnatal survival, it is reasonable to suggest that several mechanisms independently regulate surfactant synthesis.
Similar content being viewed by others


Main
The internal surfaces of lung alveoli are lined with a monolayer of pulmonary surfactant, which reduces the surface tension at the air:liquid interface. This prevents alveolar collapse at low distending pressure, sustaining alveolar volume even after expiration. Surfactant’s functional properties are specified by its phospholipid and protein components. Since the lungs are not involved in gas exchange until after birth, it is not surprising that surfactant is only formed in large amounts in the later stages of gestation. Premature birth is often accompanied by lung immaturity, including surfactant deficiency that frequently leads to neonatal respiratory distress syndrome (1,2).
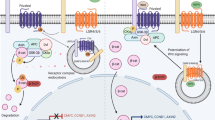
Since the pioneering work of Liggins in sheep (3), it has been established that, late in gestation of a variety of species, exogenous glucocorticoid hormones dramatically stimulate fetal lung maturation (4,5,6,7,8,9,10), including an accelerated appearance of type II pneumocytes and pulmonary surfactant. This stimulatory effect is indirect. Glucocorticoids stimulate lung fibroblasts to induce the production and release of a peptide factor(s) termed fibroblast-pneumocyte factor (FPF). FPF stimulates type II cells to mature and begin augmented synthesis of surfactant phospholipids and proteins (11,12). Smith and Fletcher (13) observed that pulmonary surfactant synthesis in fetal lung depends upon paracrine interaction between mesenchymal cells (fibroblasts) and the neighbouring type II epithelial cells. The paracrine factor was found to be a heat stable, dialyzable polypeptide with an apparent molecular weight of 5–15 kDa that stimulated type II pneumocytes to synthesize and secrete surfactant (14). Since the original description of FPF there have been numerous attempts to define its identity. Although several factors may facilitate mesenchyme-epithelial cell interactions (15,16,17,18,19,20), currently there are only three primary candidates for FPF, namely keratinocyte growth factor (KGF), leptin, and neuregulin-1β (NRG-1β).
KGF
Stimulation of Type II Cell Proliferation
KGF, a heparin-binding protein otherwise known as fibroblast growth factor 7 (FGF-7), is a product of lung fibroblasts and vascular smooth muscle cells (21). It interacts with its receptor KGF-R (FGFR2 IIIb), which is present on lung epithelial cells, and enhances their proliferation (22,23,24,25). Intratracheal and intravenous KGF each induced proliferation of lung cuboidal cells containing lamellar bodies, which are characteristic of type II pneumocytes (22). KGF also promoted lung morphological and physiological maturation (17).
KGF Induces Production of Surfactant Components
KGF induces surfactant production both in vitro and in vivo (17,26,27). Incorporation of [3H]-choline into disaturated surfactant phospholipids in fetal alveolar type II cells is significantly enhanced by KGF. This effect is time-dependent, requiring 48 h exposure for a maximal response, and also concentration-dependent ( Figure 1 ) (17). Stimulation is associated with increased activities of choline phosphate cytidylyltransferase and fatty acid synthase and elevated expression of their corresponding genes. In addition to stimulating disaturated phosphatidylcholine (DSPC) synthesis, KGF enhances both the expression (17,26,27,28) and stability (17) of SP-A, SP-B, SP-C, and SP–D mRNA. Thus, KGF elevates the production of all of the major components of surfactant. Its stimulatory effect on the expression of the four surfactant-associated proteins, especially SP-A and SP-D, may contribute to its ability to attenuate pulmonary infection (29).

Effect of keratinocyte growth factor (KGF), leptin, and NRG-1β on surfactant phospholipid synthesis. Cultured fetal rat type II cells were incubated with the indicated concentrations of KGF (filled circle; Chelly et al. (17)), leptin (filled square; Torday et al. (18)) or NRG-1β (filled triangle; King et al. (69)) for 24–48 h prior to measuring the extent of incorporation of [3H]-choline into surfactant phospholipids relative to that which occurred in control cultures.
KGF Receptors (FGFR2 IIIb) in the Lung
In mammals, fibroblast growth factors bind to alternatively spliced forms of four tyrosine kinase FGF receptors (FGFR1-4), each of which contain an intracellular ligand binding domain, a transmembrane domain and an intracellular tyrosine kinase (30,31). The Fgfr2 gene is alternatively spliced to encode two receptor proteins that have different ligand-binding specificities and tissue distributions. One of these, FGFR2 IIIb, is found mainly in epithelial cells and is activated by FGF-1, FGF-3, FGF-7 (KGF), and FGF-10, which are mainly synthesized in the mesenchyme (30,32). Thus, whereas KGF is expressed in mesenchymal cells, its receptor (KGF-R otherwise known as FGFR2 IIIb) is found predominantly in epithelia, including alveolar type II pneumocytes (17,33). KGF and its receptor are involved in both early lung branching morphogenesis and late gestation promotion of surfactant production.
The above observations indicate that mesenchymal-epithelial interactions generate KGF production by fibroblasts to regulate type II cell differentiation (34). KGF appears to be involved in the induction of surfactant synthesis by glucocorticoids (discussed below) and it has been suggested that KGF has many of the properties of FPF. However, it is larger (28 kDa) (21) than the 5–15 kDa ascribed to FPF by Smith and Post (14).
Leptin
Leptin is a 167-amino acid protein secreted by adipocytes and is essential in the regulation of energy balance within organisms (35). Leptin participates in metabolic regulation (36); it also functions in inflammatory and immune responses (37). For example, leptin expression is decreased in the bronchial epithelium of untreated asthmatics (37). Hoggard et al. (35) found high levels of leptin and its receptor in the fetal lung of mice at 14.5 d gestation suggesting that leptin acts in lung growth and development.
Leptin induces an increase in fetal lung weight relative to body weight, possibly through an increase in the number of type II pneumocytes (38). It has been reasoned that placental leptin has important functions in fetal and neonatal growth, and prevents depressed respiration in leptin-deficient mice (39,40). A 48 h antenatal exposure to leptin significantly enhanced the relative alveolar surface area and improved the lung maturity of fetal rats. Others have shown that ob/ob mice, which are leptin deficient, have reduced lung volume, diminished alveolar surface area and exhibit alveolar hyperventilation and chronic hypercapnia (40,41). Daily leptin administration overcomes these respiratory complications, indicating that leptin acts as both a growth factor in the lung and as a neurohumoral modulator of the central respiratory control mechanisms (40,42,43). These observations support the concept that leptin has a significant role in promoting normal lung maturation.
Influence of Leptin on the Production of Surfactant Components
Torday et al. (18) and Kirwin et al. (38) independently showed that during rat lung development leptin expression by fetal lung fibroblasts begins on embryonic day 17 and increases 7–10-fold by day 20. Its corresponding receptor is expressed in type II cells (44) indicative of a paracrine signaling loop in which leptin stimulates the synthesis of DSPC in the type II cells. This has been demonstrated in fetal rats (18) and rabbits (44) and, to a lesser extent, in adult human airway epithelial cells (18). Leptin stimulation of fetal type II cell DSPC is evident after 24 h exposure and appears to plateau at concentrations above 3.1 nmol/l (50 ng/ml), at which concentration it produces a 3.3-fold increase in DSPC synthesis ( Figure 1 ). Thus, it maximally stimulates surfactant phospholipid synthesis to the same extent as KGF and at a similar concentration. Although leptin stimulates DSPC synthesis, it has no stimulatory effect on surfactant phospholipid secretion from type II cells (45).
Subjecting cocultured lung fibroblasts and type II cells to stretch increases surfactant phospholipid synthesis threefold (46). This effect of stretch on alveolar fibroblast and type II cell differentiation is coordinated by parathyroid hormone-related protein (PTHrP), leptin, and their receptors (46). It involves a paracrine feedback loop in which the stretched type II cells release PTHrP, which increases fibroblast leptin release, triggering enhanced surfactant DSPC in type II cells.
PTHrP and PTH/PTHrP receptor expression is developmentally regulated in lung epithelial and mesenchymal cells, respectively. Removal of the PTHrP gene by homologous recombination produced fetal and newborn mice with delayed type II cell differentiation, reduced lamellar body formation and diminished pulmonary surfactant production (47). PTHrP enhances leptin production by fetal rat lung fibroblasts (48), leading to stimulation of de novo synthesis of DSPC in type II cells (18).
The formation and stability of surfactant is highly dependent on SP-B and SP-C (49), while SP-A and SP-D are important components of the innate immune response to microbial challenge and of additional aspects of pulmonary immune and inflammatory regulation (50). Exposure to leptin stimulates the expression of SP-A (38,39) and SP-B (18,38) in either fetal rat lung explants (18,38) or fetal growth-restricted rats (39). Leptin also enhances SP-C gene expression (38).
Leptin Receptors in the Lung
The leptin receptor is a member of the cytokine receptor superfamily (51), having at least five splice variants (Ob-R(a-e)), all derived from a single gene (52). These variants differ in the length of the intracellular domain with the Ob-Rb isoform having the longest cytoplasmic domain. Ob-Rb is considered to be the only form of the receptor that interacts with Jak2 tyrosine kinase and signals leptin’s physiological actions (53).
Ob-Rb is highly expressed in both adult mouse (35) and human (54) lung tissue, as well as in fetal lung tissue of mice (35), rats (18) and rabbits (44). Although leptin receptors are not present in fetal rat lung fibroblasts they are present in fetal rat type II cells at gestational day 18 and progressively increase until day 21 (18).
The foregoing studies showed that leptin has many of the characteristics described for FPF, including the following: the molecular weight of leptin (16 kDa) is close to the range previously reported for FPF (14); it is expressed by lung fibroblasts during fetal development; and its expression is stimulated by glucocorticoids, as discussed below (18). However, there is some controversy over the role of leptin in lung development. For example, other investigators have failed to identify an effect of leptin on fetal mouse or fetal sheep lung surfactant development, and found normal surfactant levels in ob/ob neonatal mice at all ages (55). Further, the proposed model of leptin regulating fetal lung maturation is closely tied to the function of pulmonary lipofibroblasts; while this cell type exists in rodent lungs there remains controversy regarding the existence of these cells in human lungs (56,57,58), adding uncertainty regarding the importance of leptin for development of surfactant synthesis in the human lung.
Neuregulin-1β
Neuregulin-1β (NRG-1β) is a member of a family of EGF-like growth factors expressed in multiple tissues (59,60,61). In normal human development the pulmonary concentration of NRG-1β is unchanged until 22 wk of gestation, at which time it increases sharply, reaching a sevenfold higher concentration in the lung by 33–35 wk of gestation (62). In human fetal lung explants, NRG-1β enhances both epithelial cell volume density and epithelial cell proliferation by approximately twofold (63) and induces branching morphogenesis through a PI3K signal pathway (64). The transcriptional product of the neuregulin-1 gene is subject to alternative splicing reactions leading to more than 15 distinct neuregulin-1 isoforms, some of which are membrane-bound and others released as soluble factors into the bloodstream (59,65). NRG-1β was originally described as a 44 kDa glycoprotein (66,67), however, it was subsequently shown that this soluble product is derived from a larger, membrane-bound form of the protein (68). In recent publications, a commercially available form of recombinant human neuregulin-1β, a 7.5 kDa polypeptide consisting of the EGF-domain of human NRG-1β, is sufficient to promote surfactant phospholipid synthesis (69) and secretion (45).
Expression of NRG-1β Receptors During Lung Maturation
The only receptors for NRG-1β are the transmembrane receptor tyrosine kinases ErbB3 and ErbB4, two members of the epidermal growth factor receptor family that consists of four receptors: EGF-R (epidermal growth factor receptor, or ErbB1), ErbB2, ErbB3, and ErbB4 (70,71). ErbB receptors, generally located on the basolateral membrane of type II cells (72,73), are important regulators of cell proliferation and differentiation during development of fetal organs, including the fetal lung (74,75). They exhibit diversity in signaling potential and cause different biological responses through both homo- and hetero-dimerization (73,76,77). In rat and mouse fetal lung type II cells the dimerization patterns are receptor-specific, with dimers containing ErbB4 most prominent (73,77). The ligand specificity of the ErbB receptors determines which receptor is activated. Thus, this review of NRG-1β need only consider ErbB3 and ErbB4 signaling mechanisms (19,78).
Influence of Neuregulin-1β on the Production of Surfactant Components
The stimulation of surfactant synthesis in type II cells by fibroblast-conditioned media (FCM) is mimicked in fetal mice and rats by NRG-1β (19,69) and inhibited by antibodies raised against NRG-1β (19) (see Figure 2 ). Marked stimulation by NRG-1β of DSPC synthesis in type II cells is evident after 24 h exposure and appears to plateau at concentrations above 2.7 nmol/l (20 ng/ml), producing a 3.5-fold increase in DSPC synthesis ( Figure 1 ). Thus, at a similar concentration, NRG-1β maximally stimulates surfactant phospholipid synthesis to the same extent as KGF and leptin. The response to NRG-1β was reduced at higher concentrations of the ligand (69), suggesting that the ErbB receptors to which NRG-1β binds are downregulated at these higher concentrations, possibly through receptor ubiquitination and degradation (79). NRG-1β dose–response curves have been reported in several cell types (79,80,81).

Effect of keratinocyte growth factor (KGF)- and NRG-1β-neutralizing antibodies on disaturated phosphatidylcholine (DSPC) synthesis by type II cells after induction by fibroblast-conditioned media (FCM) with and without 100 nmol/l dexamethasone. Cultured type II cells were incubated with either FCM or FCM with 100 nmol/l dexamethasone in the presence or absence of (a) KGF-neutralizing antibody or (b) NRG-1β-neutralizing antibody for 48 h. The extent of incorporation of [3H]-choline into DSPC in type II cells was determined and compared with that which occurred in control cells. Legend to Figure Bars: Open (white) bars: Control (no FCM, dexamethasone or antibody) (panel a and panel b); Gray bars: Addition of FCM alone (panel a and panel b); Black bars: Addition of FCM + KGF-neutralizing antibody (panel a) or FCM + NRG-1β-neutralizing antibody (panel b); Vertically-decreasing Gradient Bar: Addition of FCM + 100 nmol/l Dexamethasone (panel a); Dark Center Gradient Bar: Addition of FCM + 100 nmol/l Dexamethasone + KGF-neutralizing antibody (panel a). Data were taken from Chelly et al. (93) and Dammann et al. (19).
Zscheppang et al. (82) used siRNA to downregulate ErbB4 expression in fetal rat type II cells and studied the response to FCM. Downregulation through siRNA treatment reduced both the expression and phosphorylation of the ErbB4 receptor, and also decreased DSPC synthesis and cell proliferation. They concluded that ErbB4 plays an important role in regulating fetal lung maturation via mesenchyme–epithelial cell communication. This was confirmed by the observations of Liu et al. using a transgenic mouse model of ErbB4 deletion (83). Deletion of ErbB4 or neuregulin results in embryonic death due to defects in cardiac development before the onset of fetal surfactant synthesis. However, a transgenic ErbB4 deletion rescued from the cardiac lethality by directed cardiac expression of ErbB4 has been useful for studying the importance of the neuregulin-ErbB4 axis for type II cell surfactant production. Using that model of ErbB4 deletion with transgenic cardiac rescue, Liu et al. reported that although the ErbB4 knockout mice with cardiac rescue survive fetal development they suffer from diminished surfactant protein expression and phospholipid synthesis resulting in defective lung development. Further, immunoprecipitation and confocal microscopic approaches in normal cells showed that ligand activation alters both the heterodimerization and cellular localization patterns of ErbB4 receptors in fetal lung (73,77). In particular, these transmembrane receptors have a higher distribution within the nuclei of type II cells (73).
ErbB4 signal transduction is complex. NRG-1β binding to the ErbB4 extracellular domain induces the formation of both homo- and hetero-dimers with other ErbB receptors, initiating autophosphorylation within the intracellular domain. This phosphorylation activates intracellular signaling pathways such as the phosphatidylinositol-3-kinase/Akt pathway to ultimately influence gene expression (70,76,84). Alternatively, ErbB4 may undergo proteolytic processing involving two sequential cleavage processes, first by the transmembrane metalloprotease tumor necrosis factor-α converting enzyme (TACE); and second by the enzyme γ-secretase, a complex of four proteins in which the enzymatic component is either presenilin-1 or presenilin-2 (85,86). Presenilins are crucial for normal lung development (87). The cleavage of ErbB4 releases an 80 kDa fragment (4ICD) into the cytoplasm that translocates to the nuclei of type II cells where it forms complexes with transcription factors to induce transcription, enhancing SP-B and SP-C gene expression (85,88). The level of thyroid transcription factor-1, a transcription factor necessary for SP-B and possibly SP-C gene expression, is regulated by ErbB4 (88). SP-B is considered the most important surfactant protein in facilitating the reduction of surface tension by surfactant phospholipids (89). Estrogen, which upregulates surfactant protein synthesis, stimulates ErbB4 binding to the SP-B promoter (90). This estrogen effect required an interaction between its receptor (ER-β) and ErbB4. Since the authors documented that these two receptors coimmunoprecipitate, ER-β may be a nuclear chaperone for ErbB4 to enhance nuclear localization of ErbB4.
In the transgenic mouse with ErbB4 deletion and cardiac rescue by directed ErbB4 expression in cardiac cells, lack of pulmonary ErbB4 receptors leads to diminished expression of both the SP-B (83,85,86) and SP-D (74,83) genes. As described above, when NRG-1β binds to ErbB4 it leads to the formation of a transcriptional complex that enhances transcription of the SP-B gene (85) and possibly that of SP-C highlighting the importance of NRG-1β and its receptor (ErbB4) in stimulating production of these important surfactant proteins.
Neuregulin-1β Stimulates Baseline and β-Agonist-Induced Surfactant Phospholipid Secretion
Short-term, direct exposure of fetal rat type II pneumocytes to NRG-1β significantly increased surfactant phospholipid secretion in a time- and concentration-dependent manner (45). The maximal stimulatory effect on surfactant phospholipid secretion (2.4-fold) was seen at 6.7 nmol/l (50 ng/ml) ( Figure 3 ). In contrast, at similar concentrations, leptin did not stimulate surfactant phospholipid secretion. Furthermore, (__)-isoproterenol-induced secretion of surfactant phospholipids was greater (an additional 2.5-fold) after pre-exposure to NRG-1β, even though the NRG-1β was removed prior to treatment with the β-agonist (45). This NRG-1β-induced enhanced response to (__)-isoproterenol is explained by the observation that NRG-1β exposure for 24 h markedly increased β-adrenergic receptor activity in the type II cells.

Effect of leptin and NRG-1β on surfactant phospholipid secretion. Cultured fetal rat type II cells were incubated with the indicated concentrations of leptin (filled squares) or NRG-1β (filled triangles) for 3 h prior to measuring the extent of secretion of surfactant phospholipids, which had been previously labelled with [3H]-choline, and compared to that which occurred in control cultures. Data were taken from King et al. (45).
The above observations demonstrate that epithelial-mesenchymal interactions generate NRG-1β to play an important role in lung growth and development (19). NRG-1β induces surfactant phospholipid and protein synthesis (19,69), elevates the rate of phospholipid secretion and enhances the sensitivity of alveolar type II cells to (__)-isoproterenol-induced secretion of surfactant (45). The observations that NRG-1β is present in FCM (especially when the fibroblasts are exposed to glucocorticoids, see below) (19,69) and that the stimulatory effect of FCM on surfactant phospholipid synthesis can be blocked with NRG-1β antibodies (19) indicate that NRG-1β has many of the properties of FPF. Its size (44 kDa) (19) is much larger than the 5–15 kDa ascribed to FPF by Smith and Post (14), however as many isoforms of NRG-1 have been identified, it is possible that a smaller form exists in the lung that would correspond to that molecular size.
Glucocorticoids and FPF Candidates
The ability of glucocorticoids to promote surfactant synthesis in tissue culture and in clinical use raised the question of the extent to which glucocorticoids and FPF are related. In fact, the early observations that glucocorticoids promote surfactant synthesis in vitro through fibroblast—type II cell communication was a stimulus for Smith’s ground breaking discovery and definition of FPF (14,91). Numerous experiments show that glucocorticoids stimulate FPF production (11,12,13,14,92). It is therefore important to consider the relationship of glucocorticoids with FPF and FPF candidates. Here, we review the current state of knowledge regarding glucocorticoid interactions with KGF, leptin, and neuregulin activities promoting lung surfactant synthesis.
KGF and Glucocorticoid-Induced Surfactant Synthesis
The stimulation of DSPC synthesis by FCM is reduced by ~50% if the FCM is treated with anti-KGF antibodies ( Figure 2 ) (93). The stimulatory effect of FCM is greater when generated in the presence of 100 nM dexamethasone. KGF antibodies also completely blocked the effect of FCM generated in the presence of dexamethasone ( Figure 2 ). Dexamethasone enhanced KGF mRNA levels by 50% in cultured fetal rat lung fibroblasts, suggesting that dexamethasone increases the concentration of KGF in FCM. These observations led Chelly et al. (93) to conclude that KGF is a major mediator of glucocorticoid stimulation of fetal type II cell surfactant synthesis.
Leptin and Glucocorticoid-Induced Surfactant Synthesis
Leptin gene expression is elevated eightfold in lung fibroblasts following exposure to dexamethasone, resulting in a significant elevation in leptin protein synthesis and secretion (18). This is consistent with Slieker et al. (94), who demonstrated that glucocorticoids increased both leptin mRNA and secretion in mouse adipocytes. Furthermore, Ob-Rb localisation in fetal ovine lungs, including alveolar type II pneumocytes, and an elevation in Ob-Rb mRNA levels in response to glucocorticoid treatment suggest a role for glucocorticoid control of leptin signaling in prenatal lung maturation (95).
Neuregulin-1β and Glucocorticoid-Induced Surfactant Synthesis
Glucocorticoid treatment of fetal mouse type II cells affects ErbB receptor expression. In particular, glucocorticoid treatment of the fetal mouse type II cells increased the amount of ErbB4 and the phosphorylation of ErbB4 in response to NRG-1β stimulation (75).
Glucocorticoid treatment of fetal lung fibroblasts increases NRG-1β content in the FCM, however a direct induction of NRG-1β synthesis has not been conclusively demonstrated. King et al. (69) showed that exposure of lung fibroblasts to dexamethasone leads to elevated levels of NRG-1β in the FCM. However, qPCR analysis showed that NRG-1β gene expression in cultured fetal lung fibroblasts was not affected by the presence of dexamethasone. This absence of a significant increase in NRG-1β mRNA suggests that the steroid does not mediate its effect via an elevated rate of transcription of the NRG-1β gene. Given that neuregulins are produced as transmembrane precursors that generate diffusible ligands when the exodomain is cleaved (68,96), dexamethasone may stimulate the rate of cleavage of neuregulin precursors, releasing the exodomain as mature neuregulin.
Glucocorticoids and NRG-1β synergistically elevate the transcription of the β-AR gene. The β-AR gene contains a glucocorticoid response element (97), and glucocorticoids promote β-AR gene expression (98,99,100). King et al. examined the effect of NRG-1β alone and in combination with dexamethasone on β-AR gene expression using qPCR (45). Although the β-AR mRNA was not changed by exposure to NRG-1β alone, the combination of NRG-1β with dexamethasone induced a greater elevation in β-AR mRNA levels than did dexamethasone alone (45).
Conclusions
From this review, it is apparent that several factors have the potential to mediate the indirect glucocorticoid induction of surfactant synthesis in type II cells, producing the described activities of FPF ( Figure 4 ). Of the three factors that have been examined in detail in this review, each appears to reproduce the properties of FPF but only the 16 kDa peptide leptin has the predicted molecular size. However, identifying leptin with FPF is not possible because of unanswered concerns. Although leptin upregulates surfactant synthesis in rats (18,38), studies in mice and sheep have failed to show this effect (55). The source of leptin production is the pulmonary lipofibroblast, but whether humans have this cell type is controversial (56,57,58).

Direct and indirect effects of glucocorticoids on the surfactant phospholipid synthesis in and secretion from type II cells. This schematic diagram illustrates the direct stimulatory effect of glucocorticoids on the β-AR receptor activity of type II cells and their response to β-agonists. The indirect effect of the steroid is also shown to involve the production of fibroblast-pneumocyte factor, whose action may be mediated by keratinocyte growth factor, leptin or NRG-1β, or a combination of these factors. Each of the responses depicted for these agents has been shown to occur in cultured fetal type II pneumocytes.
Moreover, there is abundant evidence that KGF and NRG-1β also participate in the signaling that occurs between fetal lung fibroblasts and epithelial type II cells promoting enhanced surfactant synthesis. KGF, leptin, and NRG-1β each stimulate fetal type II cell surfactant production in the absence of added FCM known to have FPF activity. At this point, it does not appear that these factors are elements of a common pathway. There is no published evidence that KGF, leptin, or NRG-1β modifies the activity of each other or their corresponding receptors. Knockout mutations for all three of these peptides (40), or of factors involved in their action (83), demonstrate that although the mutant animals survive, close examinations show impaired lung structure, function and/or surfactant metabolism. This suggests that normal lung development and maturation is dependent upon more than one factor. From an evolutionary standpoint, it would be appropriate to have overlapping mechanisms involved in the production of a factor such as surfactant that is essential for postnatal survival. This would ensure lung development sufficient for survival even in the event of one of these mechanisms failing. Another possibility is that some of these mechanisms of surfactant homeostasis are species-specific. In other words, although leptin may well be important in stimulating lung maturation in rats, other agents (such as NRG-1β) are likely to also be involved and may play a more prominent role in other species. To clearly delineate the identity of FPF it appears that an optimal strategy to make further progress would involve at least three approaches. First would be to combine studies using antibodies to separately immunoprecipitate each candidate from FCM. Determination of levels of the other two factors and of FPF activity in immunodepleted FCM is needed. As the FPF blocking antibody has not been available for several years, it would also be necessary to raise a new antibody against FPF, perhaps by improving on Smith’s original strategy (12). Immunodepletion studies using antibodies to immunoprecipitate KGF, leptin, or NRG-1β from the FCM followed by testing the FCM for FPF activity should be done. Second, RNA deep sequencing of fetal lung fibroblasts that do not and do make FPF could be used to identify additional candidates and confirm or disprove the importance of the candidates reviewed here. Third, CHIP-seq studies of transcriptional complexes at the SP-B and SP-C promoter regions would further define important protein elements that upregulate the expression of these important surfactant proteins. The search for a more complete molecular understanding of the FPF mechanisms continues to be important, as it appears likely to lead to more advanced means of promoting lung maturation for preterm infants and possibly protect against postnatal lung injury.
Statement of Financial Support
This study was supported by National Institute of Health (Bethesda, MD, USA) HL085648 and HL09730 (H.C.N.); Peabody Foundation, Boston, MA (H.C.N.); Murdoch University (M.H.C.); and Australian Postgraduate Award (G.K.).
Disclosure
The authors have nothing to disclose regarding financial ties or conflicts of interest.
References
Clements JA, Avery ME. Lung surfactant and neonatal respiratory distress syndrome. Am J Respir Crit Care Med 1998;157(4 Pt 2):S59–66.
Avery ME. Surfactant deficiency in hyaline membrane disease: the story of discovery. Am J Respir Crit Care Med 2000;161(4 Pt 1):1074–5.
Liggins GC. Premature delivery of foetal lambs infused with glucocorticoids. J Endocrinol 1969;45:515–23.
Kotas RV, Avery ME. Accelerated appearance of pulmonary surfactant in the fetal rabbit. J Appl Physiol 1971;30:358–61.
Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics 1972;50:515–25.
Brehier A, Rooney SA. Phosphatidylcholine synthesis and glycogen depletion in fetal mouse lung: developmental changes and the effects of dexamethasone. Exp Lung Res 1981;2:273–87.
Smith BT, Sabry K. Glucocorticoid-thyroid synergism in lung maturation: a mechanism involving epithelial-mesenchymal interaction. Proc Natl Acad Sci USA 1983;80:1951–4.
Samtani MN, Pyszczynski NA, Dubois DC, Almon RR, Jusko WJ. Modeling glucocorticoid-mediated fetal lung maturation: II. Temporal patterns of gene expression in fetal rat lung. J Pharmacol Exp Ther 2006;317:127–38.
Bird AD, Choo YL, Hooper SB, McDougall AR, Cole TJ. Mesenchymal glucocorticoid receptor regulates the development of multiple cell layers of the mouse lung. Am J Respir Cell Mol Biol 2014;50:419–28.
Bird AD, McDougall AR, Seow B, Hooper SB, Cole TJ. Glucocorticoid regulation of lung development: lessons learned from conditional GR knockout mice. Mol Endocrinol 2015;29:158–71.
Post M, Barsoumian A, Smith BT. The cellular mechanism of glucocorticoid acceleration of fetal lung maturation. J Biol Chem 1986;261:2179–84.
Post M, Floros J, Smith BT. Inhibition of lung maturation by monoclonal antibodies against fibroblast-pneumonocyte factor. Nature 1984;308:284–6.
Smith BT, Fletcher WA. Pulmonary epithelial-mesenchymal interactions: beyond organogenesis. Hum Pathol 1979;10:248–50.
Smith BT, Post M. Fibroblast-pneumonocyte factor. Am J Physiol 1989;257(4 Pt 1):L174–8.
Nielsen HC. Epidermal growth factor influences the developmental clock regulating maturation of the fetal lung fibroblast. Biochim Biophys Acta 1989;1012:201–6.
Sen N, Cake MH. Enhancement of disaturated phosphatidylcholine synthesis by epidermal growth factor in cultured fetal lung cells involves a fibroblast-epithelial cell interaction. Am J Respir Cell Mol Biol 1991;5:337–43.
Chelly N, Mouhieddine-Gueddiche OB, Barlier-Mur AM, Chailley-Heu B, Bourbon JR. Keratinocyte growth factor enhances maturation of fetal rat lung type II cells. Am J Respir Cell Mol Biol 1999;20:423–32.
Torday JS, Sun H, Wang L, Torres E, Sunday ME, Rubin LP. Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am J Physiol Lung Cell Mol Physiol 2002;282:L405–10.
Dammann CE, Nielsen HC, Carraway KL 3rd . Role of neuregulin-1 beta in the developing lung. Am J Respir Crit Care Med 2003;167:1711–6.
Ito Y, Correll K, Schiel JA, Finigan JH, Prekeris R, Mason RJ. Lung fibroblasts accelerate wound closure in human alveolar epithelial cells through hepatocyte growth factor/c-Met signaling. Am J Physiol Lung Cell Mol Physiol 2014;307:L94–105.
Rubin JS, Osada H, Finch PW, Taylor WG, Rudikoff S, Aaronson SA. Purification and characterisation of a newly identified growth factor specific for epithelial cells. Biochemistry 1989;86:802–6.
Ulich TR, Yi ES, Longmuir K, et al. Keratinocyte growth factor is a growth factor for type II pneumocytes in vivo. J Clin Invest 1994;93:1298–306.
Michelson PH, Tigue M, Panos RJ, Sporn PH. Keratinocyte growth factor stimulates bronchial epithelial cell proliferation in vitro and in vivo. Am J Physiol 1999;277(4 Pt 1):L737–42.
Fehrenbach H, Kasper M, Tschernig T, et al. Keratinocyte growth factor-induced hyperplasia of rat alveolar type II cells in vivo is resolved by differentiation into type I cells and by apoptosis. Eur Respir J 1999;14:534–44.
Yano T, Mason RJ, Pan T, Deterding RR, Nielsen LD, Shannon JM. KGF regulates pulmonary epithelial proliferation and surfactant protein gene expression in adult rat lung. Am J Physiol Lung Cell Mol Physiol 2000;279:L1146–58.
Xu X, McCormick-Shannon K, Voelker DR, Mason RJ. KGF increases SP-A and SP-D mRNA levels and secretion in cultured rat alveolar type II cells. Am J Respir Cell Mol Biol 1998;18:168–78.
Gesche J, Fehrenbach H, Koslowski R, et al. rhKGF stimulates lung surfactant production in neonatal rats in vivo. Pediatr Pulmonol 2011;46:882–95.
Deterding RR, Havill AM, Yano T, et al. Prevention of bleomycin-induced lung injury in rats by keratinocyte growth factor. Proc Assoc Am Physicians 1997;109:254–68.
Pasula R, Azad AK, Gardner JC, Schlesinger LS, McCormack FX. Keratinocyte growth factor administration attenuates murine pulmonary mycobacterium tuberculosis infection through granulocyte-macrophage colony-stimulating factor (GM-CSF)-dependent macrophage activation and phagolysosome fusion. J Biol Chem 2015;290:7151–9.
Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem 2006;281:15694–700.
Matsuda Y, Ueda J, Ishiwata T. Fibroblast growth factor receptor 2: expression, roles, and potential as a novel molecular target for colorectal cancer. Patholog Res Int 2012;2012:574768.
De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 2000;127:483–92.
Orr-Urtreger A, Bedford MT, Burakova T, et al. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev Biol 1993;158:475–86.
Panos RJ, Rubin JS, Csaky KG, Aaronson SA, Mason RJ. Keratinocyte growth factor and hepatocyte growth factor/scatter factor are heparin-binding growth factors for alveolar type II cells in fibroblast-conditioned medium. J Clin Invest 1993;92:969–77.
Hoggard N, Hunter L, Duncan JS, Williams LM, Trayhurn P, Mercer JG. Leptin and leptin receptor mRNA and protein expression in the murine fetus and placenta. Proc Natl Acad Sci USA 1997;94:11073–8.
Miralles O, Sánchez J, Palou A, Picó C. A physiological role of breast milk leptin in body weight control in developing infants. Obesity (Silver Spring) 2006;14:1371–7.
Bruno A, Pace E, Chanez P, et al. Leptin and leptin receptor expression in asthma. J Allergy Clin Immunol 2009;124:230–7, 237.e1–4.
Kirwin SM, Bhandari V, Dimatteo D, et al. Leptin enhances lung maturity in the fetal rat. Pediatr Res 2006;60:200–4.
Chen H, Zhang J-B, Huang H, Wang Z-H, Cheng R, Cai W-B. Leptin promotes fetal lung maturity and upregulates SP-A expression in pulmonary alveoli type-II epithelial cells involving TTF-1 activation. PLoS One 2013;8:1–12.
Huang K, Rabold R, Abston E, et al. Effects of leptin deficiency on postnatal lung development in mice. J Appl Physiol (1985) 2008;105:249–59.
Tankersley C, Kleeberger S, Russ B, Schwartz A, Smith P. Modified control of breathing in genetically obese (ob/ob) mice. J Appl Physiol (1985) 1996;81:716–23.
O’Donnell CP, Tankersley CG, Polotsky VP, Schwartz AR, Smith PL. Leptin, obesity, and respiratory function. Respir Physiol 2000;119:163–70.
Tankersley CG, O’Donnell C, Daood MJ, et al. Leptin attenuates respiratory complications associated with the obese phenotype. J Appl Physiol (1985) 1998;85:2261–9.
Bergen HT, Cherlet TC, Manuel P, Scott JE. Identification of leptin receptors in lung and isolated fetal type II cells. Am J Respir Cell Mol Biol 2002;27:71–7.
King G, Damas JE, Cake MH, Berryman D, Maker GL. Influence of glucocorticoids, neuregulin-1β, and sex on surfactant phospholipid secretion from type II cells. Am J Physiol Lung Cell Mol Physiol 2014;306:L292–8.
Torday JS, Rehan VK. Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of PTHrP and leptin. Am J Physiol Lung Cell Mol Physiol 2002;283:L130–5.
Rubin LP, Kovacs CS, De Paepe ME, Tsai SW, Torday JS, Kronenberg HM. Arrested pulmonary alveolar cytodifferentiation and defective surfactant synthesis in mice missing the gene for parathyroid hormone-related protein. Dev Dyn 2004;230:278–89.
Torday JS, Rehan VK. Up-regulation of fetal rat lung parathyroid hormone-related protein gene regulatory network down-regulates the Sonic Hedgehog/Wnt/betacatenin gene regulatory network. Pediatr Res 2006;60:382–8.
Weaver TE, Conkright JJ. Function of surfactant proteins B and C. Annu Rev Physiol 2001;63:555–78.
Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol 2001;63:521–54.
Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995;83:1263–71.
Hoggard N, Mercer JG, Rayner DV, Moar K, Trayhurn P, Williams LM. Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem Biophys Res Commun 1997;232:383–7.
Villanueva EC, Myers MG Jr . Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 2008;32 Suppl 7:S8–12.
Tsuchiya T, Shimizu H, Horie T, Mori M. Expression of leptin receptor in lung: leptin as a growth factor. Eur J Pharmacol 1999;365:273–9.
Sato A, Schehr A, Ikegami M. Leptin does not influence surfactant synthesis in fetal sheep and mice lungs. Am J Physiol Lung Cell Mol Physiol 2011;300:L498–505.
Rehan VK, Torday JS. PPARγ signaling mediates the evolution, development, homeostasis, and repair of the lung. PPAR Res 2012;2012:289867.
Tahedl D, Wirkes A, Tschanz SA, Ochs M, Mühlfeld C. How common is the lipid body-containing interstitial cell in the mammalian lung? Am J Physiol Lung Cell Mol Physiol 2014;307:L386–94.
Ahlbrecht K, McGowan SE. In search of the elusive lipofibroblast in human lungs. Am J Physiol Lung Cell Mol Physiol 2014;307:L605–8.
Carraway KL 3rd, Weber JL, Unger MJ, et al. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature 1997;387:512–6.
Zhao YY, Sawyer DR, Baliga RR, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem 1998;273:10261–9.
Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res 2003;284:14–30.
Boucherat O, Benachi A, Chailley-Heu B, et al. Surfactant maturation is not delayed in human fetuses with diaphragmatic hernia. PLoS Med 2007;4:e237.
Patel NV, Acarregui MJ, Snyder JM, Klein JM, Sliwkowski MX, Kern JA. Neuregulin-1 and human epidermal growth factor receptors 2 and 3 play a role in human lung development in vitro. Am J Respir Cell Mol Biol 2000;22:432–40.
Liu J, Nethery D, Kern JA. Neuregulin-1 induces branching morphogenesis in the developing lung through a P13K signal pathway. Exp Lung Res 2004;30:465–78.
Lemke G. Neuregulins in development. Mol Cell Neurosci 1996;7:247–62.
Wen D, Peles E, Cupples R, et al. Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 1992;69:559–72.
Tokita Y, Keino H, Matsui F, et al. Regulation of neuregulin expression in the injured rat brain and cultured astrocytes. J Neurosci 2001;21:1257–64.
Loeb JA, Susanto ET, Fischbach GD. The neuregulin precursor proARIA is processed to ARIA after expression on the cell surface by a protein kinase C-enhanced mechanism. Mol Cell Neurosci 1998;11:77–91.
King G, Maker GL, Berryman D, Trengove RD, Cake MH. Role of neuregulin-1β in dexamethasone-enhanced surfactant synthesis in fetal type II cells. FEBS Lett 2014;588:975–80.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–37.
Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res 2003;284:66–77.
Vermeer PD, Einwalter LA, Moninger TO, et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 2003;422:322–6.
Zscheppang K, Korenbaum E, Bueter W, Ramadurai SM, Nielsen HC, Dammann CE. ErbB receptor dimerization, localization, and co-localization in mouse lung type II epithelial cells. Pediatr Pulmonol 2006;41:1205–12.
Purevdorj E, Zscheppang K, Hoymann HG, et al. ErbB4 deletion leads to changes in lung function and structure similar to bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2008;294:L516–22.
Dammann CE, Nassimi N, Liu W, Nielsen HC. ErbB receptor regulation by dexamethasone in mouse type II epithelial cells. Eur Respir J 2006;28:1117–23.
Riese DJ 2nd, van Raaij TM, Plowman GD, Andrews GC, Stern DF. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol 1995;15:5770–6.
Liu W, Zscheppang K, Murray S, Nielsen HC, Dammann CE. The ErbB4 receptor in fetal rat lung fibroblasts and epithelial type II cells. Biochim Biophys Acta 2007;1772:737–47.
Singer E, Landgraf R, Horan T, Slamon D, Eisenberg D. Identification of a heregulin binding site in HER3 extracellular domain. J Biol Chem 2001;276:44266–74.
Cao Z, Wu X, Yen L, Sweeney C, Carraway KL 3rd . Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol Cell Biol 2007;27:2180–8.
Holmes WE, Sliwkowski MX, Akita RW, et al. Identification of heregulin, a specific activator of p185erbB2. Science 1992;256:1205–10.
Wen L, Lu YS, Zhu XH, et al. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci USA 2010;107:1211–6.
Zscheppang K, Liu W, Volpe MV, Nielsen HC, Dammann CE. ErbB4 regulates fetal surfactant phospholipid synthesis in primary fetal rat type II cells. Am J Physiol Lung Cell Mol Physiol 2007;293:L429–35.
Liu W, Purevdorj E, Zscheppang K, et al. ErbB4 regulates the timely progression of late fetal lung development. Biochim Biophys Acta 2010;1803:832–9.
Liu W, Volpe MA, Zscheppang K, Nielsen HC, Dammann CE. ErbB4 regulates surfactant synthesis and proliferation in adult rat pulmonary epithelial cells. Exp Lung Res 2009;35:29–47.
Hoeing K, Zscheppang K, Mujahid S, et al. Presenilin-1 processing of ErbB4 in fetal type II cells is necessary for control of fetal lung maturation. Biochim Biophys Acta 2011;1813:480–91.
Fiaturi N, Ritzkat A, Dammann CE, Castellot JJ, Nielsen HC. Dissociated presenilin-1 and TACE processing of ErbB4 in lung alveolar type II cell differentiation. Biochim Biophys Acta 2014;1843:797–805.
Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA 1999;96:11872–7.
Marten E, Nielsen HC, Dammann CE. Interdependent TTF1 - ErbB4 interactions are critical for surfactant protein-B homeostasis in primary mouse lung alveolar type II cells. J Cell Commun Signal 2015;9:207–15.
Nogee LM. Alterations in SP-B and SP-C expression in neonatal lung disease. Annu Rev Physiol 2004;66:601–23.
Zscheppang K, Konrad M, Zischka M, Huhn V, Dammann CE. Estrogen-induced upregulation of Sftpb requires transcriptional control of neuregulin receptor ErbB4 in mouse lung type II epithelial cells. Biochim Biophys Acta 2011;1813:1717–27.
Smith BT. Lung maturation in the fetal rat: acceleration by injection of fibroblast-pneumonocyte factor. Science 1979;204:1094–5.
Smith BT. Fibroblast-pneumonocyte factor: intercellular mediator of glucocorticoid effect on fetal lung. In Stern L Oh W and Friis-Hansen B. (eds.) Intensive Care in the Newborn II. Masson: New York 1978:25–32.
Chelly N, Henrion A, Pinteur C, Chailley-Heu B, Bourbon JR. Role of keratinocyte growth factor in the control of surfactant synthesis by fetal lung mesenchyme. Endocrinology 2001;142:1814–9.
Slieker LJ, Sloop KW, Surface PL, et al. Regulation of expression of ob mRNA and protein by glucocorticoids and cAMP. J Biol Chem 1996;271:5301–4.
De Blasio MJ, Boije M, Vaughan OR, et al. Developmental expression and glucocorticoid control of the leptin receptor in fetal ovine lung. PLoS One 2015;10:e0136115.
Crovello CS, Lai C, Cantley LC, Carraway KL 3rd . Differential signaling by the epidermal growth factor-like growth factors neuregulin-1 and neuregulin-2. J Biol Chem 1998;273:26954–61.
Cornett LE, Hiller FC, Jacobi SE, Cao W, McGraw DW. Identification of a glucocorticoid response element in the rat beta2-adrenergic receptor gene. Mol Pharmacol 1998;54:1016–23.
Collins S, Caron MG, Lefkowitz RJ. Beta-adrenergic receptors in hamster smooth muscle cells are transcriptionally regulated by glucocorticoids. J Biol Chem 1988;263:9067–70.
Mak JC, Nishikawa M, Barnes PJ. Glucocorticosteroids increase beta 2-adrenergic receptor transcription in human lung. Am J Physiol 1995;268(1 Pt 1):L41–6.
Dangel V, Giray J, Ratge D, Wisser H. Regulation of beta-adrenoceptor density and mRNA levels in the rat heart cell-line H9c2. Biochem J 1996;317 (Pt 3):925–31.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
King, G., Smith, M., Cake, M. et al. What is the identity of fibroblast-pneumocyte factor?. Pediatr Res 80, 768–776 (2016). https://doi.org/10.1038/pr.2016.161
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.161
This article is cited by
-
Origins of neonatal leptin deficiency in preterm infants
Pediatric Research (2019)
-
Commentary on “What is the identity of fibroblast pneumocyte factor (FPF)?”
Pediatric Research (2017)
-
Commentary on the identity of fibroblast pneumocyte factor: rat vs. human
Pediatric Research (2017)
