
Abstract
The rate of self-fertilization (that is, selfing) is a key evolutionary parameter in hermaphroditic species, yet obtaining accurate estimates of selfing rates in natural populations can be technically challenging. Most published estimates are derived from population-level heterozygote deficiency (that is, FIS) or identity disequilibria (for example, the software RMES (robust multilocus estimate of selfing)). These indirect methods can be applied to population genetic survey data, whereas direct methods using progeny arrays require much larger data sets that are often difficult to collect in natural populations or even require captive breeding. Unfortunately, indirect methods rely on assumptions that can be problematic, such as negating biparental inbreeding, inbreeding disequilibrium and (for FIS) the presence of null alleles. The performance of indirect estimates against progeny-array estimates is still largely unknown. Here we used both direct progeny-array and indirect population-level methods to estimate the selfing rate in a single natural population of the simultaneously hermaphroditic freshwater snail Radix balthica throughout its reproductive lifespan using 10 highly polymorphic microsatellites. We found that even though progeny arrays (n=1034 field-collected embryos from 60 families) did not reveal a single selfed embryo, FIS-based selfing rates (n=316 adults) were significantly positive in all 6 sequential population samples. Including a locus with a high frequency of null alleles further biased FIS-based estimates. Conversely, RMES-based estimates were very similar to progeny-array estimates and proved insensitive to null alleles. The assumptions made by RMES were thus either met or irrelevant in this particular population, making RMES a valid, cost-efficient alternative to progeny arrays.
Similar content being viewed by others
Introduction
The rate of self-fertilization (that is, selfing) affects the population genetic and evolutionary properties of hermaphroditic species in various ways. Frequent selfing decreases heterozygosity within individuals, reduces effective population size, can lead to inbreeding depression and may ultimately impair the adaptive potential of a population (Charlesworth and Charlesworth, 1987). At the same time, selfing provides fertilization assurance, increases the transmission of genes, releases most species of costs of mating and mate acquisition and allows successful multilocus genotypes to be passed on with relatively few alterations (Fisher, 1941; Baker, 1955; Allard, 1975; Jarne and Charlesworth, 1993; Avise and Mank, 2009).
Despite the profound impact of selfing on a species, we still know relatively little about the distribution of selfing rates among hermaphroditic animals. In a survey of 142 animal species, 47% had intermediate selfing rates, ~36% were predominant outcrossers and ~17% predominant selfers (Jarne and Auld, 2006). However, 97% of these estimates were based on population-level heterozygote deficiency (that is, the inbreeding coefficient FIS), a method that has some rather strict assumptions. For example, inbreeding coefficient-based selfing rate estimates assume that there is no biparental inbreeding, that the population is not subdivided, that inbreeding is homogeneous across generations, that allele and genotype frequencies are at equilibrium and that there are no null alleles, genotyping errors or new mutations biasing the analysis. When estimating the ‘primary’ selfing rate at birth or oviposition, as opposed to a selfing rate measured in juveniles or adults (Ritland, 1990), one also needs to assume that presampling mortality has not changed the proportion of selfed to outcrossed individuals in a population (David et al., 2007; Jarne and David, 2008; Wang et al., 2012).
Not all these assumptions may be equally severe, but understandably empirical studies usually cannot produce powerful tests for all of them, or evaluate in detail which assumptions are more critical than others. Unfortunately, we can expect that violations of these assumptions often result in inflated selfing rates, rendering inbreeding coefficient-based estimates prone to error (David et al., 2007; Jarne and David, 2008; Escobar et al., 2011). Fortunately, a method has been developed that uses correlations of heterozygosity among loci (that is, identity disequilibria) to estimate selfing rates (David et al., 2007). The method has been implemented in a software package named ‘robust multilocus estimate of selfing’ (RMES) and promises a significant improvement of population-level estimates because its estimates are thought to be immune to the distorting effect of null alleles. However, RMES-based selfing rate estimates also do share some of the assumptions of FIS-based estimates, for example, that biparental inbreeding and, when estimating ‘primary’ selfing rates, in- or outbreeding depression before sampling can be ignored, and that the population is in inbreeding equilibrium (Wang et al., 2012). In comparisons where both methods were used on the same molecular data many species classified as outcrossers by RMES were classified as having intermediate selfing rates (that is, as being mixed-maters) by the inbreeding coefficient (David et al., 2007; Escobar et al., 2011). In contrast, the bias introduced by null alleles into FIS-based estimates was less noticeable in highly selfing species, in which null alleles often occur as easily detectable null homozygotes (David et al., 2007; Escobar et al., 2011).
Alternatively, selfing rates can be estimated directly using progeny arrays (Ritland and Jain, 1981). In progeny-array analysis, selfing events are identified directly by comparing a mother’s genotype to the genotypes of her offspring. Although substantially more cost- and labor-intensive, selfing rates based on progeny arrays are almost free of problematic assumptions, especially when computed using progenies collected in the field (to ensure fertilization events are free of laboratory artifacts) and, if ‘primary’ selfing rates are to be estimated, when progenies are genotyped at a very early stage in life (to minimize bias because of presampling mortality caused by in- or outbreeding depression) (Jarne and David, 2008; Wang et al., 2012). Progeny-array methods also offer the possibility to examine variation in selfing rates among individuals (Jarne and David, 2008; Wang et al., 2012), as well as the certainty that all identified selfing events must have occurred in the last generation. In contrast, population-level estimates assess the frequency of selfing and biparental inbreeding over several generations (David et al., 2007; Jarne and David, 2008; Wang et al., 2012). To yield reliable estimates, progeny-array methods require the use of genetic markers with sufficient resolution to unequivocally identify outcrossed and selfed individuals (Bernatchez and Duchesne, 2000; Jarne and David, 2008). This is particularly important when genotyping field-collected progenies with unknown mothers, making it much harder to infer parenthood than when mothers can be genotyped along with their offspring, as is usually the case in laboratory studies. Allozyme polymorphism, used for estimating 97% of the selfing rates compiled by Jarne and Auld (2006), unfortunately often fails to provide the required resolution, and the same is true for small sets of weakly polymorphic microsatellite loci.
Presuming that technical problems such as null alleles are absent and that statistical power is adequate, we therefore expect the comparison of progeny-array and population-level estimates to provide complementary insights into the history of a studied mating system. Nevertheless, for the most part we do not know how often indirect population-level estimates deviate from direct progeny-array estimates, be that for technical or biological reasons, as both types of methods have very rarely been applied simultaneously to the same population with sufficient statistical power. In 14 studies that estimated both FIS- and progeny-array-based selfing rates in the same populations of hermaphroditic animals, the mean absolute difference (±s.d.) between both types of estimates was 0.11±0.13 (Table 1, full references provided in the Supplementary Material). In seven studies FIS-based estimates and in five studies progeny-array-based estimates were higher. In two studies both estimates were identical, one of them analyzing a purely outcrossing population and one two purely selfing populations. This finding tallies with the expectation that differences between methods should be smallest at the distribution’s extremes. Only one study also estimated selfing rates using RMES, and found RMES- and FIS-based estimates to be very similar and slightly lower than progeny-array estimates (Kupfernagel et al., 2010). Although these results seem reassuring, it should be noted that almost all these studies exclusively used progenies produced in the laboratory, in most cases by adult individuals caught in the field and then kept in isolation. Apart from potentially altering natural patterns of fertilization, the isolation treatment assumes that mothers had obtained sufficient amounts of allosperm before being captured and can store it long enough for successful allofertilization. If not met, these assumptions may lead to artificially increased selfing rates in progenies, potentially masking the true differences between methods.
As a consequence, our current knowledge about the distribution of selfing rates among hermaphroditic animals, and especially about the proportion and identity of mixed maters, is still far from complete (Jarne and Auld, 2006; Jarne and David, 2008; Escobar et al., 2011). Biased selfing rate estimates will also result in incorrect inferences drawn about the population genetic and evolutionary properties of individual populations and species.
Here we estimated both ‘primary’ selfing rates and selfing rates among adult individuals in a natural population of the annual, simultaneously hermaphroditic freshwater snail Radix balthica. We used progeny arrays, the inbreeding coefficient and RMES, and covered the reproductive lifespan from beginning to end. R. balthica has been suspected of being a mixed mater (Coutellec-Vreto et al., 1997; Wiehn et al., 2002; Jokela et al., 2006; Pfenninger et al., 2011; Haun et al., 2012; but see also Jarne and Delay, 1990), but for reasons mentioned above these selfing rate estimates may come with some uncontrolled bias. We used 10 highly polymorphic microsatellite loci as genetic markers and ensured that selfing rates were free of laboratory artifacts, the influence of in- or outbreeding depression, errors caused by incomplete sampling and contamination with samples from other species. The comparison of indirect population-level estimates both affected and unaffected by null alleles with direct individual-level estimates based on progeny arrays enabled us to quantify the potential bias introduced by unmet assumptions and technical problems (that is, null alleles) in the former two methods. In addition, we assessed the magnitude of bias caused by the presence of known null alleles in a single locus directly by running all analyses both with and without this locus. We found that selfing rates estimated using RMES were very similar to progeny-array estimates, making this method a valid, cost-efficient alternative, whereas inbreeding coefficient-based estimates suffered heavily from the presence of null alleles.
Materials and methods
Study system
R. balthica is a diploid, simultaneously hermaphroditic freshwater snail that lives in the shallow littoral zone of large water bodies throughout Europe from Iceland to Mediterranean Countries (Cordellier and Pfenninger, 2009; Pfenninger et al., 2011; Lawton et al., 2015). In Lake Zurich, Switzerland, R. balthica hatches from eggs in spring and reaches sexual maturity by the end of the year. The reproductive season starts in late February or early March and lasts until May. Adult snails die after they reproduce and by late May the adult cohort is completely replaced by juveniles. Thus, generations are non-overlapping and generation time is ∼1 year. During the egg-laying period (March to May), snails may copulate repeatedly in both sexual roles and lay hundreds of eggs in distinct egg clutches (A Bürkli, unpublished data). The population studied here (Uerikon, Lake Zurich, ∼4700 m2 of suitable habitat) has been surveyed for parasitological studies, population genetic studies and teaching purposes occasionally since almost 20 years, during which population size has remained constant and fairly large (>10 000 individuals, J Jokela, unpublished data).
As the species is phenotypically very variable (see, for example, Brönmark et al., 2011; Schniebs et al., 2011; Ahlgren et al., 2013), the taxonomic identification of Radix snails is notoriously difficult (Pfenninger et al., 2006; Schniebs et al., 2011; Lawton et al., 2015). We therefore verified that only individuals of R. balthica were included in this study by sequencing the mitochondrial cytochrome oxidase subunit I (COI) gene, shown to be well suited for phylogenetic species delineation in Radix (Lawton et al., 2015), and by microsatellite genotyping (for details see Supplementary Methods 1).
Collection of egg clutches and adult snails
We collected adult snails and egg clutches from a relatively large area (∼900 m2) using snorkelling equipment. Adult snails served as candidate parents in parentage analyses of progeny arrays and were used to estimate indirect population-level selfing rates at the level of adults, whereas egg clutches served for estimating ‘primary’ selfing rates based on progeny arrays. Adult snails were caught at peak breeding season in three consecutive years (24 April 2013, 11 April 2014 and 13 April 2015) to be able to test for potential temporal differences in estimated selfing rates. Adults were also caught on three additional dates in 2014, spanning the reproductive season from its very beginning (06 March 2014) to its very end (23 May 2014). Egg clutches were collected on the same four dates in 2014 on which adult snails were caught, resulting in four and six samples for progeny-array and population-level analyses, respectively. Egg clutches were gently detached from boulders using a plastic spoon, and adult snails were collected by hand. All samples were brought to the laboratory in Eawag-Dübendorf, where adults were killed and preserved for genotyping at −80 °C, either immersed in 70% ethanol or dry. Egg clutches were placed in individual, water-filled 40 cl plastic cups (room temperature 18 °C) and allowed to develop until hatching was imminent, at which point they were transferred to individual 1.5 ml plastic tubes and stored at −80 °C as well. Later, egg clutches were thawed and the number of developed and undeveloped embryos was counted in each clutch using a dissection microscope.
Genetic analysis
We estimated the selfing rate from 15 egg clutches and from 53±20 (mean±s.d.) adult snails per sampling day. We genotyped 15 embryos per clutch irrespective of clutch size, the only exception being small clutches with <15 embryos, of which all embryos were genotyped. When present, we also genotyped undeveloped embryos. This was done to evaluate the sampling bias caused by a potential over- or underrepresentation of selfed offspring among undeveloped embryos. However, undeveloped embryos were very rare (30/1034 (2.9%) genotyped embryos, and 105/3038 (3.5%) eggs present in the 60 clutches). Sample sizes of adult snails and embryos are listed in Tables 2 and 3, respectively. All samples were genotyped for 10 microsatellite markers (GenBank accession nos. KX830983–KX830992) that had been newly developed for this population by Ecogenics (Zurich, Switzerland). We tried to minimize scoring errors by running an extensive pilot study and performing independent repeatability tests using newly extracted DNA on 17% of adult genotypes (Pompanon et al., 2005). Details of markers and genotyping routines are provided in Table 4 and Supplementary Methods 2.
Screening for null alleles
We screened all loci for the presence of null alleles using 2044 multilocus genotypes of snails originating from the Uerikon population. This included all samples genotyped for this study, but we also used all additional material we had available. For each locus, we counted the number of genotypes that could not be scored, either because no microsatellite peak was present (that is, ‘missing peaks’), more than two peaks were present or peaks were ambiguous in other ways (for example, lacking the locus-specific shape). Overall, 1392/20 440 (6.8%) locus-specific genotypes could not be scored, and most of them (75.6%) were because of missing peaks. Missing peaks can result from amplification failure caused by mutations in the primer region, and may thus be indicative of null allele homozygotes (Dabrowski et al., 2015). We therefore computed locus-specific frequencies of null alleles from the frequency of genotypes with missing peaks (following Dabrowski et al., 2015), and frequencies of other types of genotyping errors based on the frequency of genotypes that could not be scored for other reasons. All locus-specific error rates are listed in Table 4. In addition, we compared the genotypes of 59 maternal and 1320 juvenile snails reared in the laboratory to identify mother–offspring mismatches (data not shown).
Selfing rates based on progeny arrays
Parentage analyses were performed using COLONY version 2.0.5.9 (Jones and Wang, 2010). COLONY does not require previous knowledge of maternal genotypes but rather reconstructs them based on the offspring genotypes present in an egg clutch, making it possible to estimate selfing rates of field-collected clutches with unknown mothers. After reconstructing maternal genotypes, COLONY returns for each embryo the probability of being self-fertilized. This kind of progeny-array approach has the advantage of estimating the natural selfing rate at oviposition (that is, the ‘primary’ selfing rate in the population), but comes at the disadvantage of complicating the inference of parenthood. Parentage was inferred simultaneously for clutches collected on the same day. Adult genotypes from the same sampling day were entered into COLONY both as candidate mothers and fathers, and clutch identities were specified as maternal sibships. As parentage assignments can be distorted by both null alleles (Dakin and Avise, 2004; Dabrowski et al., 2015), known to be common in molluscs (see, for example, Kopp et al., 2012), and genotyping errors, we included the previously computed locus-specific error rates due to null alleles and due to other types of genotyping errors (see above) in all parentage analyses (Jones and Wang, 2010).
The selfing rate of an egg clutch will only be known without error if every single embryo present in the clutch is genotyped. To estimate the error introduced by genotyping only a subset of embryos, we genotyped seven egg clutches of different sizes (36.3±16.3 eggs (mean±s.d.)) as comprehensively as possible (91.4±5.2% of all eggs genotyped). Five of these clutches were collected at peak and two at the end of the breeding season, when we expected selfing rates to be lowest and the potential error caused by sampling incompleteness to be highest.
Mean selfing rates and associated 95% confidence intervals (CIs) for each of the four sampling days were computed after pooling the probabilities of being selfed for all embryos collected on the same sampling day—regardless of clutch identities. However, means and confidence intervals that were computed from the residuals of a linear model of probability of being selfed versus clutch identity, thus correcting for potential differences among clutches, were very similar. To make sure that statistical power among sampling days and egg clutches was identical when computing means and confidence intervals, we restricted the data set to 15 randomly chosen embryos in clutches where >15 embryos had been genotyped. Even so, including all genotyped embryos did not change estimates noticeably.
Selfing rates based on adult genotypes
We estimated the number of polymorphic loci, the mean number of alleles per locus and the observed and expected heterozygosity, the latter calculated without bias (Nei, 1978), for each of the six groups of adults collected on the same day using Genetix, version 4.05.2 (Belkhir et al., 1996–2004). The same software was used to compute population-level estimates of the inbreeding coefficient FIS over all loci according to Weir and Cockerham (1984) with a 95% CI based on 10 000 bootstrap iterations. The inbreeding coefficient-based population-level selfing rate (s(FIS)) was then obtained from the relationship s=2FIS/(1+FIS) (Wright, 1969). The 95% CI for s(FIS) was computed based on the variance of s(FIS) and served to assess the significance of s(FIS). Var(s(FIS)) was estimated using equation 2 in Jarne and David (2008), modified to accommodate multiple loci by substituting the single-locus sampling variance of FIS with the multilocus sampling variance estimated in Genetix using the jackknife (Belkhir et al., 1996–2004), and the single-locus FIS with the multilocus FIS.
In addition, we computed selfing rates that should be unaffected by the presence of null alleles using the software RMES (David et al., 2007). RMES implements two methods for estimating selfing rates: one that uses two-locus heterozygosity disequilibrium values (g2, with associated selfing rate s(g2)) and one that maximizes the log-likelihood of the multilocus heterozygosity structure of a sample (s(ML)). As a significance test for s(g2) RMES computes the probability that there is no selfing (g2=s=0), here obtained from 10 000 iterations of randomly reassorting single-locus heterozygosities among individuals. A 95% CI for s(g2) was obtained by computing a 95% CI for g2 and transforming it back to the scale of s using equation 9 in David et al. (2007). s(ML) was calculated with a precision for the log-likelihood of 0.00001 and a maximum of 10 generations of selfing allowed for a heterozygote (kmax=10). A 95% CI for s(ML) was provided by RMES (David et al., 2007) and was also used to judge the significance of s(ML).
Results
Presence of null alleles
Missing microsatellite peaks occurred in varying frequency at all loci, suggesting that all loci suffered from null alleles, but some loci more so than others. The frequency of null alleles ranged from 2.8% (locus Rb_8) to 8.2% (locus Rb_3) with an average of 5.1% (Table 4). Tellingly, locus Rb_3 also showed the highest frequency of ambiguous peaks (4.7%) and caused numerous mismatches between maternal and offspring genotypes in lab-reared snails (data not shown). We therefore computed all selfing rates twice, once without locus Rb_3 to obtain estimates that were largely unaffected by null alleles, and once including it to quantify the bias introduced by it.
Allelic diversity
The number of alleles observed per locus in the 9 loci that were largely free from null alleles ranged from 7 to 29 with an average of 16.9 (Table 4). Within groups of 15 embryos, each sampled from a separate clutch on the same day, the mean number of alleles per locus (±s.d.) was 7.1±3.2 (Table 4). The number of alleles was very similar within groups of 15 randomly chosen adults sampled on the same day (7.6±3.0, Table 4). Such substantial allelic richness was reflected in high values of observed single-locus heterozygosity, both among embryos (mean±s.d.: 0.64±0.27) and adults collected on the same day (0.66±0.20, Table 4). Including locus Rb_3, which showed an increased frequency of null alleles, did not change these numbers substantially (Table 4). We also estimated the probability of identity, which is the probability that a multilocus genotype is shared by randomly drawn adult snails (P(ID)unbiased from Waits et al., 2001). When including locus Rb_3, this probability is between 6.8 × 10−14 and 3.8 × 10−12 for all six sampling days. When excluding the locus, probabilities of identity are slightly higher, but still extremely low (between 1.8 × 10−11 and 2.7 × 10−10). Statistical power in our data set should thus be both adequate and sufficient to estimate selfing rates without any bias caused by a lack of allelic diversity.
No selfing in field-collected egg clutches
Among 1034 successfully genotyped embryos from a total of 60 egg clutches, not a single embryo was more likely to be selfed than to be outcrossed (Table 3). Seven of these egg clutches were genotyped as comprehensively as possible so as not to overlook very rare selfing events, but still revealed no selfed embryos. As a consequence, mean selfing rates based on all embryos collected on a sampling day were zero or extremely close to zero on all four sampling days, regardless of the inclusion of locus Rb_3 (that is, mean selfing rates 0.000–0.003, Figure 1, Table 3 and Supplementary Table 1). When excluding locus Rb_3, thereby restricting analyses to 9 loci largely free from null alleles, just 3 embryos had slightly elevated individual probabilities of being selfed (that is, probabilities 0.06, 0.26 and 0.39), whereas all other embryos were selfed with probabilities of <0.01 (Table 3). The three deviant embryos all came from different clutches and were collected either at the begin or at the end of the reproductive season (Table 3). When including locus Rb_3, heavily affected by null alleles, the number of embryos with slightly elevated individual probabilities of being selfed increased to 10 (that is, probabilities 0.01–0.44, mean 0.11; Table 3). These probabilities are below the threshold of 0.5, indicating that all embryos were still more likely to be outcrossed than to be selfed. One out of 30 undeveloped embryos (3.3%) and, depending on the inclusion of locus Rb_3, 2 to 9 out of 1004 developed embryos (0.2–0.9%) had an elevated probability to be selfed (Table 3). As the 95% CIs of these proportions are overlapping, the difference between undeveloped and developed embryos is not statistically significant (0.0008–0.17 vs 0.0002–0.007 or 0.004–0.017, respectively, Zar, 1996).

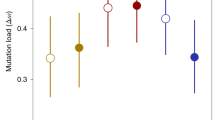
Selfing rates in a natural population of the freshwater snail R. balthica, estimated using one direct and three indirect methods and separately assessed on 6 sampling days spanning 3 years. Error bars show 95% confidence intervals. Selfing rates were computed using nine highly polymorphic microsatellite loci that are largely free from null alleles (a), and including an additional locus with a high frequency of null alleles (b). Selfing rates were obtained directly from field-collected egg clutches (progeny-array method yielding ‘primary’ selfing rates; 15 egg clutches with a total of 259±34 (mean±s.e.) embryos for each of four sampling days) and indirectly from field-collected adult snails (population-level methods; 52.7±8.1 adults for each of 6 sampling days). Population-level selfing rates were estimated in three ways: from the multilocus inbreeding coefficient FIS (Weir and Cockerham, 1984) using the relationship s=2FIS/(1+FIS) (Wright, 1969), from two-locus heterozygosity disequilibrium values (g2) using RMES (David et al., 2007) and by maximizing the log-likelihood of the multilocus heterozygosity structure of the sample (ML), also using RMES.
Apparent selfing in field-collected adults due to null-allele bias
Multilocus inbreeding coefficients (FIS) across the 9 loci that were largely free from null alleles were low (0.039–0.109, mean 0.071), but significantly positive on 4 of the 6 sampling days (Table 2). Accordingly, population-level selfing rates based on the inbreeding coefficient were relatively low yet significantly different from zero, ranging from 7.4% to 19.7% with an average of 13.1% (Figure 1 and Supplementary Table 1). Including locus Rb_3 almost doubled FIS-values (0.120–0.170, mean 0.133), rendering them significantly positive on all 6 sampling days (Table 2). As a consequence, mean s(FIS) increased to 23.4% (21.5–29.0%; Figure 1 and Supplementary Table 1).
Selfing rates computed based on the excess of doubly heterozygous genotypes, a method not sensitive to the distorting effect of null alleles (David et al., 2007), were considerably lower, only averaging 2.9% (s(g2)) and 1.7% (s(ML)), respectively (Figure 1 and Supplementary Table 1). Only 1 of the 12 date- and method-specific selfing rate estimates was marginally significant (s(ML) on sampling day 22 March 2014). The inclusion of locus Rb_3 did not change these estimates markedly (mean 2.0% (s(g2)) and mean 1.3% (s(ML)), Figure 1 and Supplementary Table 1), nor did it change their statistical significance.
There were no significant temporal differences in selfing rates when considering s(g2) and s(ML), independent of the inclusion of locus Rb_3, as confidence intervals on all sampling days were overlapping (Figure 1 and Supplementary Table 1). The selfing rate based on the inbreeding coefficient, on the other hand, did show some significant variation over time. According to FIS, selfing was most common at the beginning of 2014 (both with and without locus Rb_3), and least common in 2015 (only without locus Rb_3, Figure 1 and Supplementary Table 1).
Comparison of selfing rates between methods
On all sampling days and irrespective of the inclusion of locus Rb_3, selfing rates based on progeny arrays were lowest and could be estimated with the highest precision (Figure 1 and Supplementary Table 1). In contrast, inbreeding coefficient-based estimates significantly exceeded progeny-array estimates in all direct comparisons (Figure 1 and Supplementary Table 1). Meanwhile, selfing rates based on a point estimate of the second-order heterozygosity disequilibrium (g2), computed using the software RMES, were never significantly different from progeny-array estimates, as all CIs of s(g2) included zero (Figure 1 and Supplementary Table 1). Similarly, maximum-likelihood estimates of the selfing rate, also provided by RMES, overlapped with progeny-array estimates in 6/8 (75.0%) direct comparisons, the sole exception being sampling day 22 March 2014 independent of the inclusion of locus Rb_3 (Figure 1 and Supplementary Table 1).
Discussion
Our study shows that inbreeding coefficient-based selfing rate estimates can be significantly positive even though progeny-array analysis indicates complete outcrossing. We found that most of this upward bias seems to be caused by null alleles, highlighting the importance of a detailed technical assessment of marker performance as part of the study. Had we estimated selfing rates solely based on inbreeding coefficients, we would have erroneously concluded that our study population is moderately mixed-mating. We would have been more confident about this erroneous conclusion had we failed to exclude the single locus with a high frequency of null alleles. This finding demonstrates how severely null alleles can distort selfing rate estimates that rely on heterozygote deficiency (David et al., 2007; Wang et al., 2012), and how carefully one needs to examine the properties of the available molecular markers. Given how difficult it is to develop genetic markers that are both sufficiently powerful and entirely free of null alleles, it is probably safest to stop using the inbreeding coefficient in its current form for estimating selfing rates with this type of markers.
Our study also shows that selfing rate estimates obtained from the software RMES, which deploys identity disequilibria-based algorithms that are not sensitive to null alleles (David et al., 2007; Wang et al., 2012), effectively corrected the shortcomings of the inbreeding coefficient-based method and yielded estimates very similar to those from progeny arrays. It appears that the assumptions made by RMES about the lack of biparental inbreeding, inbreeding disequilibrium and cryptic population subdivision were largely met in the particular population studied here. Apart from corroborating the general absence of selfing, the similarity of the two types of estimates provides additional insights into the studied mating system. First, it suggests that not only selfing but also biparental forms of inbreeding are rare, seeing that population-level estimates, which would be inflated by biparental inbreeding, are very low. Second, it shows that selfing was mostly absent not only in the last generation, as documented by the progeny-array estimates, but also in the few generations before, as attested by the population-level estimates that integrate selfing events over several generations (David et al., 2007; Jarne and David, 2008; Wang et al., 2012). Third, the sampled snails are most likely part of a single, coherent population, given that population-level selfing rate estimates do not suffer from a Wahlund effect. Fourth, our progeny-array estimates reflect ‘primary’ selfing rates estimated in embryos, whereas the population-level estimates were computed using adult individuals that are, by definition, the surviving subset of all the snails originally present in the population. In principle, finding that ‘primary’ and adult-level selfing rate estimates are very similar thus indicates that in- or outbreeding depression is absent or weak (Ritland, 1990). However, as all four embryonic cohorts were devoid of selfed individuals, no inference can be made about the presence of in- or outbreeding from our study.
We conclude that RMES proves to be a valid alternative to the progeny-array approach for estimating selfing rates under a number of conditions, listed without a claim to completeness in Table 5 (see also Jarne and David, 2008; Wang et al., 2012). From this list it is evident that using RMES as a fully equivalent method to progeny arrays puts relatively high requirements on the prior knowledge available about the population to be studied (conditions 1–5). Studies that unwittingly fail to meet one or several of these requirements run the risk of falsely inferring either selfing or its absence, depending on the assumptions that are not met. Progeny arrays should thus be used for estimating the selfing rate in populations that have not been studied before in sufficient detail, or if conditions 6 or 7 are not fulfilled. If all conditions are met, however, the savings in terms of time and money associated with using a population-level method are substantial, as for example in our study population-level estimates were computed using 5.7 × fewer genotypes than progeny-array estimates (181 vs 1034).
To the best of our knowledge, ours is only the third study in which selfing rates have been estimated simultaneously using the population-level methods implemented in RMES and using progeny arrays. In four populations of the terrestrial snail Arianta arbustorum, analyzed using four polymorphic microsatellite loci in 6–7 laboratory-bred families per population (mean 42 hatchlings per family), both methods yielded similar values, with progeny-array estimates exceeding RMES estimates only by 0.01, 0.03, 0.07 and 0.11, respectively (Kupfernagel et al., 2010). In addition, in three populations of the coral Favia fragum, both methods have been applied (Carlon and Lippé, 2011). Unfortunately, statistical power was very low in this data set, caused by extremely low levels of heterozygosity within mothers (only 4/22 mothers heterozygous for ⩾1 locus) despite an average of 13.7 polymorphic microsatellite loci per population. Undoubtedly, more studies are necessary until the biological and technical conditions will be identified under which RMES estimates can replace progeny-array estimates. Past studies on mating systems in hermaphroditic animals showed much variability among selfing rates (Jarne and Auld, 2006), and our empirical, field-based results support the previously voiced belief that unfortunately a significant part of this variation may stem from technical problems associated with the performance of the molecular markers that are available (Jarne and Auld, 2006; Jarne and David, 2008; Escobar et al., 2011). Below we highlight this issue by commenting on previous studies evaluating the mating system of R. balthica, some of which were done in our own research group.
Based on our study this natural snail population is fully outcrossing and does not show any variation in selfing rates, neither among families, nor during the reproductive lifespan within a single generation, nor across at least three consecutive generations. We accounted for a wide range of potential confounding effects, including biases due to null alleles, and ensured that the statistical power of our genetic markers was sufficient. We are thus confident in concluding that the mating system of R. balthica in the study population is, at least for the time being, stably outcrossing. This finding is largely in accordance with an early, FIS-based analysis of four populations in Lake Geneva (Jarne and Delay, 1990).
The mating system of snail populations in Lake Zurich has already been studied before in our research group and the data have implied mixed mating (Wiehn et al., 2002; Jokela et al., 2006). In the year 2000, the same population was reported to be mixed-mating based on both progeny arrays and heterozygote deficiency in adults (Jokela et al., 2006). Selfing had also been described to be widespread in four additional populations of R. balthica in Lake Zurich (Wiehn et al., 2002). Clearly, these results are in stark contrast to our findings. Several reasons for the discrepancy can be imagined. Differences to earlier, laboratory-based studies could have arisen where in the laboratory setting some isolated mothers ran out of allosperm. Alternatively, there could be true temporal or spatial differences in the mating system, for instance caused by bottlenecks and the ensuing scarcity of mating partners. At least for the study population this explanation appears unlikely, as population density has remained constant during the past two decades (J Jokela, unpublished data). A long population history and ample time for accumulating mutations are also suggested by discontinuing allele size distributions at several microsatellite loci (for example, loci Rb_1–3, Rb_8 and Rb_10; Table 4). Theoretically, discontinuing allele size distributions could also have arisen through hybridization of closely related Radix species, but the evident purity of the study population (see Supplementary Methods 1) speaks against this scenario. We thus consider it likely that two methodological issues led, together or separately, to an overestimation of selfing rates. The earlier studies, even if they used state-of-the-art methods at that time, relied on a small number of weakly polymorphic allozyme markers, resulting in insufficient statistical power for detecting outcrossed offspring (Bernatchez and Duchesne, 2000; Jarne and David, 2008). The low number of loci and low degree of polymorphism also led to difficulties in detecting null alleles that might have led to heterozygote deficiency (that is, upwards biased FIS values) (David et al., 2007; Jarne and David, 2008; Wang et al., 2012).
There is also little evidence for selfing to be common in R. balthica in two more recent studies of molecularly identified, adult snails genotyped at eight microsatellite loci and analyzed using RMES. Non-zero selfing rate estimates were found in 16/25 European populations (Pfenninger et al., 2011) and in 2/2 populations in western Switzerland (Haun et al., 2012) that had sample sizes of at least 20 genotyped individuals (mean s(g2) across all 27 populations: 0.16, range: 0.00–0.53, mean sample size per population: 28.0). As no CIs are reported for these estimates, however, we do not know whether some of them are significantly different from zero. The occurrence of mixed mating in other populations of R. balthica thus cannot be confirmed until more populations are analyzed using both reliable methods and adequate sample sizes.
In conclusion, our study emphasizes the technical challenges in mating system analysis. The positive note is that at present powerful methods are available for both family- and population-level analyses that should allow for an accurate estimation of mating systems if used appropriately. Both approaches may also be applied fruitfully using next-generation sequencing data such as large numbers of single-nucleotide polymorphisms. Unmet conditions such as the presence of biparental inbreeding when using indirect methods will cause the same difficulties independent of whether single-nucleotide polymorphisms or microsatellites are used, especially when mixed maters are studied. However, the high resolution provided by millions of single-nucleotide polymorphisms will undoubtedly facilitate progeny arrays. Unfortunately, our results also indicate the problems that may have gone undetected in earlier published assessments of selfing rates. Science is self-correcting, and therefore we hope that our study motivates reanalysis and reassessment of selfing rates in cases where we need to know the mating system of our study species and populations.
Data archiving
Sequence data have been submitted to GenBank accession nos. KX832428–KX832513. Microsatellite genotype data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.2sk82.
References
Ahlgren J, Yang X, Hansson LA, Bronmark C . (2013). Camouflaged or tanned: plasticity in freshwater snail pigmentation. Biol Lett 9: 20130464.
Allard RW . (1975). The mating system and microevolution. Genetics 79: 115–126.
Avise JC, Mank JE . (2009). Evolutionary perspectives on hermaphroditism in fishes. Sex Dev 3: 152–163.
Baker HG . (1955). Self-compability and establishment after ‘long-distance’ dispersal. Evolution 9: 347–349.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F . (1996–2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II: Montpellier, France.
Bernatchez L, Duchesne P . (2000). Individual-based genotype analysis in studies of parentage and population assignment: how many loci, how many alleles? Can J Fish Aquat Sci 57: 1–12.
Brönmark C, Lakowitz T, Hollander J, Fenton B . (2011). Predator-induced morphological plasticity across local populations of a freshwater snail. PLoS One 6: e21773.
Carlon D, Lippé C . (2011). Estimation of mating systems in Short and Tall ecomorphs of the coral Favia fragum. Mol Ecol 20: 812–828.
Charlesworth D, Charlesworth B . (1987). Inbreeding depression and its evolutionary consequences. Annu Rev Ecol Syst 18: 237–268.
Cordellier M, Pfenninger M . (2009). Inferring the past to predict the future: climate modelling predictions and phylogeography for the freshwater gastropod Radix balthica (Pulmonata, Basommatophora). Mol Ecol 18: 534–544.
Coutellec-Vreto MA, Madec L, Guiller A . (1997). Selfing and biparental inbreeding: a mating system analysis in Lymnaea peregra (Gastropoda: Lymnaeidae). Heredity 79: 277–285.
Dabrowski MJ, Bornelöv S, Kruczyk M, Baltzer N, Komorowski J . (2015). ‘True’ null allele detection in microsatellite loci: a comparison of methods, assessment of difficulties and survey of possible improvements. Mol Ecol Resour 15: 477–488.
Dakin EE, Avise JC . (2004). Microsatellite null alleles in parentage analysis. Heredity 93: 504–509.
David P, Pujol B, Viard F, Castella V, Goudet J . (2007). Reliable selfing rate estimates from imperfect population genetic data. Mol Ecol 16: 2474–2487.
Escobar J, Auld J, Correa A, Alonso J, Bony Y, Coutellec M-A et al. (2011). Patterns of mating-system evolution in hermaphroditic animals: correlations among selfing rate, inbreeding depression, and the timing of reproduction. Evolution 65: 1233–1253.
Fisher RA . (1941). Average excess and average effect of a gene substitution. Ann Eugen 11: 53–63.
Haun T, Salinger M, Pachzelt A, Pfenninger M . (2012). On the processes shaping small-scale population structure in Radix balthica (Linnaeus 1758). Malacologia 55: 219–233.
Jarne P, Auld JR . (2006). Animals mix it up too: the distribution of self-fertilization among hermaphroditic animals. Evolution 60: 1816–1824.
Jarne P, Charlesworth D . (1993). The evolution of the selfing rate in functionally hermaphrodite plants and animals. Annu Rev Ecol Syst 24: 441–466.
Jarne P, David P . (2008). Quantifying inbreeding in natural populations of hermaphroditic organisms. Heredity 100: 431–439.
Jarne P, Delay B . (1990). Population genetics of Lymnaea peregra (Müller) (Gastropoda: Pulmonata) in Lake Geneva. J Molluscan Stud 56: 317–322.
Jokela J, Wiehn J, Kopp K . (2006). Among- and within-population variation in outcrossing rate of a mixed-mating freshwater snail. Heredity 97: 275–282.
Jones O, Wang J . (2010). COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10: 551–555.
Kopp K, Wolff K, Jokela J . (2012). Natural range expansion and human-assisted introduction leave different genetic signatures in a hermaphroditic freshwater snail. Evol Ecol 26: 483–498.
Kupfernagel S, Rusterholz HP, Baur B . (2010). Variation in multiple paternity and sperm utilization patterns in natural populations of a simultaneous hermaphrodite land snail. Biol J Linn Soc 99: 350–361.
Lawton SP, Lim RM, Dukes JP, Kett SM, Cook RT, Walker AJ et al. (2015). Unravelling the riddle of Radix: DNA barcoding for species identification of freshwater snail intermediate hosts of zoonotic digeneans and estimating their inter-population evolutionary relationships. Infect Genet Evol 35: 63–74.
Nei M . (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Pfenninger M, Cordellier M, Streit B . (2006). Comparing the efficacy of morphologic and DNA-based taxonomy in the freshwater gastropod genus Radix (Basommatophora, Pulmonata). BMC Evol Biol 6: 100.
Pfenninger M, Salinger M, Haun T, Feldmeyer B . (2011). Factors and processes shaping the population structure and distribution of genetic variation across the species range of the freshwater snail Radix balthica (Pulmonata, Basommatophora). BMC Evol Biol 11: 135.
Pompanon F, Bonin A, Bellemain E, Taberlet P . (2005). Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6: 847–846.
Ritland K . (1990). Inferences about inbreeding depression based on changes of the inbreeding coefficient. Evolution 44: 1230–1241.
Ritland K . (2002). Extensions of models for the estimation of mating systems using n independent loci. Heredity 88: 221–228.
Ritland K, Jain S . (1981). A model for the estimation of outcrossing rate and gene frequencies using n independent loci. Heredity 47: 35–52.
Schniebs K, Gloer P, Vinarski MV, Hundsdoerfer AK . (2011). Intraspecific morphological and genetic variability in Radix balthica (Linnaeus 1758) (Gastropoda: Basommatophora: Lymnaeidae) with morphological comparison to other European Radix species. J Conchol 40: 657–678.
Waits L, Luikart G, Taberlet P . (2001). Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10: 249–256.
Wang JL, El-Kassaby YA, Ritland K . (2012). Estimating selfing rates from reconstructed pedigrees using multilocus genotype data. Mol Ecol 21: 100–116.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wiehn J, Kopp K, Rezzonico S, Karttunen S, Jokela J . (2002). Family-level covariation between parasite resistance and mating system in a hermaphroditic freshwater snail. Evolution 56: 1454–1461.
Wright S . (1969) Evolution and the Genetics of Populations: The Theory of Gene Frequency v. 2. University of Chicago Press: Chicago, IL, USA.
Zar JH . (1996) Confidence limits for population proportions. In: Fisher S, Snavely SL (eds), Biostatistical Analysis, 3 edn. Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, p 525.
Acknowledgements
We thank the editor and three anonymous reviewers for helpful comments on earlier versions of this manuscript, Kirstin Kopp for assistance with the establishment of a fast, cost-effective genotyping routine and Patrice David for support when using RMES. DNA fragments were analyzed for length polymorphisms using a 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) situated at the Genetic Diversity Centre (GDC), ETH Zurich (Zurich, Switzerland).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Bürkli, A., Sieber, N., Seppälä, K. et al. Comparing direct and indirect selfing rate estimates: when are population-structure estimates reliable?. Heredity 118, 525–533 (2017). https://doi.org/10.1038/hdy.2017.1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.1
