
Abstract
Cottonwoods are foundation riparian species, and hybridization among species is known to produce ecological effects at levels higher than the population, including effects on dependent species, communities and ecosystems. Because these patterns result from increased genetic variation in key cottonwood traits, novel applications of genetic tools (for example, QTL mapping) could be used to place broad-scale ecological research into a genomic perspective. In addition, linkage maps have been produced for numerous species within the genus, and, coupled with the recent publication of the Populus genome sequence, these maps present a unique opportunity for genome comparisons in a model system. Here, we conducted linkage analyses in order to (1) create a platform for QTL and candidate gene studies of ecologically important traits, (2) create a framework for chromosomal-scale perspectives of introgression in a natural population, and (3) enhance genome-wide comparisons using two previously unmapped species. We produced 246 backcross mapping (BC1) progeny by crossing a naturally occurring F1 hybrid (Populus fremontii × P. angustifolia) to a pure P. angustifolia from the same population. Linkage analysis resulted in a dense linkage map of 541 AFLP and 111 SSR markers distributed across 19 linkage groups. These results compared favorably with other Populus linkage studies, and addition of SSR loci from the poplar genome project provided coarse alignment with the genome sequence. Preliminary applications of the data suggest that our map represents a useful framework for applying genomic research to ecological questions in a well-studied system, and has enhanced genome-wide comparisons in a model tree.
Similar content being viewed by others
Introduction
Over the past decade, advances in DNA marker technology have provided a glimpse into the genetic bases of ecological processes, expanding our knowledge of ecological genetics (reviewed in Via, 2002) and leading to the emerging field of community and ecosystem genomics (see Feder and Mitchell-Olds, 2003; Thomas and Klaper, 2004; Mauricio, 2005; Whitham et al., 2006). The ability to identify and characterize quantitative trait loci (QTL) associated with traits of ecological significance continues to be an important task and has contributed significantly to an understanding of ecological processes from a genomic perspective (Jackson et al., 2002). Marker techniques such as amplified fragment length polymorphisms (AFLP, Vos et al., 1995) have made linkage and QTL mapping possible for virtually any organism by overcoming the major barrier to such studies in the past (that is, lack of sufficient markers, see review by Doerge (2002)). In addition to AFLP based maps, over 4000 SSR markers have been developed for the genus Populus (http://www.ornl.gov/sci/ipgc/ssr_resource.htm), many of which have been used for genetic mapping in both Populus (discussed below) and the related genus Salix (Hanley et al., 2006, see also the above website). These markers provide a unique opportunity for wide-ranging comparative genomic studies across diverse taxa.
Beginning with Keim et al. (1989) and Whitham (1989), 18 years of research on a cottonwood hybrid zone (Populus fremontii × P. angustifolia) in northern Utah has revealed numerous relationships between genetic variation (via hybridization) in a foundation tree and higher order processes. Genetically based variation in cottonwood phytochemical (for example, Driebe and Whitham, 2000; Schweitzer et al., 2004; Bailey et al., 2004, 2005; Rehill et al., 2006), morphological (for example, Floate and Whitham, 1995; Larson and Whitham, 1997) and phenological traits (for example, Floate et al., 1993) has been shown to affect populations, communities and ecosystem processes at multiple scales including individual trees, stands, rivers and the western US region (Bangert et al., 2006a, 2006b). These patterns suggest that novel applications of population and quantitative genetic tools may provide unprecedented opportunities to link genetic factors (for example, QTL and genes) with ecological patterns. This would help fulfill a major goal of placing community and ecosystem ecology within an evolutionary framework (Mitton, 2003; Whitham et al., 2003, 2006).
In addition to its potential as a model organism for ecological research, Populus has recently emerged as a premier model for forest tree biology and improvement (Taylor, 2002; Wullschleger et al., 2002). Characteristics such as ease of vegetative and seed propagation, fast growth rate, small genome size, and conservation of chromosome number across the genus (n=19) make Populus ideal for experimental research, and mapping populations have been produced for numerous geographically and ecologically distinct species from diverse sections within the genus (see Table 2 below). These experimental populations provide a unique opportunity for comparative linkage mapping in a model system (for example, Yin et al., in review)—an opportunity that has been further enhanced by the recent publication of the P. trichocarpa genome sequence (Tuskan et al., 2006).
Here, we have created a high-density AFLP linkage map from a segregating interspecific backcross population of hybrid cottonwoods (P. fremontii × P. angustifolia). We chose a backcross design for four reasons: first, because few codominant markers had been developed for Populus at the beginning of our study, we used a dominant marker system (AFLP) which is best served by a backcross design (that is, few repulsion phase markers); second, our study was aimed at identifying QTL of ecological importance in a hybrid system where F2's (but not backcrosses) are apparently rare (Keim et al., 1989); third, introgression in the natural population occurs unidirectionally (P. fremontii alleles to P. angustifolia) (Keim et al., 1989; Martinsen et al., 2001); and fourth, F1 × F1 crosses in the greenhouse showed decreased success relative to backcrosses, suggesting negative interactions in the F2 generation (G Martinsen, unpublished data). We aligned our map with Yin et al.'s (2004) map using SSR markers from the poplar genome sequence project (Tuskan et al., 2006) in order to link our data with the poplar genome sequence. The specific objectives of this study were to (1) create a linkage map for future QTL and candidate gene studies of ecologically important traits, (2) provide a framework for a chromosomal scale perspective of introgression in a natural system, and (3) to enhance comparisons of genome structure among multiple species within the genus.
Materials and methods
Mapping pedigree and DNA extraction
Parents for a segregating backcross mapping population were chosen from a naturally occurring hybrid zone on the Weber River in northern Utah. Parental species/hybrid class was determined using preliminary marker data from 33 nuclear RFLP loci (detailed in Martinsen et al., 2001). Using the technique of Stanton and Villar (1996), we crossed a P. angustifolia female clone (#996) with a male F1 hybrid (P. fremontii × P. angustifolia, clone WSU-6) resulting in 246 full-sib backcross progeny. The seed progeny were germinated under a misting bench within 2 weeks of dehiscence and planted in standard potting mix. Cuttings of the parental clones were made at the same time. Cuttings from the parent clones and hybrid progeny were grown in a greenhouse for 2 years under uniform conditions. Fresh leaves were collected from parents and progeny at the height of the growing season, frozen on dry ice, and in some cases lyophilized. DNA was extracted as per Martinsen et al. (2001), or using the Qiagen DNeasy plant miniprep kit (Qiagen, Helden, Germany). Reanalysis of RFLP markers subsequent to the cross confirmed WSU-6 as an F1 hybrid, but showed P. angustifolia clone #996 to likely be an advanced backcross hybrid/introgressant (see Martinsen et al., 2001) heterozygous for P. fremontii and P. angustifolia alleles at a single locus (RFLP probe p1254, Bradshaw and Stettler, 1993).
AFLP analysis
AFLP analysis was performed using the method of Vos et al. (1995) with modifications from Travis et al. (1996). Preselective amplification was conducted using adenine (A) as the first selective base in all cases. Forty-five 3+3 primer combinations (EcoRI+AXX/MseI+AXX) were chosen at random, and used to generate marker data. Marker names include the second and third selective bases for the EcoRI enzyme followed by the second and third MseI selective bases, and finally by the approximate marker size in base pairs. For example, GGCC150 represents a 150 bp marker generated from an EcoRI+AGG/MseI+ACC primer combination.
SSR analysis
A subset of 46 individuals from our mapping population were screened with 341 SSR markers that were derived from the Populus trichocarpa whole-genome sequencing project, and mapped in a P. trichocarpa × P. deltoides (TD) pedigree to enhance genome assembly (Tuskan et al., 2006). These SSR markers were selected at regular intervals throughout the genome to allow integration of the P. angustifolia × P. fremontii map with the whole-genome sequence, and to enhance comparisons of genome structure among multiple members of the genus. Initial screening was conducted with both parents and six progeny, and loci that appeared to be segregating in both parents were selected for mapping. SSR amplification and genotyping was performed as described elsewhere (Yin et al., 2004), except loci were analyzed on an ABI3730 automated capillary electrophoresis instrument, and amplification was performed with 10 pmol fluorescein 12-dUTP (Roche Diagnostics, Indianapolis, IN, USA), rather than end-labeled primers.
Marker segregation and map construction
Linkage analysis was restricted exclusively to markers with expected segregation ratios of 1:1 (that is, testcross markers where the F1 parent was +/− and the P. angustifolia parent −/−, see Supplementary Table S1, electronic Supplementary Material). Segregation distortion in the testcross markers was assessed using a χ2 analysis, and was identified as significant (P<0.05) deviation from expected Mendelian segregation. Distorted markers were not excluded from the linkage analysis (discussed below). Species origins of markers were inferred primarily on the assumption that markers that were homozygous null in P. angustifolia and heterozygous in the F1 were likely fixed absent or rare in P. angustifolia and present in P. fremontii. Furthermore, markers that were putatively derived from the same species were consistently in the same linkage phase, and were generally fixed in wild populations (M Zinkgraf, S Woolbright and G Allan, unpublished data), thus lending support to our assumptions.
The linkage map was created using MAPMAKER 3.0 (Lander et al., 1987). Given the number of framework markers from preliminary results (Woolbright, 2001), the estimated genome size from Bradshaw et al. (1994) and whole-genome sequence assembly (Tuskan et al., 2006), and simulations from Yin et al. (2004), we chose an LOD score of 8.0 for linkage analysis. We then determined the appropriate recombination fraction (rf=0.37) using the relationship between LOD score and population size described in Cervera et al. (2001) (see also Yin et al., 2004). Using these values as the ‘default linkage criteria’, preliminary linkage groups were identified with the ‘groups’ command. Marker data were then inverted for the entire dataset in order to place possible repulsion phase markers. Once initial groups were identified, one or two anchor loci were chosen to begin map construction. Markers within each group were ordered using the ‘Order’ command, and initial orders checked using the ‘ripple’ command again with an LOD threshold of 8.0. Additional markers were added using the ‘build’ command and checked with the ‘ripple’ command. When markers could not be ordered unambiguously, the marker with the least amount of missing data was usually chosen as a framework marker and the rest added as accessory markers using the ‘try’ command. Occasionally, markers that resulted in the least number of likely scoring errors or in the least amount of map expansion were chosen for the framework map.
SSR were also placed in the framework AFLP map using the ‘try’ command. Because SSR markers were mapped using a much smaller population size, distances between framework AFLP markers were fixed, and SSR positions were determined by interpolation between framework positions. Codominant SSR were used to infer alignment with other Populus maps, and each linkage group was reoriented and assigned a name according to the convention of the International Populus Genome Consortium (http://www.ornl.gov/sci/ipgc/) and Cervera et al. (2001).
Once linkage groups were characterized, the size of blocks showing segregation distortion in favor of a particular allele was estimated as per Yin et al. (2004). These values were then used to calculate the ratio of distorted regions to total length of the chromosome.
Marker distribution
The distribution of markers among linkage groups was calculated using the method from Remington et al. (1999). Using the Poisson distribution, we evaluated the probabilities P(m⩽λ) and P(m⩾λ) at α⩽0.05 where m and λ are the total and expected marker numbers, respectively, for each linkage group.
We also looked for regions of clustering and dispersion within each linkage group using the method from Yin et al. (2004). By sliding along each linkage group, ‘windows’ for clustering analysis were identified as consecutive intervals where marker spacing was less than the average spacing for the entire map. Windows for testing marker dispersion were defined by consecutive intervals with spacing greater than the average. The number of markers within each window was counted, and compared to the null expectation for evenly spaced markers for a particular window size. Significant departures from expectation were tested under a cumulative Poisson distribution using a one-tailed test (α⩽0.05).
Genome length and coverage
Observed genome length was calculated as the sum (cM) of all linkage groups for both the complete (all markers) and framework maps. Only framework markers were used to estimate genome length in order to avoid problems associated with marker clustering (see Cervera et al., 2001). Estimated genome length was calculated using the method from Hulbert et al. (1988), which provides an estimate based on partial linkage data. We also used the method from Nelson et al. (1994), which incorporates information from all linked and unlinked markers.
Observed map coverage was calculated as the ratio of observed map length to the estimated map length (Ge from Hulbert et al., 1988) for both the complete and framework maps. Theoretical map coverages were estimated for the framework map as per Lange and Boenke (1982), which accounts for chromosomal ends; and using the method from Bishop et al. (1983), which accounts for linear chromosomes.
Results
Marker analysis
Forty-five AFLP primer combinations yielded a total of 809 scorable polymorphic markers, with an average of 18 polymorphisms per primer combination. Of these, 564 were ‘pseudo-testcross’ markers with the F1 parent heterozygous (+/−), and the recurrent parent carrying only the null allele (−/−). Of the remaining markers, 97 were ‘intercross’ markers (+−/+−), and 148 were heterozygous in the recurrent parent (−−/+−, see Supplementary Table S1, ESM). These were excluded from linkage analysis. A total of 790 monomorphic fragments (average 17.6 per primer combination) were also identified. Of the 341 SSR markers tested, 89 failed to amplify, 35 were monomorphic, 24 were intercross informative, 86 were paternally informative, 32 were maternally informative and 75 were both paternally and maternally informative.
Segregation distortion
Chi-square analysis of the raw AFLP marker data revealed significant (P⩽0.05) deviation from the expected 1:1 segregation pattern in 113 of the 541 (21%) mapped AFLP testcross markers. Table 1 summarizes genome-wide segregation distortion at the level of individual linkage groups. Fifteen distinct regions or blocks of distortion occurred on 11 of 20 linkage groups, with two linkage groups (XVIII and XIX) exhibiting distortion across more than half their lengths. The size of the distorted regions varied among linkage groups. Distortion occurred more often in the direction of the recurrent allele (P. angustifolia), with 276.1 cM distorted (13.4% of the genome) vs 112.6 cM for the donor allele (P. fremontii 5.5% of the genome).
Map construction genome length, and coverage
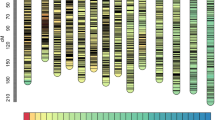
MAPMAKER grouped the 564 AFLP testcross markers into 19 linkage groups, one triplet and nine unlinked markers. Twenty-four markers were removed from the analysis due to unnecessary map expansion or linkage to multiple groups. This was most often caused by the inclusion of faint markers that were difficult to score, and/or by extreme segregation distortion that may have been the result of comigration of separate loci. One hundred eleven SSR markers were placed in interpolated positions (see Materials and methods), including a minimum of 2 and a maximum of 11 markers on each of the 19 Populus linkage groups (Figure 1).




Genetic linkage map of male clone WSU-6, a Populus. fremontii × P. angustifolia F1 hybrid, as determined by 246 progeny from a backcross to P. angustifolia clone #996. Linkage maps were drawn using the MapChart software (Voorrips, 2002). Linkage groups were compared with groups from Yin et al. (2004) (shown at right of each pair) using homologous SSR markers and named as per Cervera et al. (2001). Bars between each pair of linkage groups show the relative position of homologous SSR. Numbers at left of each group show absolute marker position in Kosambi map units. Marker names are to the right. Accessory markers are in italics. Names ending in ‘r’ represent inverted markers. Markers in square brackets, [], indicate microsatellite loci used to compare the two maps. Markers in brackets,{}, indicate possible translocations and include the alternate linkage group. Shaded regions indicate blocks of distortion. Light gray shading indicates distortion toward the recurrent (P. angustifolia) allele, dark gray distortion toward the donor (P. fremontii) allele.
A total of 328 framework AFLP markers were identified and used to create a framework map spanning a distance of 2030.6 cM. Table 2 summarizes the results of our linkage and genome analyses, comparing them with results from other recent Populus mapping efforts. All results are within the range reported from other studies, except for Bishop et al.'s (1983) method for theoretical map coverage, which was slightly higher than the others.
Table 3 summarizes AFLP marker distribution when all markers are considered. Three linkage groups (XIII, XV, and XIX) contained more markers than expected, and four (IX, XI, XII and XVIII) contained less. At the level of individual linkage groups, significant clustering occurred within all but one (VII) major linkage group.
Discussion
Our experimental design yielded a genetic map of comparable quality to other Populus maps (see Table 2). The number of major linkage groups was equal to that of the haploid chromosome number in Populus (n=19). Our estimate of genome length falls within the range observed from other studies and is near the original estimate of 2400–2800 cM set by Bradshaw et al. (1994), which has been verified through simulation studies (Yin et al., 2004). Observed map length was also within the range of other Populus maps but lower than our estimated length, and lower than the more robust map of Yin et al. (2004). The discrepancy between estimated and observed lengths has been observed in other studies (Table 2), and can be explained by problems with marker clustering or dispersion due to map expansion caused by cosegregation of AFLP bands and other genotyping errors.
Marker clustering and dispersion were also comparable to other Populus maps (for example, Yin et al., 2004). Explanations for dispersion include regions of increased recombination and missing markers that have not been identified, perhaps due to gaps in occurrence of restriction enzyme recognition sites (Supplementary Figure S1, ESM). The addition of multiallelic SSR markers has helped to alleviate these problems and the availability of a map-linked genome sequence (Tuskan et al., 2006) will allow future targeted design of SSR and single nucleotide polymorphic (SNP) markers specifically for dispersed regions.
Targeted SSR and SNP markers should also be useful for characterizing problem areas arising from the presence of P. fremontii alleles in the genome of the P. angustifolia parent (for example, RFLP p1254, Martinsen et al., 2001). The presence of introgressed (that is, intercross, Supplementary Table S1, ESM) fragments are indistinguishable from shared parental alleles and both likely lead to ‘blind spots’ when using dominant markers to search for ecologically relevant QTL. If introgression is the result of positive selection, ecologically important regions of the genome could therefore be missed due to poor linkage data when codominant markers are unavailable. Future addition of evenly distributed, targeted SSRs, combined with introgression studies in natural populations should help alleviate this problem.
In contrast to dispersed regions, we observed marker clusters for all but one (VII) of the 19 major linkage groups. Some marker clusters (that is, 5% at α=0.05) are expected due to random chance. Clustering also occurs in regions of the genome with reduced recombination and has been used to describe structural features of chromosomes. Young et al. (1999) were able to identify likely positions of centromeres in soybean linkage groups by comparing the distribution of a methylation sensitive (PstI) vs an insensitive restriction enzyme (EcoR1). In the analysis, PstI markers were underrepresented in marker clusters thought to occur in cytosine methylated heterochromatic regions surrounding the centromere. Thus, enzyme choice in AFLP analyses might be used to produce more uniformly distributed maps. Finally, clustering may arise from problems with meiotic pairing due to divergence of parental chromosomes, particularly when using interspecific crosses between highly divergent species.
Map comparisons
Our map showed a high degree of marker colinearity with the map of Yin et al. (2004); however, map alignment using SSR markers identified two putative inversions, and eight putative translocations (Figure 1). Given the divergent species used in the comparison, chromosomal rearrangements are not necessarily unexpected. Alternatively, the inversions on linkage groups XV and XVIII could be the result of errors in map order due to the small sample size used in the SSR analysis. Marker translocations could also be the result of multiple and divergent SSR priming sites arising from recent or ancient genome duplications (see Tuskan et al., 2006). These issues are being addressed by enhancing the resolution of our SSR map, by performing comparative analyses with additional Populus genetic maps (Yin et al., in review), and by resequencing and reassembling problematic areas of the genome (GA Tuskan, personal communication).
The absence of major chromosomal rearrangements in this and other comparative mapping (for example, Cervera et al., 2001), coupled with the shared areas of segregation distortion and recombination repression (Yin et al., in review) suggest that genic interactions are mostly responsible for species barriers between P. fremontii and P. angustifolia. These barriers likely resulted in decreased success observed in experimental F1 × F1 crosses and backcrosses to P. fremontii (G Martinsen, unpublished data), as well as unidirectional introgression in the natural system (Keim et al., 1989; Martinsen et al., 2001). Similar patterns have been observed in other species from sections Tacamahaca and Aigeiros (Floate, 2004), and likely indicate shared barriers at the section level. Molecular data have contributed to the characterization of such barriers by revealing ‘hallmarks’ such as segregation distortion (discussed below) and recombination repression. Linkage analyses, QTL studies and candidate gene surveys have been useful for identifying traits and genes underlying these phenomena (for example, Bradshaw and Stettler, 1994; Cervera et al., 2001; Yin et al., 2004).
Our sample size allowed for only coarse map alignment, and reliable statistical tests for shared segregation distortion were not feasible. However, we did notice large areas of shared distortion favoring Tacamahaca alleles on at least two linkage groups (IV and XIX). One of these (XIX) was used in a recent study by Yin et al. (in review) that identified similarities in recombination repression and segregation distortion across multiple families. These patterns have provided insight into potential species barriers (for example, R genes), and suggest the evolution of a primitive Populus sex chromosome (Yin et al., in review). Thus, the data revealing segregation distortion in P. fremontii × P. angustifolia hybrids have contributed to our understanding of Populus at levels exceeding our original intention (that is, section vs species).
In contrast to genic interactions contributing to species barriers, genetic admixture may also lead to the adaptive introgression of alleles, an important but largely understudied aspect of plant evolution (Grant, 1971; Martinsen et al., 2001; Whitney et al., 2006). Recently, Lexer et al. (2007) used map-based SSR to avoid tightly linked markers when surveying for introgression and linkage disequilibrium in European hybrid zones of P. alba and P. tremula. Primers for the loci they used are known to amplify in multiple species across several sections within the genus (see also Rahman and Rajora, 2002 and citations therein). SSR markers developed for Populus have also been used for mapping in Salix (Hanley et al., 2006). Thus, map-based genetic markers provide a unique (but untested) opportunity for comparative studies of introgression across multiple taxonomic levels. Furthermore, these studies demonstrate how research questions aimed at specific populations or species can contribute to a larger focus (that is, evolution in the Salicacae), arguing for the continued use of map-based markers across broad areas of inquiry.
Segregation distortion
Segregation distortion is common in mapping studies of forest trees and has been documented in most if not all Populus mapping efforts. While distortion can influence map construction (Zhang et al., 2002 and citations theirin) and may affect QTL detection through spurious associations, exclusion of distorted markers is not necessarily warranted as they may be linked to genes or traits of interest. For example, both Cervera et al. (2001) and Yin et al. (2004) found that segregation distortion in some markers may have resulted from susceptibility to Melamspora rust or other selective forces acting during generation of the hybrid pedigree. Bradshaw and Stettler (1994) found that a recessive pollen lethal allele tightly linked to a mapped RFLP marker (p1054) in a P. trichocarpa × P. deltoides cross was the most likely cause of distortion in their mapping population. These results suggest markers showing segregation distortion due to linkage with genes under selection may have important ecological consequences, and should therefore be included in mapping studies of natural populations. Caution should be exercised however, when making conclusions involving QTL linked to distorted markers.
Assigning species status to dominant marker alleles is problematic given the difficulty distinguishing introgression from coancestry. Assuming most alleles segregate in both species, we would have expected to see a more-or-less equal distribution of coupling- vs repulsion phase (that is, inverted markers). In our study, most mapped markers (n=495 or 92%) were in coupling phase, and were likely donated by P. fremontii chromosomes carried by the F1. Furthermore, in a survey of individuals from multiple populations of each species, 71 of 100 mapped markers (71%) were fixed absent or rare (allele frequencies ⩽0.05) in P. angustifolia relative to P. fremontii (M Zinkgraf, S Woolbright and G Allan unpublished data). Similarly, Martinsen et al. (2001) found that P. fremontii-specific alleles at 26 of 33 RFLP markers (78.8%) were absent from the P. angustifolia zone. Given these data, the difference in marker phase likely reflects a high level of divergence among the species, and AFLP alleles segregating in both species could indicate introgression.
Conclusions and future research
Given the extensive amount of ecological research on the Weber River hybrid zone (Whitham et al., 2003, 2006), our map represents a unique opportunity to combine long-term ecological research with map-based genetic techniques. For example, we have begun to identify QTL associated with a number of ecologically important traits such as condensed tannins, which have important community and ecosystem phenotypes (Woolbright, 2001; Whitham et al., 2003). Foliar condensed tannin concentrations have been linked to arthropod communities (Whitham et al., 2006), aquatic and terrestrial litter decomposition (Schweitzer et al., 2005; LeRoy et al., 2006), root production (Fischer et al., 2006) and nutrient cycling (Schweitzer et al., 2004). Using the recently completed Populus genome sequence (Tuskan et al., 2006), we have begun to build candidate gene lists for a number of ecologically relevant QTL. The ability to link genetic-level factors with community composition and ecosystem-level processes is unprecedented, and demonstrates the potential of genetic mapping in ecological genetic/genomic research.
The original aim of our study was to describe broad-scale ecological processes in terms of the genetic variation within a foundation species. Historically, P. fremontii and P. angustifolia have played little if any commercial role, and have been studied primarily for their ecological importance. Here, we have shown that research focusing on specific ecological questions in two largely overlooked species contributes to much larger questions relating to evolution in a model system, and studies such as those by Rahman and Rajora (2002); Hanley et al. (2006)and Lexer et al. (2007) demonstrate the potential for comparative studies across even broader taxonomic levels. In light of these results, future genetic studies of Populus and its relatives should capitalize on the availability of shared SSR and other markers.
References
Bailey JK, Deckert R, Schweitzer JA, Rehill BJ, Lindroth RL, Gehring C et al. (2005). Host plant genetics affect hidden ecological players: links among Populus, condensed tannins, and fungal endophyte infection. Can J Bot 83: 356–361.
Bailey JK, Schweitzer JA, Rehill BJ, Lindroth RL, Martinsen GD, Whitham TG (2004). Beavers as molecular geneticists: a genetic basis to the foraging of an ecosystem engineer. Ecology 85: 603–608.
Bangert RK, Lonsdorf EV, Wimp GM, Shuster SM, Fischer D, Schweitzer JA et al. (2006a). Genetic structure of a foundation species: scaling community phenotypes from the individual to the region. Heredity (advance online publication, 18 October 2006; doi:10.1038/sj.hdy.6800914).
Bangert RK, Turek RJ, Rehill B, Wimp GM, Meneses N, Martinsen GD et al. (2006b). From genes to geography: a genetic similarity rule for arthropod community structure at multiple geographic scales. Mol Ecol 15: 4215–4228.
Bishop DT, Cannings C, Skolnick M, Williamson JA (1983). The number of polymorphic DNA clones required to map the human genome. In: BS Weir (ed). Statistical Analysis of DNA Sequence Data. Dekker: New York. pp 181–200.
Bradshaw Jr HD, Stettler RF (1993). Molecular genetics of growth and development in Populus I. Triploidy in hybrid poplars. Theor Appl Genet 86: 301–307.
Bradshaw Jr HD, Stettler RF (1994). Molecular genetics of growth and development in Populus II. Segregation distortion due to genetic load. Theor Appl Genet 89: 551–558.
Bradshaw Jr HD, Villar M, Watson BD, Otto KG, Stewart S, Stettler RF (1994). Molecular genetics of growth and development in Populus. III. A genetic linkage map of a hybrid poplar composed of RFLP, STS, and RAPD markers. Theor Appl Genet 89: 167–178.
Cervera M, Storme V, Ivens B, Gusmao J, Liu BH, Hostyn V et al. (2001). Dense genetic linkage maps of three Populus species (Populus deltoides, P. nigra and P. trichocarpa) based on AFLP and microsatellite markers. Genetics 158: 787–809.
Doerge RW (2002). Multifactorial genetics: mapping and analysis of quantitative trait loci in experimental populations. Nature Rev Genet 3: 43–52.
Driebe EM, Whitham TG (2000). Cottonwood hybridization affects tannin and nitrogen content of leaf litter and alters decomposition. Oecologia 123: 99–107.
Feder ME, Mitchell-Olds T (2003). Evolutionary and ecological functional genomics. Nature Rev Genet 4: 649–655.
Fischer DG, Hart SC, Rehill BJ, Lindroth RL, Keim P, Whitham TG (2006). Do high-tannin leaves require more roots? Oecologia 149: 668–675.
Floate KD (2004). Extent and patterns of hybridization among the three species of Populus that constitute the riparian forest of southern Alberta Canada. Can J Bot 82: 253–264.
Floate KD, Kearsley MJC, Whitham TG (1993). Elevated herbivory in plant hybrid zones: Chrysomela confluens, Populus and phenological sinks. Ecology 7: 2056–2065.
Floate KD, Whitham TG (1995). Insects as traits in plant systematics: their use in discriminating between hybrid cottonwoods. Can J Bot 73: 1–13.
Frewen BE, Chen THH, Howe GT, Davis J, Rhode A, Boerjan W et al. (2000). Quantitative trait loci and candidate gene mapping of bud set and bud flush in Populus. Genetics 154: 837–845.
Grant V (1971). Plant Speciation. Columbia University Press: New York.
Hanley SJ, Mallot MD, Karp A (2006). Alignment of a Salix linkage map to the Populus genomic sequence reveals macrosynteny between willow and poplar genomes. Tree Genet Genomes 3: 35–48.
Hulbert SH, Ilott TW, Legg EJ, Lincoln SE, Lander ES, Michelmore RW (1988). Genetic analysis of the fungus, Bremia lactucae, using restriction fragment length polymorphisms. Genetics 120: 947–958.
Jackson RB, Linder CR, Lynch M, Purugganan M, Somerville S, Thayer SS (2002). Linking molecular insight and ecological research. Trends Ecol Evol 17: 409–414.
Keim P, Paige KN, Whitham TG, Lark KG (1989). Genetic analysis of an interspecific hybrid swarm of Populus: occurrence of unidirectional introgression. Genetics 123: 557–565.
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln S et al. (1987). Mapmaker: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1: 174–181.
Lange K, Boenke M (1982). How many polymorphic genes will it take to span the human genome? Am J Hum Genet 34: 842–845.
Larson KC, Whitham TG (1997). Competition between gall aphids and natural plant sinks: plant architecture affects resistance to galling. Oecologia 109: 575–582.
LeRoy CJ, Whitham TG, Keim P, Marks JC (2006). Plant genes link forests and streams. Ecology 87: 255–261.
Lexer C, Buerkle CA, Joseph JA, Heinze B, Fay MF (2007). Admixture in European Populus hybrid zones makes feasible the mapping of loci that contribute to reproductive isolation and trait differences. Heredity 98: 74–84.
Martinsen GD, Whitham TG, Turek RJ, Keim P (2001). Hybrid populations selectively filter gene introgression between species. Evolution 55: 1325–1335.
Mauricio R (2005). Can ecology help genomics: the genome as ecosystem? Genetica 123: 1–2.
Mitton JB (2003). The union of ecology and evolution: extended phenotypes and community genetics. BioScience 53: 208–209.
Nelson CD, Kubisiak TL, Stine M, Nance WL (1994). A genetic linkage map of longleaf pine (Pinus palustris Mill) based on random amplified polymorphic DNAs. J Hered 85: 433–439.
Rahman MH, Rajora OP (2002). Microsatellite DNA fingerprinting, differentiation, and genetic relationships of clones, cultivars, and varieties of six poplar species from three sections of the genus Populus. Genome 45: 1083–1094.
Rehill B, Whitham TG, Martinsen GD, Schweitzer JA, Bailey JK, Lindroth RL (2006). Developmental trajectories in cottonwood phytochemistry. J Chem Ecol 32: 2269–2285.
Remington DL, Whetten RW, Liu B-H, O'Malley DM (1999). Construction of an AFLP genetic map with nearly complete genome coverage in Pinus taeda. Theor Appl Genet 98: 1279–1292.
Schweitzer JA, Bailey JK, Hart SC, Wimp GM, Chapman SK, Whitham TG (2005). The interaction of plant genotype and herbivory decelerate leaf litter decomposition and alter nutrient dynamics. Oikos 110: 133–145.
Schweitzer JA, Bailey JK, Rehill BJ, Martinsen GD, Hart SC, Lindroth RL et al. (2004). Genetically based trait in a dominant tree affects ecosystem processes. Ecol Lett 7: 127–134.
Stanton BJ, Villar M (1996). Controlled reproduction of Populus. In: Stettler RF, Bradshaw JDH, Heilman PE, and Hinkley TM (eds). Biology of Populus and its Implications for Management and Conservation. NRC Research Press: Ottawa. pp 113–138.
Taylor G (2002). Populus: Arabidopsis for forestry. Do we need a model tree? Ann Bot 90: 681–689.
Thomas MA, Klaper R (2004). Genomics for the ecological toolbox. Trends Ecol Evol 19: 439–445.
Travis SE, Maschinkski J, Keim P (1996). An analysis of genetic variation in Astragalus cremnophylax var. cremnophylax, a critically endangered plant, using AFLP markers. Mol Ecol 5: 735–745.
Tuskan GA, DiFazio SP, Jansson S, Bohlmann J, Grigoriev I, Hellsten U et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596–1604.
Via S (2002). The ecological genetics of speciation. Am Nat 159: S1–S7.
Voorrips RE (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93: 77–78.
Vos P, Hogers R, Bleeker M, Reijans M, Van De Lee T, Hornes M et al. (1995). AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res 23: 4407–4414.
Whitham TG (1989). Plant hybrid zones as sinks for pests. Science 244: 1490–1493.
Whitham TG, Bailey JK, Schweitzer JA, Shuster SM, Bangert RK, LeRoy CJ (2006). A framework for community and ecosystem genetics: from genes to ecosystems. Nat Rev Genet 7: 510.
Whitham TG, Young WP, Martinsen GD, Gehring CA, Schweitzer JA, Shuster SM et al. (2003). Community and ecosystem genetics: a consequence of the extended phenotype. Ecology 84: 559–573.
Whitney KD, Randell RA, Rieseberg LH (2006). Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. Am Nat 167: 794–807.
Woolbright SA (2001). Genetic analyses of a synthetic population of hybrid cottonwoods with implications for community-level processes. MSc Thesis, Northern Arizona University, AZ.
Wu RL, Han YF, Hu JJ, Fang JJ, Li L, Li ML et al. (2000). An integrated map of Populus deltoides based on amplified fragment length polymorphisms. Theor Appl Genet 100: 1249–1256.
Wullschleger SD, Jansson S, Taylor G (2002). Genomics and forest biology: Populus emerges as the perennial favorite. Plant Cell 14: 2651–2655.
Yin T, Zhang X, Huang M, Wang M, Zhuge Q, Tu S et al. (2002). Molecular linkage maps of the Populus genome. Genome 45: 541–555.
Yin TM, DiFazio SP, Gunter LE, Riemenschneider D, Tuskan GA (2004). Large-scale heterospecific segregation distortion in Populus revealed by a dense genetic map. Theor Appl Genet 109: 451–463.
Yin T-M, DiFazio SP, Gunter LE, Zhang X, Sewell MM, Woolbright SA et al. (in review). Genome structure and chromosomal diversification revealed by comparative mapping in Populus. Genome Res (submitted).
Young WP, Schupp JM, Keim P (1999). DNA methylation and AFLP marker distribution in the soybean genome. Theor Appl Genet 99: 785–790.
Zhang DQ, Zhang ZY, Yang K, Tian L (2002). Segregation of AFLP markers in a (Populus tomentosa × P. bolleana) × P. Tomentosa Carr. BC1 family. For Stud China 4: 21–26.
Acknowledgements
We thank the National Science Foundation FIBR (DEB-0425908) and IRCEB (DEB-0078280) grants and the Phoenix chapter of the Achievement Rewards for College Scientists (ARCS) Foundation for financial support, and the Ogden Nature Center, Utah Department of Natural Resources, and the Bureau of Reclamation for common garden facilities. We also thank Jen Schweitzer, Joe Bailey, and Adrian Stone for their critical reviews; Bill Young, Mark Miller, Joe Busch, and Jim Schupp for their assistance in the laboratory; Matt Zinkgraf and Gancho Slavov for sharing data; Stephanie Woolbright for assisting in the laboratory and greenhouse; and the assistance of Brad Blake and the NAU research greenhouse staff.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Woolbright, S., DiFazio, S., Yin, T. et al. A dense linkage map of hybrid cottonwood (Populus fremontii × P. angustifolia) contributes to long-term ecological research and comparison mapping in a model forest tree. Heredity 100, 59–70 (2008). https://doi.org/10.1038/sj.hdy.6801063
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6801063
Keywords
This article is cited by
-
Investigation of genome-wide InDel distribution and segregation in Populus with restriction-site associated DNA sequencing data
Tropical Plant Biology (2022)
-
Identification of recombination events in outbred species with next-generation sequencing data
BMC Genomics (2018)
-
High density linkage mapping of genomic and transcriptomic SNPs for synteny analysis and anchoring the genome sequence of chickpea
Scientific Reports (2015)
-
Construction of a genetic linkage map and mapping of drought tolerance trait in Indian beveragial tea
Molecular Breeding (2015)
-
Genetic analysis of post-mating reproductive barriers in hybridizing European Populus species
Heredity (2011)

{kind=link}