
Abstract
Obesity and type 2 diabetes are the most frequent metabolic disorders, but their causes remain largely unclear. Insulin resistance, the common underlying abnormality, results from imbalance between energy intake and expenditure favouring nutrient-storage pathways, which evolved to maximize energy utilization and preserve adequate substrate supply to the brain. Initially, dysfunction of white adipose tissue and circulating metabolites modulate tissue communication and insulin signalling. However, when the energy imbalance is chronic, mechanisms such as inflammatory pathways accelerate these abnormalities. Here we summarize recent studies providing insights into insulin resistance and increased hepatic gluconeogenesis associated with obesity and type 2 diabetes, focusing on data from humans and relevant animal models.
Similar content being viewed by others

Main
Over the past 50 years, the prevalence of diabetes mellitus has continued to increase, spreading from western countries to the western Pacific, Asia and Africa. Current projections estimate an increase of more than 50% between 2017 and 2045, leading to around 693 million people suffering from diabetes, with estimated healthcare costs of about US$850 billion per year1. This epidemic mainly results from an increase in the incidence of type 2 diabetes (T2D), a heterogeneous disease characterized by deficient insulin secretion by pancreatic islet β-cells in the context of impaired insulin sensitivity, termed insulin resistance. A genome-wide association study (GWAS) found more than 400 T2D-associated gene variants—mostly related to islet function, but the roles of the individual genes are minor and explain less than 20% of overall disease risk2. Lifestyle-modification studies demonstrating T2D remission underline the predominant role of acquired alterations3,4, including intake of highly palatable, energy-dense refined food, sedentary behaviour and other factors (for example, environmental pollution, socioeconomic and psychosocial conditions, smoking and sleep deprivation)5. Moreover, parental lifestyle, intrauterine programming and early postnatal metabolic alterations may influence the risk profile6 via DNA methylation7. The roles of these mechanisms and of the gut metagenome are controversial in humans and beyond the scope of this Review. This Review focuses on studies in humans and relevant rodent models to provide an outlook on future precision medicine for T2D by better understanding its pathogenesis.
Fed-to-fasting transition and insulin resistance
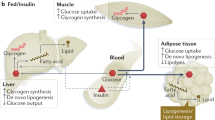
Fasted humans display impaired insulin-stimulated glucose disposal and elevation of certain, mainly branched-chain, amino acids and nonesterified fatty acids (NEFA) in plasma despite low-to-normal glycaemia and hypoinsulinaemia8. While initially hepatic glycogenolysis and gluconeogenesis maintain normoglycaemia9, the shift from carbohydrate to fatty acid oxidation preserves glucose for obligate glucose utilizers (such as the brain, red blood cells and renal medulla) and essential protein stores, which would otherwise be used for gluconeogenesis (Fig. 1a). Stimulation of gluconeogenesis has mostly been attributed to decreasing plasma insulin and increasing plasma glucagon concentrations favouring gluconeogenic enzyme transcription. Recent studies in rats have demonstrated a critical role of the leptin–hypothalamic–pituitary–adrenal (HPA) axis in mediating the fed-to-fasting transition via glucocorticoid regulation of white adipose tissue (WAT) lipolysis10,11,12,13—similar to mechanisms that operate in uncontrolled diabetes14. These studies show that the early postabsorptive decline in hepatic glycogenolysis is predominantly responsible for the fall in plasma insulin and glucose concentrations, resulting in approximately 50% reduction in plasma leptin concentrations. Thus, leptin acts as important fuel gauge for energy stored as triacylglycerol (TAG) in WAT and as glycogen in liver, signalling to the brain when both energy depots are depleted10. The fall in leptin to less than 1 ng ml−1 stimulates the HPA axis, ultimately increasing plasma corticosterone concentrations, which, during hypoinsulinaemia, stimulates WAT lipolysis, and release of NEFA and glycerol, with a switch towards fatty acid oxidation. Increased fatty acid flux to the liver increases hepatic β-oxidation and acetyl-CoA content (Fig. 1b). This allosterically activates pyruvate carboxylase flux, which, along with increased glycerol flux to the liver, is essential for maintaining hepatic gluconeogenesis and endogenous glucose production (EGP) during starvation. Simulation of fasting conditions also increased the contribution of gluconeogenesis to EGP by up to 75%, probably owing to lipid-dependent control of hepatic glycogen stores15. Starvation also promotes hepatic accumulation of TAG and diacylglycerol (DAG)10, which can occur independently of the direct action of hepatic insulin on de novo lipogenesis (DNL)16. Subsequently, the novel protein kinase C isoform ε (PKCε) is translocated to the plasma membrane, where it binds to and phosphorylates Thr1160 of the insulin receptor (IR), thereby inhibiting IR kinase activity17 (Fig. 1b). Prolonged fasting also resulted in 60% lower rates of glucose–alanine cycling, with a 50% reduction in hepatic mitochondrial oxidation, demonstrating an interorgan link between liver and muscle during the fed-to-fasting transition in both rats10 and humans18,19. Unbiased metabolomic analysis suggests that the same sequence of events occurs in ten-day fasted humans and reveals discrete starvation phases with gluconeogenic amino acid consumption and subsequent surge in lipids with a high degree of unsaturation and chain length12,13, reflecting increased adipocyte NEFA release. This study also reported a rapid fall of around 50% in circulating leptin and an early rise in plasma glucocorticoids, similar to that occurring in fasting20 and anorexia nervosa21. Although circulating leptin tightly reflects WAT mass, fasting-induced hypoleptinaemia can occur independently and initiate neuroendocrine adaptation22,23.

a, Under physiological postprandial conditions, insulin rapidly stimulates lipid storage by inhibiting lipolysis via adipose triglyceride lipase (ATGL and CGI-58), phosphodiesterase 3B (PDE3B) and protein kinase A (PKA)-controlled hormone-sensitive lipase (HSL) and perilipins (PLINs), and stimulating lipogenesis (1). Lower NEFA–glycerol flux decreases hepatic acetyl-CoA and glycerol, thereby acutely diminishing gluconeogenesis (GNG), reflecting indirect insulin action (2). Direct hepatic IR activation stimulates glycogen synthesis (GLY) (3) and chronic transcriptional control via decreased FOXO1 to downregulate gluconeogenic enzymes and upregulate glucokinase (GK), increasing glucose-6-phosphate (G6P) levels (4). Inhibition of serine phosphorylation of GSK3 activates and increases glycogen synthase (GS) flux (3). Furthermore, glucose-6-phosphate allosterically activates glycogen synthase and inhibits glycogen phosphorylase (GP), resulting in glycogen storage and suppressed glucose production. Insulin further participates in protein synthesis via mTORC1 (5) and DNL via FOXO1, carbohydrate and sterol response element binding proteins (ChREBP and SREBP1c) (6). b, In insulin-resistant states (obesity and T2D), adipocyte dysfunction—for example, owing to relative hypoxia induced by saturated fat-stimulated mitochondrial ANT2 triggering the transcription factor HIF-1α—leads to chemokine secretion, attracting macrophages (MΦ) (1). Immune cell infiltration inhibits adipocyte insulin sensitivity by mechanisms that ultimately increase lipolysis and NEFA and glycerol flux to the liver. Here, acetyl-CoA allosterically stimulates pyruvate carboxylase flux (PC), subsequently raising fasting glucose production (2). Increased lipid re-esterification generates DAG, thereby activating PKCε translocation and inhibitory Thr1160 phosphorylation of the IR (3) and increasing production of ceramides (4) and ROS (5), collectively promoting insulin resistance. Inhibiting direct hepatocellular insulin action acutely favours glycogenolysis and chronically upregulates gluconeogenesis with postprandial and finally continuous hyperglycaemia (6). In parallel, hepatic TAG deposition increases, not only from augmented lipid availability and DNL, partly controlled by insulin and FOXO1, but also via nutrient-sensitive pathways (ChREBP, SREBP1c and mTORC1) (7). Finally, endoplasmic reticulum (ER)-derived factors (PKR-like eukaryotic initiation factor 2α kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating-transcription factor 6 (ATF6)) can induce an unfolded protein response, which may stimulate lipogenesis, supported by X-box binding protein 1 (XBP1) and inflammatory pathways. All of these mechanisms accelerate TAG accumulation and NAFLD progression. Dotted lines represent regulation, that is, stimulation or inhibition; thicker lines represent pathways with increased flux, thinner lines represent pathways with decreased flux. FAS, fatty acid synthase; PEPCK, phosphoenolpyruvate carboxykinase; FBPase, fructose-1,2-bis-phosphatase; LPK, l-pyruvate kinase. P, phosphorylation.
This mechanism may have been important for survival during periods of famine and explain the high evolutionary conservation of the Thr1160 residue in the catalytic loop of the IR. Likewise, the blind cave fish Astyanax mexicanus—which endures infrequent limited nutrient availability—develop hyperglycaemia, steatosis and insulin resistance owing to a mutation in its IR gene24, similar to the one observed in some patients with Rabson–Mendenhall syndrome25. Despite abnormal energy metabolism, these fish show delayed senescence, further supporting the survival benefit of limited insulin-dependent glucose disposal. Thus, DAG and novel PKC (nPKC)-induced insulin resistance may have served a key evolutionary role to promote survival during starvation, while favouring metabolic syndrome and T2D during overnutrition.
Transition from normoglycaemia to hyperglycaemia
Longitudinal studies have demonstrated that people who later develop T2D display gradually increasing fasting and postprandial glycaemia26,27,28. Insulin sensitivity, which is predominately dependent on age, sex and weight gain, declines decades before T2D onset, represents one of the earliest pathogenic events and can be mostly ascribed to reduced nonoxidative glucose metabolism29 resulting from impaired insulin-stimulated storage of ingested carbohydrate as muscle glycogen30 (Fig. 2b). Initially, β-cells compensate for insulin resistance by secreting more insulin, resulting in hyperinsulinaemia, which promotes hepatic DNL, steatosis, hyperlipidaemia and WAT expansion30. WAT dysfunction, due to insulin resistance and inflammation14,31,32, stimulates lipolysis, further aggravating hepatic insulin resistance and non-alcoholic fatty liver disease (NAFLD) (Fig. 1b). Additionally, increased NEFA and/or glycerol flux to the liver stimulates gluconeogenesis. Combined with declining β-cell function—at least partly due to glucolipotoxicity—typically occurring just before T2D onset27,28, this leads to fasting and postprandial hyperglycaemia. There appear to be population-specific differences (for example, quicker decline in β-cell function in African Americans33 and increased hepatic lipid accumulation and muscle insulin resistance despite lower bodyweight in Asian Indians28,34). Nevertheless, without weight loss, insulin resistance and β-cell dysfunction occur simultaneously and continuously, increasing the risk of comorbidities even before glycaemia exceeds current criteria defining diabetes.

a, Under physiological postprandial conditions, insulin stimulates autophosphorylation of its receptor, tyrosine phosphorylation of IRS-1 and PI3K and serine/threonine phosphorylation of AKT2, causing several post-receptor events (1). Inhibition of AS160 threonine phosphorylation and RAC1 activation accelerates trafficking of GLUT4-containing storage vesicles (GSV) to the plasma membrane (2), enabling increases in glucose uptake via GLUT4 (3) and in intracellular glucose-6-phosphate levels via hexokinase II. Inhibition of serine phosphorylation of GSK3 activates glycogen synthase flux (4). Glucose-6-phosphate allosterically activates glycogen synthase (5) and inhibits glycogen phosphorylase, resulting in postprandial glycogen storage. b, In insulin-resistant states (obesity or T2D), overnutrition increases NEFA uptake and TAG storage (1). Starting from fatty acyl-CoA (FA-CoA), lipid synthesis occurs in different subcellular compartments and yields DAGs (2), which accumulate in the plasma membrane and stimulate translocation of novel PKC isoforms (PKCθ and PKCε) to the plasma membrane (3), thereby inducing inhibitory serine phosphorylation of IRS1 (4). Via serine-palmitoyl transferase (SPT), saturated fatty acids (SFA) can undergo synthesis of ceramides, which may also arise via sphingomyelin and salvage pathways (5). Ceramides activate protein phosphatase (PP2A) and PKCζ inhibiting AKT2 phosphorylation. Amino acids (AA) inhibit IRS1 activation via the mTOR–p70S6 serine kinase (S6K) pathway (6). Independently, inherited and acquired factors can lead to abnormal mitochondrial function (7) accelerating accumulation of DAG and, potentially, acylcarnitine (from incomplete β-oxidation) (8). Finally, chronically elevated reactive oxygen species (ROS) can inhibit IRS1 phosphorylation via NF-κB and JNK pathways (9). These effects combine to decrease glucose transport and glycogen synthesis, thereby contributing to postprandial hyperglycaemia. Dotted lines represent regulation, that is, stimulation or inhibition; thicker lines represent increased (flux through) pathways, thinner lines represent decreased (flux through) pathways. CY, cytosol; ER, endoplasmic reticulum; LD, lipid droplet; MT, mitochondria; PM, plasma membrane; G3P, glycerol-3-phosphate; LPA, lysophosphatidic acid; PA, phosphatidic acid; GPAT, acyl-CoA:glycerol-3-phosphate acyltransferase, AGPAT: acyl-CoA:1-acyl-glycerol-3-phosphate acyltransferase; DGAT2; DAG-O-acyltransferase; PAP; phosphatidate phosphatase; MGL, monoacylglycerol lipase.
Identifying distinct diabetes phenotypes
Recent studies challenge the traditional concept of T2D as single entity, as patients already exhibit a broad variability in insulin secretion and sensitivity at diagnosis35. Unbiased cluster analyses discriminated subgroups with different degrees of insulin deficiency and moderate obesity-related, moderate age-related or severe insulin resistance35,36. Whereas no known diabetes gene variants were associated with all clusters, a TCF7L2 variant related to insulin deficiency and a TM6SF2 variant related to the severely insulin-resistant cluster predicted nephropathy, cardiovascular disease36 and NAFLD35. Soft clustering analyses point to further gene–phenotype associations37 underlining different pathogenic mechanisms.
Postprandial hepatic metabolite fluxes
Fasting hyperglycaemia in T2D results from increased rates of hepatic gluconeogenesis and EGP and from hepatic insulin resistance, characterized by reduced ability of insulin to suppress this process38,39,40,41. This may be because of direct IR-mediated cell-autonomous or indirect effects (substrate availability, allosteric regulation or redox status)42 (Fig. 1b). Recent studies showed that these indirect effects probably result from insulin action on WAT and mainly account for acute suppression of gluconeogenesis and EGP during postprandial hyperinsulinaemia14. Consistent with a minor role for direct hepatic effects of insulin, rodent models with altered hepatic insulin signalling exhibit relatively normal glucose tolerance and compensatory hyperinsulinaemia, with reduced hepatic glycogen synthesis as the only indication of disrupted insulin signalling14,43,44,45,46,47.
Direct assessment of glycogen synthesis by 13C magnetic resonance spectroscopy demonstrated lower rates of postprandial and insulin-regulated hepatic glycogen synthesis in people with T2D38,39. The higher half-maximal effective concentration and lower maximum effect of insulin on hepatic glycogen synthesis39 indicate impaired IR activation with subsequent posttranslational modifications of the glycogen synthetic machinery and transcriptional regulation of glucokinase (Fig. 1b). Whereas other insulin effects, such as transcriptional DNL activation via sterol receptor enhancing binding protein-1c (SREBP1c), would be expected to be blunted, hepatic insulin resistance is generally associated with increased hepatic TAG and NAFLD. Accordingly, it has been proposed that only the FOXO1-dependent, but not the SREBP1c-dependent branch of insulin signalling, is defective, suggesting selective hepatic insulin resistance48. This hypothesis relies on the assumption that DNL is the major source of hepatic TAG and on experiments showing different roles of insulin receptor substrate (IRS)-1 and IRS-2, substrate-specific AKT phosphorylation or intrinsic pathway sensitivities to insulin. Conversely, NEFA re-esterification probably accounts for the majority of hepatic lipogenesis and very low-density lipoprotein (VLDL) secretion49,50,51. Decreased insulin-stimulated hepatic IR kinase activity suggests a common proximal abnormality in T2D52. Furthermore, DNL upregulation is not dependent exclusively on IR kinase activity, but can also occur through activation of carbohydrate receptor enhancing binding protein (ChREBP)53, mTORC1–SREBP1c54 and fructose-stimulated pathways55 (Fig. 1b). A recent study found that fatty acid esterification to TAG is mostly dependent on NEFA delivery to the liver and independent of hepatic insulin signalling16. This alternative hypothesis also explains the development of NAFLD through increased NEFA flux derived from increased lipolysis by insulin-resistant WAT.
In addition to caloric overload, macronutrients exert specific effects by modulating enteroendocrine secretion and, in turn, pancreatic islet and brain function before reaching the splanchnic bed to directly stimulate insulin secretion and entering the liver. Only around 33% of dietary carbohydrates enter the liver, and dietary fat is considered to amount to only 10–20% of the hepatic fatty acid pool49. Nevertheless, macronutrients can deliver substrates for the hepatic acetyl-CoA pool, which allosterically stimulates gluconeogenesis or activates nutrient-sensitive pathways (ChREBP, mTORC and SREBP) to collectively stimulate the transcriptional DNL program. Elevated hepatic acyl-CoA favours production of sn-1,2-DAG, sphingolipids and TAG. In obese humans with NAFLD, the sn-1,2-DAG–PKCε pathway tightly correlates with hepatic insulin resistance56,57,58,59,60, whereas ceramide–JUN N-terminal kinase (JNK) correlates more with hepatic oxidative stress and inflammation58,61,62 (Fig. 1b). In this context, lowering cellular ceramide by ablating dihydroceramide desaturase 1 increased mitochondrial oxygen flux and improved steatosis and glucose metabolism in insulin-resistant mice63. Conversely, mitochondrial C16:0 ceramide, generated by overexpression of ceramide synthase 6 (CerS6), interacts with mitochondrial fission factor (MFF) to promote mitochondrial fragmentation, insulin resistance and steatosis64. Silencing of MFF prevented CerS6-dependent metabolic abnormalities despite elevated C16:0 ceramide. This suggests that the effects of ceramides on insulin-stimulated glucose metabolism might result indirectly from impaired mitochondrial function with lower fatty acid oxidation, giving rise to other metabolites, for example, sn-1,2-DAG or acetyl-CoA, rather than from direct ceramide interference with insulin signalling. Recent studies indicate a critical role of molecular compartmentation of sn-1,2-DAGs, specifically in the plasma membrane, in inducing nPKC translocation and insulin resistance. Mice treated with CGI-58 antisense oligonucleotide exhibit elevated hepatic TAG and DAG in lipid droplets, are protected from lipid-induced hepatic insulin resistance and show reductions in plasma membrane DAG and PKCε translocation65.
Alvarez-Hernandez et al. monitored the earliest diet-induced metabolic alterations by examining the effect of a single oral saturated fat load in healthy humans66. This study revealed that saturated fat simultaneously induces insulin resistance in liver, skeletal muscle and WAT, and is associated with 70% higher rates of hepatic gluconeogenesis and 20% lower rates of net hepatic glycogenolysis. Similar studies in mice found upregulated expression of toll-like receptor (TLR) and inflammatory pathways, which might contribute to progression of NAFLD, including non-alcoholic steatohepatitis (NASH)66. Of note, chronic overfeeding also increased levels of intestine-derived endotoxins promoting TLR4-induced cytokine release by Kupffer cells67,68. Other intestinal functions also affect glycaemia and diabetes risk: integrin β7-knockout mice, which lack natural small-intestinal intraepithelial T lymphocytes, are metabolically hyperactive and resistant to obesity and diabetes69. Finally, dietary habits may affect the gut microbiota, modulating intestinal metabolite release and insulin sensitivity70. Humans with T2D and NAFLD show distinct metagenomic signatures along with increased branched-chain amino acids71,72 and decreased short-chain NEFA73, which may affect body weight and metabolism.
In summary, overnutrition and WAT dysfunction lead to increased WAT lipolysis, which promotes insulin-independent hepatic lipogenesis resulting in increased ectopic lipid deposition and increased hepatic gluconeogenesis owing to increased increased acetyl-CoA stimulation of pyruvate carboxylase as well as increased glycerol conversion to glucose. This mechanism obviates the previously reported need to invoke selective hepatic insulin resistance to explain the discordance of increased hepatic lipogenesis occurring simultaneously with increased gluconeogenesis48 (Fig. 1b). This is in line with recent studies showing that weight loss caused by very-low caloric diets rapidly normalizes hepatic steatosis and insulin resistance in liver, but not intramyocellular lipid content or muscle insulin resistance in individuals with T2D3,11,74.
Insulin resistance in skeletal muscle
Studies using 13C and 31P magnetic resonance spectroscopy identified impaired insulin-stimulated glycogen synthesis as the major factor responsible for insulin resistance in muscle and reduced insulin-stimulated glucose transport activity as the rate-controlling step that underlies lower glycogen synthesis in patients with T2D and their insulin-resistant relatives75,76,77,78,79 (Fig. 2a). Reduced insulin-stimulated glucose transport can be mainly attributed to defective insulin signalling at the level of IR and IRS-1-associated PI3K, which has been observed in one study to occur without altered AKT phosphorylation80. Whereas the majority of studies in humans point to proximal defects in insulin signalling, some experimental models provide evidence for distal abnormalities81,82. Glycogen synthase can also be stimulated via insulin regulation of glycogen synthase kinase-3 (GSK3) or independently via allosteric activation by glucose-6-phosphate83 in skeletal muscle75,76,77,84 and liver38,85, but its activity does not appear to regulate insulin-stimulated glucose disposal (Figs. 1, 2).
Lean first-degree relatives of patients with T2D present with predominantly muscle insulin resistance76. Ingestion of two high-carbohydrate meals revealed their early metabolic abnormalities: ingested carbohydrates were diverted from muscle glycogen synthesis to the liver, where augmented carbohydrate availability and compensatory hyperinsulinaemia promoted hepatic DNL, hepatic TAG synthesis and VLDL secretion, hypertriglyceridaemia and reduced plasma high-density lipoproteins30. The critical importance of skeletal muscle is illustrated by the observation that a single bout of exercise, which activates AMP kinase, promotes translocation of the glucose transporter GLUT4 and glucose uptake independently of insulin86, completely reversed these abnormalities76,87. Insulin-resistant individuals also exhibit reduced muscle mitochondrial density, gene expression and function, which impedes lipid oxidation. This, combined with augmented hepatic TAG release, contributes to muscle lipid accumulation. Collectively, these findings suggest a specific phenotype, whereby genetic and/or acquired reductions in muscle mitochondrial function predispose these individuals to sn-1,2-DAG accumulation, activation of PKCθ and PKCε and insulin resistance in muscle, which can be enhanced further by excessive production of reactive oxygen species88 (Fig. 2b). Such selective muscle insulin resistance also increases cardiometabolic risk owing to increased TAG and VLDL production and subsequent dyslipidaemia. The association between muscle insulin resistance and abnormal mitochondrial function represents a frequently observed feature of the elderly and people prone to or with overt T2D79,89.
There is increasing evidence supporting a hypothesis whereby gene variants in mitochondrial DNA (mtDNA) and mitochondrial-function-related nuclear DNA contribute to insulin resistance and T2D90 or abnormal exercise-induced responses91. Gene variants in mitochondrial-function-related nuclear DNA lead to relatively mildly impaired mitochondrial function, whereas classical mtDNA gene variants typically cause a severe reduction in mitochondrial function with neurological deficits and β-cell failure. In contrast to genetic and acquired alterations that lead to mild impairments in mitochondrial activity and a predisposition to ectopic lipid accumulation and insulin resistance, alterations that lead to severe reductions in mitochondrial activity (for example, mtDNA variants) result in increased dependency on anaerobic glycolysis, hyperlactaemia and increased glucose metabolism92,93,94. In support of this hypothesis, a recent European GWAS reported that a nonsynonymous variant of N-acetyltransferase 2 (NAT2) is associated with insulin resistance and related traits as well as with decreased adipocyte differentiation, insulin-mediated glucose uptake and increased WAT lipolysis95. Silencing or knocking down the mouse NAT2 orthologue, NAT1, induces insulin resistance, glucose intolerance and exercise intolerance96,97 and is associated with ectopic accumulation of TAG and DAG accumulation and activation of hepatic PKCε and muscle PKCθ97. Nat1−/− mice also display mild reductions in mitochondrial function and altered morphology, demonstrating another genetic link between reduced mitochondrial function, TAG and DAG deposition and nPKC-induced liver and muscle insulin resistance97. Further supporting a relevant role of mitochondria for the development of insulin resistance, mice with muscle-specific knockout of sarcolipin, which is required for mitochondrial sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) uncoupling and lipid oxidation, develop obesity and DAG–nPKC-mediated muscle insulin resistance, whereas sarcolipin overexpression prevents obesity-induced insulin resistance98. Other gene variants may also predispose humans to muscle insulin resistance and T2D independently from altered mitochondrial function, such as the AKT2 partial loss-of-function mutation that results in lower insulin-stimulated muscle and adipose glucose uptake99, whereas an activating mutation causes fasting hypoglycaemia100. AS160 (also known as TBC1D4) gene variants suggest links between insulin signalling and glucose transport leading to muscle insulin resistance, postprandial hyperinsulinaemia and hyperglycaemia101,102. Furthermore, RAC1-mediated glucose transport can become dysregulated in insulin-resistant murine and human skeletal muscle103 (Fig. 2a,b). These genotypes may have been advantageous for preserving glucose for other tissues in the prehistoric arctic environment.
Adipose tissue dysfunction
Similar to skeletal muscle, insulin-resistant humans exhibit reductions in membrane IR content, IR tyrosine kinase activity and insulin-stimulated glucose uptake in adipose tissue104,105,106,107. Although WAT accounts for less than 5% of postprandial glucose disposal, it has a disproportionate role in regulating whole-body glucose metabolism through its ability to alter rates of hepatic gluconeogenesis through NEFA and glycerol release10,14,104. Furthermore, increased WAT glucose uptake can enhance ChREBP-mediated lipogenesis, provide glycerol-3-phosphate for NEFA esterification or serve as signal for adipokines108. Other pathways, such as insulin-regulated β-adrenergic lipolysis or cytokine interaction, may also contribute to dysregulated WAT metabolism31,32,109. Insulin resistance in WAT shows temporal differences in the direction of net NEFA flux across the capillary bed between fasting and postprandial states, indicating lower fluctuations in obese men with impaired net adipose fat storage110. Finally, compartment-specific differences with higher WAT lipolysis and lower lipogenesis in visceral versus subcutaneous adipose tissue could enhance portal lipid delivery to the liver and contribute to metabolic dysregulation107.
Despite the association between body fat mass and insulin resistance, there is accumulating evidence that abnormal adipocyte function as well as liver and muscle lipid metabolites (including sn-1,2-DAG and ceramide), but not fat mass per se underlie common insulin resistance. In addition to lifestyle, GWAS studies suggest that genetic variants may affect the association of body fat mass or ectopic fat distribution with glycaemia and insulin resistance111,112. Some gene loci recently identified to associate with insulin resistance are associated with insulin resistance at a given level of adiposity and modulate insulin sensitivity via adipocyte differentiation113, supporting the concept that limited WAT storage capacity rather than overall fat mass is the main contributor to insulin resistance and associated diseases. Epigenetic adipose tissue modifications may further influence these interactions114.
Enlarged WAT mass and adipocyte size has been linked with inadequate vascularization, hypoxia, fibrosis and/or macrophage infiltration with low-grade inflammation108 (Fig. 1b). High-fat diet and obesity may activate saturated fatty acid-stimulated adenine nucleotide translocase 2 (ANT2), an inner mitochondrial membrane protein, which results in relative adipocyte hypoxia and triggers the transcription factor hypoxia-inducible factor-1α (HIF-1α), setting off adipose dysfunction and inflammation115. Adipocyte-specific Ant2 (also known as Slc25a5) deletion improves obesity-induced adipocyte hypoxia by lowering oxygen demand—despite unchanged mitochondrial mass—and, in turn, inflammation and insulin resistance116. This suggests that fatty acid-mediated ANT2 activation may be an early event in adipocyte dysfunction and a possible target for novel insulin sensitizers or anti-obesity drugs. Other early events comprise mechanical stress on membranes and extracellular matrix, causing dynamic adaptation until adipocyte death, apoptosis or de-differentiation108. During this process, release of chemotactic signals attracts bone marrow-derived pro-inflammatory M1 macrophages, leading to adipose remodelling by a wide range of activities including PPARγ-driven lipid storage, extracellular matrix modification, lysosomal clearance of dead adipocytes and cytokine release (Fig. 3). Compared with acute clinical inflammation, metabolic macrophage activation exhibits distinct activation modes117, involving chemokine monocyte chemotactic protein 1 (MCP-1) and its receptor CCR2118, B2 lymphocytes119 and/or interferon-γ and tumour necrosis factor (TNF), produced by natural killer cells in visceral adipose tissue120,121. Local cytokine release (TNF, interleukins (IL)-1β and IL-6) within WAT can suffice to induce adipose insulin resistance without overflow-related effects in distant tissues, usually associated with so-called subclinical or low-grade inflammation. In line with these results, insulin-resistant obese adolescents displayed more than 20-fold higher IL-6 levels in adipose tissue than in plasma, and increased adipose tissue expression of CGI-58 protein, similar to observations in high-fat fed rats14. Of course, continued adipose tissue enlargement and concomitant stress will lead to cytokine overflow, creating an imbalance between insulin-sensitizing (including adiponectin and leptin) and pro-inflammatory adipokines (including RBP4, resistin, IL-6 and TNF). These endocrine effects have been intensively studied over the past decades, demonstrating inhibition of IR kinase activity or activation of JNK, oxidative and ER stress. However, the concentrations required to achieve inhibitory effects are often orders of magnitude higher than those measured in plasma from patients with insulin resistance, and pharmacological agents are not generally specific for inflammatory pathways. Recent clinical trials examining IL-1β inhibition with canakinumab, despite causing large reductions in C-reactive protein and IL-6, did not reduce incidence of chronic hyperglycaemia in T2D122. Inhibiting the obesity-related kinases, IKKβ and TBK1, with amlexanox slightly reduced glycaemia, albeit with a paradoxical increase in serum IL-6123. Of note, insulin resistance can exist without any relevant adipose tissue inflammation; this is supported by lipodystrophy models in human and mice95. Moreover, rats rapidly develop WAT insulin resistance after just several days of high-fat feeding associated with liver and muscle lipid deposition124, whereas adipocyte death and macrophage infiltration are detectable only after four weeks125,126. Similarly, healthy humans exhibit adipose tissue insulin resistance within hours after saturated fat loading without alteration of circulating anti- or pro-inflammatory markers66.

a, Overnutrition leads to adipose tissue hypertrophy and hyperplasia and ectopic TAG deposition, mainly in muscle and liver—key features of insulin resistance. b, Adipose dysfunction, possibly due to local hypoxia ultimately resulting in apoptosis and cell death, recruits and transforms macrophages to release, for example, TNF and interleukins (IL-1β and IL-6). Local inflammatory reaction increases lipolysis directly or via inhibiting insulin signalling with subsequent release of NEFA and glycerol from WAT. Chronically, adipose dysfunction alters adipocytokine secretion favouring systemic low-grade inflammation. c, In liver, glycerol as substrate and NEFA-derived acetyl-CoA, allosterically activating pyruvate carboxylase flux (VPC), stimulate gluconeogenesis and fasting glucose production. NEFA and glycerol, as substrates of TAG accumulation, initiate NAFLD in the absence of adequate mitochondrial function and generate lipotoxic metabolites that inhibit insulin signalling. d, Hepatocellular insulin resistance not only chronically upregulates gluconeogenesis, but also decreases insulin-stimulated net glycogen synthesis and glucose uptake, in turn raising postprandial glucose production. e, In muscle, increased NEFA availability, accelerated by—possibly inherited—inadequate mitochondrial fat oxidation, also favours lipid synthesis, inhibiting insulin-stimulated glucose transport and glycogen synthesis. This, combined with lower non-insulin-mediated glucose uptake due to sedentary lifestyle, contributes to the postprandial glucose rise. f, These different mechanisms, along with direct stimulation by nutrients and enteroendocrine signals (such as GLP-1 and GIP) increase the insulin:glucagon secretion ratio, resulting in normoglycaemia at the expense of hyperinsulinaemia. g, Chronically, both acquired and inherited factors impair insulin secretion, with subsequent postprandial and fasting hyperglycaemia. The brain may also contribute to regulation of peripheral metabolism via afferent (for example, leptin from WAT (orange dashed line)) and efferent (for example, to liver (grey dashed line)) signalling.
Together, these findings indicate that metabolic changes leading to ectopic lipid accumulation are relatively early events in the pathogenesis of insulin resistance and T2D, whereas WAT inflammation with cytokine spillover represent chronic alterations that occur later, promoting progression to fasting and postprandial hyperglycaemia in conjunction with reduced β-cell function. Recent studies have also implicated other adipose-derived factors, for example, circulating exosomal miRNAs, which may contribute to gene expression in distant tissues and glucose tolerance, as demonstrated in mice lacking the miRNA-processing enzyme Dicer and in lipodystrophic humans127.
Cerebral regulation of hepatic metabolism
High energy requirements and limited energy storage capacity in the brain may explain why cerebral energy supply by glucose and ketones is completely dependent on the liver and, to some extent, kidney, during starvation and nearly independent of direct endocrine regulation. Conversely, cerebral insulin action may affect appetite control, mood, cognitive function and possibly peripheral glucose metabolism128. In mice, insulin and leptin act directly on the hypothalamic arcuate nucleus to activate proopiomelanocortin and inhibit Agouti-related protein neurons, whereas adipostatic signals stimulate melanocortin 4-expressing paraventricular neurons to induce satiety and energy expenditure129. Hypothalamic inflammation, reflected by higher mediobasal hypothalamic gliosis in obese rodents and humans130, has been suggested to lead to chronic central insulin and leptin resistance, which would promote excessive food intake and bodyweight gain. In rodents, central insulin action lowered EGP, hepatic gluconeogenesis, WAT lipolysis and glucagon secretion, but increased muscle glucose uptake131,132,133,134,135 (Fig. 3). However, carefully controlled studies failed to confirm similar brain insulin action to regulate hepatic glucose fluxes in awake dogs136,137. In humans, intranasal insulin application did not affect fasting EGP, slightly decreased hepatic fat and increased ATP content in glucose-tolerant individuals, but not in people with T2D138. Similarly, KATP-channel activation decreased EGP only in glucose-tolerant humans139,140. Some studies suggested that cerebral insulin action results in parasympathomimetic IL-6 secretion by Kupffer cells to inhibit hepatic gluconeogenesis133,141. All of these studies are limited by experimental conditions such as application and dosing of insulin, spillover of intranasally delivered insulin into systemic circulation and suitable metabolic control. Nevertheless, the brain can be involved in other aspects of interorgan crosstalk, orchestrated by metabolites11, adipokines (leptin) or enteroendocrine circuits (such as glucagon-like peptide 1, gastric inhibitor peptide, ghrelin, cholecystokinin or fibroblast growth factor (FGF)-19). The hypoleptinaemia-mediated stimulation of the HPA axis with subsequent stimulation of WAT lipolysis10 might be an example of how the human brain could indirectly regulate hepatic gluconeogenesis and EGP during starvation12.
A unifying concept of the development of T2D
Recent studies assessing rates of hepatic pyruvate carboxylase flux, palmitate turnover and hepatic acetyl-CoA content in an awake rat model of T2D, revealed a key mechanism by which insulin acutely suppresses hepatic gluconeogenesis and how increased WAT lipolysis, owing to macrophage infiltration with localized inflammation, can increase rates of hepatic gluconeogenesis and cause fasting hyperglycaemia14. During hyperinsulinaemic–normoglycaemic clamps, nondiabetic rats exhibited a sequence of events starting with a 90% fall in circulating NEFA and glycerol within 5 min, followed by a 50% reduction in hepatic acetyl-CoA and 70% suppression of EGP within 10 min without affecting hepatic glycogen, circulating lactate or glucagon concentrations. Furthermore, insulin-mediated suppression of WAT lipolysis, leading to reductions in hepatic acetyl-CoA could entirely explain acute insulin-induced suppression of hepatic gluconeogenesis. In line with these results, rodents lacking canonical hepatocellular insulin signalling (AKT1-, AKT2-, FOXO1- or IR-antisense oligonucleotide treatment) showed intact insulin-mediated EGP suppression14,45,47,48. Together, these studies demonstrate that—in contrast to the effects of insulin to stimulate hepatic glycogen synthesis through direct stimulation of hepatic insulin signaling—insulin acutely suppresses hepatic gluconeogenesis, mostly via an indirect mechanism through suppression of WAT lipolysis (Fig. 3).
Conversely, rats on a four-week high-fat diet developed fasting hyperglycaemia along with 25% higher rates of EGP, owing to increases in hepatic pyruvate carboxylase flux and glycerol-to-glucose conversion14. During clamps, impaired EGP suppression, along with higher rates of pyruvate carboxylase flux, could be attributed to increased hepatic acetyl-CoA content resulting from greater WAT lipolysis. Inhibition of lipolysis in atglistatin-treated rat models of T2D or high-fat fed adipose triglyceride lipase (ATGL)-knockout mice reversed these abnormalities. Furthermore, this study depicted distinct time-dependent alterations in WAT biology, starting with adipocyte hypertrophy, followed by increased levels of macrophage-secreted granulocyte– macrophage colony-stimulating factor (GM-CSF) and IL-6 in plasma and WAT. Consistent with the potential role for localized macrophage-induced WAT lipolysis, IL-6 infusion stimulated, whereas anti-IL-6 treatment or macrophage-specific JNK knockout ameliorated WAT lipolysis, hepatic insulin resistance and gluconeogenesis. Translating these studies to humans, obese insulin-resistant adolescents also exhibited increased fasting EGP, impaired insulin-mediated suppression of lipolysis, EGP and macrophage infiltration, and 50% higher IL-6 concentrations in WAT14. Taken together, these studies indicate that macrophage-induced cytokine-mediated WAT lipolysis raises hepatic acetyl-CoA content and pyruvate carboxylase activity and flux, probably serving as a molecular mechanism linking WAT inflammation to both fasting and postprandial hyperglycaemia (Fig. 3). These data also challenge the canonical view of inflammation-mediated hepatic insulin resistance occurring through activation of the NF-κB–JNK–ceramide biosynthetic pathways and explain the relatively mild metabolic phenotype in rodents with abrogated hepatic insulin action.
Nevertheless, hepatic insulin resistance, concomitant increases in EGP and hyperglycaemia in T2D are probably multifactorial in nature and not exclusively due to increased WAT lipolysis. Supporting this view, a three-day very low-calorie diet reversed hyperglycaemia in a rat model of uncontrolled T2D, not only via reductions in hepatic acetyl-CoA with lower rates of hepatic gluconeogenesis, but also via reductions of hepatic DAG–PKCε-mediated hepatic insulin resistance and lower rates of hepatic glycogenolysis11. Of note, these effects occurred independently of any changes in hepatic ceramides, cytokines, plasma branched-chain amino acids, glucagon, corticosterone or FGF-21.
Holistically, adaptation of metabolic fluxes during fasting and obesity-related diabetes represents the response of WAT to altered substrate supply, which would prevent distant insulin-dependent tissues from substrate oversupply and provide sufficient vital substrates to brain. We postulate that the biology of fasting and postprandial hyperglycaemia depends on dysregulated WAT lipolysis (and possibly contributions from intrahepatic lipolysis) driving hepatic gluconeogenesis through allosteric hepatic acetyl-CoA activation and altered substrate signalling, preferably via the sn-1,2-DAG–nPKC pathway (Fig. 3). This concept also highlights important targets for future T2D treatment.
Outlook
The idea that metabolic flux adaptation in liver and, to some extent, skeletal muscle is largely orchestrated by WAT in health and disease is supported by a series of studies in humans and model organisms. Nevertheless, several aspects still require confirmation both on a molecular–cellular level and in humans under specific metabolic conditions. First, the initial events leading to adipocyte dysfunction and the factors responsible for ectopic lipid deposition in skeletal muscle are not fully understood in humans. Second, the subcellular distribution of different lipid mediators in various compartments and their interactions with nPKC activation and other downstream factors require additional translational studies. In this context, certain intracellular lipids may be linked with insulin sensitivity to varying degrees in sedentary and strongly in physically active humans142. Moreover, genetically modified animals such as adipose- and liver-specific PKCε-knockout mice143, are helpful for exploring cellular pathways, but require detailed analysis of the experimental conditions and ultimately testing of their relevance in humans. Third, the rapidly growing body of multi-omics data might contribute to a better understanding of the cooperative action of metabolites to modify flux rates and insulin signalling. Along these lines, the relevance of metagenomics and epigenomics for the initiation, amplification or reversal of insulin resistance in humans is still largely unclear, despite recently gained insights into the dynamic regulation of insulin sensitivity following metabolic surgery144. Recent detection of different T2D phenotypes35,36,37 will reinforce investigation of gene variants, metabolites and neuro-immune-endocrine signals for interorgan communication regulating insulin sensitivity145. We anticipate that future studies will yield the mechanisms that underlie insulin resistance and β-cell dysfunction, which will guide precision medicine towards more effective treatments for T2D and related disorders such as NAFLD, including NASH and the metabolic syndrome.
References
Cho, N. H. et al. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 138, 271–281 (2018).
Mahajan, A. et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 50, 559–571 (2018).
Petersen, K. F. et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54, 603–608 (2005).
Lean, M. E. J. et al. Durability of a primary care-led weight-management intervention for remission of type 2 diabetes: 2-year results of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 7, 344–355 (2019).
Bellou, V., Belbasis, L., Tzoulaki, I. & Evangelou, E. Risk factors for type 2 diabetes mellitus: an exposure-wide umbrella review of meta-analyses. PLoS One 13, e0194127 (2018).
Barrès, R. & Zierath, J. R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat. Rev. Endocrinol. 12, 441–451 (2016).
Wahl, S. et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 541, 81–86 (2017).
Cahill, G. F. Jr. Starvation in man. N. Engl. J. Med. 282, 668–675 (1970).
Rothman, D. L., Magnusson, I., Katz, L. D., Shulman, R. G. & Shulman, G. I. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 254, 573–576 (1991).
Perry, R. J. et al. Leptin mediates a glucose-fatty acid cycle to maintain glucose homeostasis in starvation. Cell 172, 234–248 (2018). This study describes the physiological response to starving, which integrates hormone secretion and metabolic fluxes to promote the shift from carbohydrate to fat metabolism for maintaining gluconeogenesis, reflecting key features of insulin resistance.
Perry, R. J. et al. Mechanisms by which a very-low-calorie diet reverses hyperglycemia in a rat model of type 2 diabetes. Cell Metab. 27, 210–217 (2018).
Steinhauser, M. L. et al. The circulating metabolome of human starvation. JCI Insight 3, e121434 (2018).
Fazeli, P. K. et al. FGF21 and the late adaptive response to starvation in humans. J. Clin. Invest. 125, 4601–4611 (2015).
Perry, R. J. et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160, 745–758 (2015).
Roden, M. et al. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 49, 701–707 (2000).
Vatner, D. F. et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl Acad. Sci. USA 112, 1143–1148 (2015).
Petersen, M. C. et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest. 126, 4361–4371 (2016).
Peterson, K. F., Dufour, F., Cline, G. W. & Shulman, G. I. Regulation of hepatic mitochondrial oxidation by glucose-alanine cycling during starvation in humans. J. Clin. Invest. 129, 4671–4675 (2019).
Sarabhai, T. & Roden, M. Hungry for your alanine: when liver depends on muscle proteolysis. J. Clin. Invest. 129, 4563–4566 (2019).
Pasiakos, S. M., Caruso, C. M., Kellogg, M. D., Kramer, F. M. & Lieberman, H. R. Appetite and endocrine regulators of energy balance after 2 days of energy restriction: insulin, leptin, ghrelin, and DHEA-S. Obesity 19, 1124–1130 (2011).
Schorr, M. & Miller, K. K. The endocrine manifestations of anorexia nervosa: mechanisms and management. Nat. Rev. Endocrinol. 13, 174–186 (2017).
Ravussin, Y., Leibel, R. L. & Ferrante, A. W. Jr. A missing link in body weight homeostasis: the catabolic signal of the overfed state. Cell Metab. 20, 565–572 (2014).
Friedman, J. The long road to leptin. J. Clin. Invest. 126, 4727–4734 (2016).
Riddle, M. R. et al. Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 555, 647–651 (2018). Certain cave-adapted fish populations develop diminished insulin signalling in a nutrient-restricted environment, which protects them from blood glucose decline, reflecting a beneficial effect of insulin resistance.
Carrera, P. et al. Substitution of Leu for Pro-193 in the insulin receptor in a patient with a genetic form of severe insulin resistance. Hum. Mol. Genet. 2, 1437–1441 (1993).
Abdul-Ghani, M. A. & DeFronzo, R. A. Plasma glucose concentration and prediction of future risk of type 2 diabetes. Diabetes Care 32, S194–S198 (2009).
Tabák, A. G. et al. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet 373, 2215–2221 (2009).
Ohn, J. H. et al. 10-year trajectory of β-cell function and insulin sensitivity in the development of type 2 diabetes: a community-based prospective cohort study. Lancet Diabetes Endocrinol. 4, 27–34 (2016).
DeFronzo, R. A. & Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32, S157–S163 (2009).
Petersen, K. F. et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl Acad. Sci. USA 104, 12587–12594 (2007).
Saltiel, A. R. & Olefsky, J. M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4 (2017).
Guilherme, A., Henriques, F., Bedard, A. H. & Czech, M. P. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat. Rev. Endocrinol. 15, 207–225 (2019).
Umpierrez, G. E., Smiley, D. & Kitabchi, A. E. Narrative review: ketosis-prone type 2 diabetes mellitus. Ann. Intern. Med. 144, 350–357 (2006).
Petersen, K. F. et al. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc. Natl Acad. Sci. USA 103, 18273–18277 (2006).
Zaharia, O. P. et al. Clusters of patients with recent-onset diabetes show different risk profiles for diabetes-associated diseases during a 5-year follow-up. Lancet. Diabetol. Endocrinol. 7, 684–694 (2019). A subgroup of people with diabetes exhibit severe insulin resistance along with higher ectopic fat accumulation and increased risk of comorbidities, which require specific attention for precise prevention and treatment.
Ahlqvist, E. et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 6, 361–369 (2018).
Udler, M. S. et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 15, e1002654 (2018).
Magnusson, I., Rothman, D. L., Katz, L. D., Shulman, R. G. & Shulman, G. I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J. Clin. Invest. 90, 1323–1327 (1992).
Krssak, M. et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 53, 3048–3056 (2004).
Rizza, R. A. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes 59, 2697–2707 (2010).
Gastaldelli, A. et al. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 49, 1367–1373 (2000).
Rebrin, K., Steil, G. M., Mittelman, S. D. & Bergman, R. N. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J. Clin. Invest. 98, 741–749 (1996).
Buettner, C. et al. Severe impairment in liver insulin signaling fails to alter hepatic insulin action in conscious mice. J. Clin. Invest. 115, 1306–1313 (2005).
Cherrington, A. D. The role of hepatic insulin receptors in the regulation of glucose production. J. Clin. Invest. 115, 1136–1139 (2005).
Lu, M. et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med. 18, 388–395 (2012).
O-Sullivan, I. et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat. Commun. 6, 7079 (2015).
Titchenell, P. M., Chu, Q., Monks, B. R. & Birnbaum, M. J. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat. Commun. 6, 7078 (2015). This mouse study showed that in the absence of FOXO1, insulin signals independently of the hepatic insulin receptor–AKT–FOXO1 axis via an intermediary extrahepatic tissue to regulate hepatic glucose production.
Brown, M. S. & Goldstein, J. L. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96 (2008).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 (2005).
Albert, J. S. et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 370, 2307–2315 (2014).
Ali, A. H., Mundi, M., Koutsari, C., Bernlohr, D. A. & Jensen, M. D. Adipose tissue free fatty acid storage in vivo: effects of insulin versus niacin as a control for suppression of lipolysis. Diabetes 64, 2828–2835 (2015).
Caro, J. F. et al. Studies on the mechanism of insulin resistance in the liver from humans with noninsulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. J. Clin. Invest. 78, 249–258 (1986).
Abdul-Wahed, A., Guilmeau, S. & Postic, C. Sweet sixteenth for ChREBP: established roles and future goals. Cell Metab. 26, 324–341 (2017).
Li, S., Brown, M. S. & Goldstein, J. L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl Acad. Sci. USA 107, 3441–3446 (2010).
Herman, M. A. & Samuel, V. T. The sweet path to metabolic demise: fructose and lipid synthesis. Trends Endocrinol. Metab. 27, 719–730 (2016).
Kumashiro, N. et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl Acad. Sci. USA 108, 16381–16385 (2011).
Magkos, F. et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142, 1444–1446 (2012).
Luukkonen, P. K. et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 64, 1167–1175 (2016).
ter Horst, K. W. et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. J. Hepatol. 64, 1167–1175 (2016).
Ruby, M. A. et al. Human carboxylesterase 2 reverses obesity-induced diacylglycerol accumulation and glucose intolerance. Cell Rep. 18, 636–646 (2017).
Apostolopoulou, M. et al. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care 41, 1235–1243 (2018).
Koliaki, C. et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21, 739–746 (2015).
Chaurasia, B. et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365, 386–392 (2019).
Hammerschmidt, P. et al. CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in besity. Cell 177, 1536–1552 (2019).
Cantley, J. L. et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc. Natl Acad. Sci. USA 110, 1869–1874 (2013).
Hernández, E. Á. et al. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J. Clin. Invest. 127, 695–708 (2017).
Parks, E., Yki-Järvinen, H. & Hawkins, M. Out of the frying pan: dietary saturated fat influences nonalcoholic fatty liver disease. J. Clin. Invest. 127, 454–456 (2017).
Luukkonen, P. K. et al. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care 41, 1732–1739 (2018).
He, S. et al. Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature 566, 115–119 (2019).
Ussar, S. et al. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 22, 516–530 (2015).
Pedersen, H. K. et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381 (2016).
Hoyles, L. et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 24, 1070–1080 (2018).
Sanna, S. et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 51, 600–605 (2019).
Taylor, R. et al. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for β cell recovery. Cell Metab. 28, 547–556 (2018).
Cline, G. W. et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 341, 240–246 (1999).
Perseghin, G. et al. Increased glucose transport–phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N. Engl. J. Med. 335, 1357–1362 (1996).
Roden, M. et al. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 97, 2859–2865 (1996).
Dresner, A. et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 103, 253–259 (1999).
Szendroedi, J. et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 4, e154 (2007).
Kim, Y. B., Nikoulina, S. E., Ciaraldi, T. P., Henry, R. R. & Kahn, B. B. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Invest. 104, 733–741 (1999).
Fazakerley, D. J., Krycer, J. R., Kearney, A. L., Hocking, S. L. & James, D. E. Muscle and adipose tissue insulin resistance: malady without mechanism? J. Lipid Res. 60, 1720–1732 (2019).
Czech, M. P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 23, 804–814 (2017).
Wan, M. et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 18, 99–105 (2013).
Bouskila, M. et al. Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab. 12, 456–466 (2010).
von Wilamowitz-Moellendorff, A. et al. Glucose-6-phosphate-mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes 62, 4070–4082 (2013).
Musi, N. et al. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50, 921–927 (2001).
Rabøl, R., Petersen, K. F., Dufour, S., Flannery, C. & Shulman, G. I. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc. Natl Acad. Sci. USA 108, 13705–13709 (2011).
Ruegsegger, G. N., Creo, A. L., Cortes, T. M., Dasari, S. & Nair, K. S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Invest. 128, 3671–3681 (2018).
Petersen, K. F. et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300, 1140–1142 (2003).
Kraja, A. T. et al. Associations of mitochondrial and nuclear mitochondrial variants and genes with seven metabolic traits. Am. J. Hum. Genet. 104, 112–138 (2019).
Kacerovsky-Bielesz, G. et al. Short-term exercise training does not stimulate skeletal muscle ATP synthesis in relatives of humans with type 2 diabetes. Diabetes 58, 1333–1341 (2009).
Holloszy, J. O. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62, 1036–1040 (2013).
Pospisilik, J. A. et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131, 476–491 (2007).
Koh, J. H. et al. TFAM enhances fat oxidation and attenuates high fat diet induced insulin resistance in skeletal muscle. Diabetes 68, 1552–1564 (2019).
Lotta, L. A. et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet. 49, 17–26 (2017).
Knowles, J. W. et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J. Clin. Invest. 125, 1739–1751 (2015).
Chennamsetty, I. et al. Nat1 deficiency is associated with mitochondrial dysfunction and exercise intolerance in mice. Cell Rep. 17, 527–540 (2016).
Maurya, S. K. et al. Sarcolipin signaling promotes mitochondrial biogenesis and oxidative metabolism in skeletal muscle. Cell Rep. 24, 2919–2931 (2018).
Latva-Rasku, A. et al. A partial loss-of-function variant in AKT2 is associated with reduced insulin-mediated glucose uptake in multiple insulin-sensitive tissues: a genotype-based callback positron emission tomography study. Diabetes 67, 334–342 (2018).
Hussain, K. et al. An activating mutation of AKT2 and human hypoglycemia. Science 334, 474 (2011).
Dash, S. et al. A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc. Natl Acad. Sci. USA 106, 9350–9355 (2009).
Moltke, I. et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512, 190–193 (2014).
Sylow, L. et al. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes 62, 1865–1875 (2013).
Kahn, C. R. Insulin resistance, insulin insensitivity, and insulin unresponsiveness: a necessary distinction. Metabolism 27, 1893–1902 (1978).
Freidenberg, G. R., Reichart, D., Olefsky, J. M. & Henry, R. R. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin-dependent diabetes mellitus. Effect of weight loss. J. Clin. Invest. 82, 1398–1406 (1988).
Kahn, B. B. & Flier, J. S. Obesity and insulin resistance. J. Clin. Invest. 106, 473–481 (2000).
Bódis, K. & Roden, M. Energy metabolism of white adipose tissue and insulin resistance in humans. Eur. J. Clin. Invest. 48, e13017 (2018).
Scherer, P. E. The many secret lives of adipocytes: implications for diabetes. Diabetologia 62, 223–232 (2019).
Zeng, X. et al. Innervation of thermogenic adipose tissue via a calsyntenin 3β–S100b axis. Nature 569, 229–235 (2019).
McQuaid, S. E. et al. Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes 60, 47–55 (2011).
Manning, A. K. et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 44, 659–669 (2012).
Shungin, D. et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015).
Camporez, J. P. et al. Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl Acad. Sci. USA 114, E11285–E11292 (2017).
Orozco, L. D. et al. Epigenome-wide association in adipose tissue from the METSIM cohort. Hum. Mol. Genet. 27, 1830–1846 (2018).
Lee, Y. S. et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352 (2014).
Seo, J. B. et al. Knockdown of ANT2 reduces adipocyte hypoxia and improves insulin resistance in obesity. Nat. Metab. 1, 86–97 (2019). Using mouse models, this study shows that adipocyte oxygen demand rather than oxygen supply or angiogenesis is the key determinant of intracellular hypoxia, which may be the initial event leading to adipose tissue inflammation.
Kratz, M. et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 20, 614–625 (2014).
Sartipy, P. & Loskutoff, D. J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl Acad. Sci. USA 100, 7265–7270 (2003).
Ying, W. et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J. Clin. Invest. 127, 1019–1030 (2017).
Lee, B.-C. et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab. 23, 685–698 (2016).
Wensveen, F. M. et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 16, 376–385 (2015).
Everett, B. M. et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J. Am. Coll. Cardiol. 71, 2392–2401 (2018).
Oral, E. A. et al. Inhibition of IKKε and TBK1 improves glucose control in a subset of patients with type 2 diabetes. Cell Metab. 26, 157–170 (2017).
Samuel, V. T. et al. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest. 117, 739–745 (2007).
Nishimura, S. et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 15, 914–920 (2009).
Strissel, K. J. et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56, 2910–2918 (2007).
Thomou, T. et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455 (2017).
Kullmann, S. et al. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 96, 1169–1209 (2016).
Jais, A. & Brüning, J. C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Invest. 127, 24–32 (2017).
Thaler, J. P. et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Invest. 122, 153–162 (2012).
Obici, S., Zhang, B. B., Karkanias, G. & Rossetti, L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 8, 1376–1382 (2002).
Pocai, A. et al. Hypothalamic KATP channels control hepatic glucose production. Nature 434, 1026–1031 (2005).
Inoue, H. et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 3, 267–275 (2006).
Gelling, R. W. et al. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 3, 67–73 (2006).
Scherer, T. et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 13, 183–194 (2011).
Ramnanan, C. J., Edgerton, D. S. & Cherrington, A. D. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation of hepatic glucose production. Cell Metab. 15, 656–664 (2012). This perspective discusses the evidence that the brain can sense insulin and regulate hepatic glucoregulatory enzyme expression, although the action of cerebral insulin is not essential for the rapid insulin-mediated suppression of glucose production.
Winnick, J. J. et al. Hepatic glycogen can regulate hypoglycemic counterregulation via a liver–brain axis. J. Clin. Invest. 126, 2236–2248 (2016).
Gancheva, S. et al. Intranasal insulin lowers hepatic fat accumulation and improves energy metabolism in humans. Diabetes 64, 1966–1975 (2015).
Kishore, P. et al. Activation of KATP channels suppresses glucose production in humans. J. Clin. Invest. 121, 4916–4920 (2011).
Esterson, Y. B. et al. Central regulation of glucose production may be impaired in type 2 diabetes. Diabetes 65, 2569–2579 (2016).
Kimura, K. et al. Central insulin action activates Kupffer cells by suppressing hepatic vagal activation via the nicotinic alpha 7 acetylcholine receptor. Cell Rep. 14, 2362–2374 (2016).
Perreault, L. et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 3, e96805 (2018).
Brandon, A. E. et al. Protein kinase C epsilon deletion in adipose tissue, but not in liver, improves glucose tolerance. Cell Metab. 29, 183–191 (2019).
Gancheva, S. et al. Dynamic changes of muscle insulin sensitivity after metabolic surgery. Nat. Commun. 10, 4179 (2019).
Parker, B. L. et al. An integrative systems genetic analysis of mammalian lipid metabolism. Nature 567, 187–193 (2019).
Acknowledgements
This research is supported by grants from the German Federal Ministry of Health and Ministry of Culture and Science of the state North Rhine-Westphalia to DDZ, the German Federal Ministry of Education and Research to DZD, European Funds for Regional Development (EFRE-0400191), EUREKA Eurostars-2 (E! 113230 DIA-PEP) and the German Science Foundation (CRC/SFB 1116/2 B12) (to M.R.) and by grants from the US Public Health Service (R01 DK-113984, R01 DK114793, R01 DK116774, R01 DK119968, P30 DK-045735) (to G.I.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
M.R. and G.I.S. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.R. is on the scientific advisory boards of Bristol-Myers Squibb, Eli Lilly, Gilead Sciences, NovoNordisk, Servier Laboratories, Target Pharmasolutions and Terra Firma and receives investigator-initiated support from Boehringer Ingelheim, Nutricia/Danone and Sanofi–Aventis. G.I.S. is on the scientific advisory boards of Merck, NovoNordisk, Gilead Sciences, AstraZeneca, Aegerion, iMBP, Janssen Research and Development and receives investigator-initiated support from Gilead Sciences, Merck and AstraZeneca.
Additional information
Peer review information Nature thanks Kei Sakamoto and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Rights and permissions
About this article

Cite this article
Roden, M., Shulman, G.I. The integrative biology of type 2 diabetes. Nature 576, 51–60 (2019). https://doi.org/10.1038/s41586-019-1797-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1797-8
This article is cited by
-
Inter-organ crosstalk during development and progression of type 2 diabetes mellitus
Nature Reviews Endocrinology (2024)
-
Genome-wide cross-disease analyses highlight causality and shared biological pathways of type 2 diabetes with gastrointestinal disorders
Communications Biology (2024)
-
Liver ACOX1 regulates levels of circulating lipids that promote metabolic health through adipose remodeling
Nature Communications (2024)
-
Role of steatotic liver disease in prediction and prevention of cardiometabolic diseases
Nature Reviews Gastroenterology & Hepatology (2024)
-
Increased monocytic myeloid-derived suppressor cells in type 2 diabetes correlate with hyperglycemic and was a risk factor of infection and tumor occurrence
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
