
Article
|
Open Access
Featured
-
-
Article
| Open Access Modeling uniquely human gene regulatory function via targeted humanization of the mouse genome
Modeling uniquely human gene regulatory function via targeted humanization of the mouse genome
Human Accelerated Regions (HARs) are prime candidates for driving the evolution of uniquely human traits. Using humanized mice, the authors show how one HAR alters gene expression during embryonic development, yielding insight into HAR function.
- Emily V. Dutrow
- , Deena Emera
- & James P. Noonan
-
Article
| Open Access Large-scale genome-wide study reveals climate adaptive variability in a cosmopolitan pest
Large-scale genome-wide study reveals climate adaptive variability in a cosmopolitan pest
The diamondback moth is a cosmopolitan pest of significant economic importance. Here the authors analyse globally distributed genomic data to find evidence of climate-associated adaptive variation, and use an ecogenetic index to predict that it will maintain a global pest status under climate change.
- Yanting Chen
- , Zhaoxia Liu
- & Shijun You
-
Article
| Open Access Aborting meiosis allows recombination in sterile diploid yeast hybrids
Aborting meiosis allows recombination in sterile diploid yeast hybrids
Hybrids are often considered evolutionary dead ends because they do not generate viable offspring. Here, the authors show that sterile yeast hybrids generate genetic diversity through meiotic-like recombination by aborting meiosis and return to asexual growth.
- Simone Mozzachiodi
- , Lorenzo Tattini
- & Gianni Liti
-
Article
| Open Access Refining models of archaic admixture in Eurasia with ArchaicSeeker 2.0
Refining models of archaic admixture in Eurasia with ArchaicSeeker 2.0
Existing methods to identify the presence of DNA from other hominin species can be limited in the ability to accurately estimate introgression waves, or can only be applied to specific populations. Here, the authors have developed a generalizable method to identify introgression in multi-wave situations.
- Kai Yuan
- , Xumin Ni
- & Shuhua Xu
-
Article
| Open Access Thousands of Qatari genomes inform human migration history and improve imputation of Arab haplotypes
Thousands of Qatari genomes inform human migration history and improve imputation of Arab haplotypes
Arab populations are relatively understudied, especially their genetic architecture and historical relationship with early founders of the ancient Near East. Here, the authors examine 6,218 Qatari whole genomes, revealing insights on migration, population history and genetic structure of populations across the Middle Eastern region.
- Rozaimi Mohamad Razali
- , Juan Rodriguez-Flores
- & Younes Mokrab
-
Article
| Open Access Elucidating gene expression adaptation of phylogenetically divergent coral holobionts under heat stress
Elucidating gene expression adaptation of phylogenetically divergent coral holobionts under heat stress
As corals struggle to survive under climate change, it is crucial to know whether they can withstand increasing seawater temperatures. Using a controlled thermal stress experiment across three divergent coral holobionts, this study examines metatranscriptomic responses to heat stress corresponding to the coral host, photosymbionts and associated microbiota.
- Viridiana Avila-Magaña
- , Bishoy Kamel
- & Mónica Medina
-
Article
| Open Access Captivity and the co-diversification of great ape microbiomes
Captivity and the co-diversification of great ape microbiomes
Here the authors sequence 16S rRNA and the more variable gyrase B protein-coding gene to profile the gut microbiome of captive great apes, which together with analysis of wild apes and humans, reveal a displacement of bacterial strains normally restricted to their wild conspecifics with those that are otherwise restricted to humans.
- Alex H. Nishida
- & Howard Ochman
-
Article
| Open Access Incipient diploidization of the medicinal plant Perilla within 10,000 years
Incipient diploidization of the medicinal plant Perilla within 10,000 years
Perilla is a young allotetraploid species within the mint family Lamiaceae. Here, the authors assemble the genomes of a tetraploid species and its diploid progenitor, characterize the incipient diploidization of the tetraploid, conduct population genetics analyses, and identify loci associate with pigmentation and oil content.
- Yujun Zhang
- , Qi Shen
- & Shilin Chen
-
Article
| Open Access Different historical generation intervals in human populations inferred from Neanderthal fragment lengths and mutation signatures
Different historical generation intervals in human populations inferred from Neanderthal fragment lengths and mutation signatures
Historical interbreeding between Neanderthals and humans should leave signatures of historical demographics in modern human genomes. Analysing the size distribution of Neanderthal fragments in non-African genomes suggests consistent differences in the generation interval across Eurasia, and that this could explain mutational spectrum variation.
- Moisès Coll Macià
- , Laurits Skov
- & Mikkel Heide Schierup
-
Article
| Open Access Developmental genetics of color pattern establishment in cats
Developmental genetics of color pattern establishment in cats
Intricate color patterns are a defining aspect of morphological diversity in the Felidae. Here the authors apply morphological and single-cell gene expression analysis to fetal skin of domestic cats to identify when, where, and how, during fetal development, felid color patterns are established.
- Christopher B. Kaelin
- , Kelly A. McGowan
- & Gregory S. Barsh
-
Article
| Open Access Changes in the distribution of fitness effects and adaptive mutational spectra following a single first step towards adaptation
Changes in the distribution of fitness effects and adaptive mutational spectra following a single first step towards adaptation
Analyses of both natural and experimental evolution suggest that adaptation depends on the evolutionary past and adaptive potential decreases over time. Here, by tracking yeast adaptation with DNA barcoding, the authors show that such evolutionary phenomena can be observed even after a single adaptive step.
- Dimitra Aggeli
- , Yuping Li
- & Gavin Sherlock
-
Article
| Open Access Evidence for opposing selective forces operating on human-specific duplicated TCAF genes in Neanderthals and humans
Evidence for opposing selective forces operating on human-specific duplicated TCAF genes in Neanderthals and humans
Duplications of gene segments can allow novel physiological adaptations to evolve. A detailed analysis of the TCAF gene family in primates and archaic humans suggest rapid duplication and diversification in this gene family is associated with cold or dietary adaptations.
- PingHsun Hsieh
- , Vy Dang
- & Evan E. Eichler
-
Article
| Open Access Clinal genomic analysis reveals strong reproductive isolation across a steep habitat transition in stickleback fish
Clinal genomic analysis reveals strong reproductive isolation across a steep habitat transition in stickleback fish
How ecological divergence causes reproductive isolation between populations in close contact remains poorly understood at the genomic level. This study presents a clinal investigation based on whole-genome sequencing to characterize reproductive isolation between threespine stickleback adapted to contiguous but ecologically different lake and stream habitats.
- Quiterie Haenel
- , Krista B. Oke
- & Daniel Berner
-
Article
| Open Access The evolution of hematopoietic cells under cancer therapy
The evolution of hematopoietic cells under cancer therapy
The mutational effects of chemotherapies on healthy cells are unclear. Here, the authors show that the mutational signature of platinum-based drugs -but not 5-fluorouracil- is detectable in secondary acute myeloid leukemia, implying that the clonal expansion begins after the start of therapy.
- Oriol Pich
- , Albert Cortes-Bullich
- & Nuria Lopez-Bigas
-
Article
| Open Access Quantifying the contribution of Neanderthal introgression to the heritability of complex traits
Quantifying the contribution of Neanderthal introgression to the heritability of complex traits
We lack a comprehensive understanding of how Neanderthal ancestry influences human traits. This study finds that regions with Neanderthal ancestry are broadly depleted of trait-associated variation; yet, introgressed variants likely contributed to human adaptation in a few traits, like skin color and immune response modulation.
- Evonne McArthur
- , David C. Rinker
- & John A. Capra
-
Article
| Open Access Engineered reproductively isolated species drive reversible population replacement
Engineered reproductively isolated species drive reversible population replacement
There exist only a handful of methods to engineer reproductive barriers in eukaryotes. Here the authors use CRISPR to engineer multiple barriers in D. melanogaster and model their spread.
- Anna Buchman
- , Isaiah Shriner
- & Omar S. Akbari
-
Article
| Open Access Genetic architecture and lifetime dynamics of inbreeding depression in a wild mammal
Genetic architecture and lifetime dynamics of inbreeding depression in a wild mammal
Without understanding the genetic architecture of inbreeding depression, its effects are hard to pinpoint. Long-term data from wild Soay sheep shows that inbreeding manifests in long runs of homozygosity, which made up nearly half of the genome in the most inbred individuals with severe fitness consequences.
- M. A. Stoffel
- , S. E. Johnston
- & J. M. Pemberton
-
Article
| Open Access Selective sweep for an enhancer involucrin allele identifies skin barrier adaptation out of Africa
Selective sweep for an enhancer involucrin allele identifies skin barrier adaptation out of Africa
Selection on alleles contributing to human evolution is not well understood. Here, the authors investigate positive selection on skin barrier adaptation, identifying a selective sweep on involucrin alleles associated with migration out of Africa, and confirming enhancer regulatory effects with functional assays.
- Mary Elizabeth Mathyer
- , Erin A. Brettmann
- & Cristina de Guzman Strong
-
Article
| Open Access Chromosome-scale assembly and analysis of biomass crop Miscanthus lutarioriparius genome
Chromosome-scale assembly and analysis of biomass crop Miscanthus lutarioriparius genome
The genus Miscanthus has great potential for bio-energy production due to its high biomass yield and strong stress resistance. Here, the authors report the genome assembly of the diploid M. lutarioriparius, showing it has an allotetraploid origin and an expanded number of genes in families related to stress resistance.
- Jiashun Miao
- , Qi Feng
- & Bin Han
-
Article
| Open Access Genome editing reveals fitness effects of a gene for sexual dichromatism in Sulawesian fishes
Genome editing reveals fitness effects of a gene for sexual dichromatism in Sulawesian fishes
The mutations underlying sexually selected traits like the red fins on a male medaka fish can be hard to pinpoint. Using a new genome, transcriptomics and gene editing, Ansai et al. find that the gene csf1 causes male fins to be red, which attracts females and, surprisingly, is less attractive to predators.
- Satoshi Ansai
- , Koji Mochida
- & Jun Kitano
-
Article
| Open Access Retroviral integrations contribute to elevated host cancer rates during germline invasion
Retroviral integrations contribute to elevated host cancer rates during germline invasion
Koalas are susceptible to neoplasms, which are related to infection with the Koala retrovirus. Here, the authors use DNA sequencing to show that the retroviral insertion sites cluster near known cancer genes and demonstrate a high mutational load associated with the germline invasion of the virus.
- Gayle K. McEwen
- , David E. Alquezar-Planas
- & Alex D. Greenwood
-
Article
| Open Access Genome assembly and population genomic analysis provide insights into the evolution of modern sweet corn
Genome assembly and population genomic analysis provide insights into the evolution of modern sweet corn
Sweet corn is one of the most important vegetables in North America and has undergone different selection pressures than non-sweet cultivars. Here, the authors report its genome assembly and reveal the evolutionary history of modern sweet corn through population genomic analyses.
- Ying Hu
- , Vincent Colantonio
- & Marcio F. R. Resende Jr.
-
Article
| Open Access Population-specific causal disease effect sizes in functionally important regions impacted by selection
Population-specific causal disease effect sizes in functionally important regions impacted by selection
Trans-ethnic genetic correlation is significantly less than 1 for many diseases. Here, the authors stratify this correlation by genomic annotations, finding that loci whose causal disease effect sizes differ between ethnicities are likely impacted by selection, particularly positive selection.
- Huwenbo Shi
- , Steven Gazal
- & Alkes L. Price
-
Article
| Open Access Asymmetric introgression reveals the genetic architecture of a plumage trait
Asymmetric introgression reveals the genetic architecture of a plumage trait
Hybrid zones are windows into the evolutionary process. Semenov et al. find that the head plumage differences between white wagtail subspecies have a simple genetic basis involving two small genetic regions, in which partially dominant and epistatic interactions help to explain how this sexual signal has become decoupled from other plumage traits.
- Georgy A. Semenov
- , Ethan Linck
- & Scott A. Taylor
-
Article
| Open Access Multiple mechanisms drive genomic adaptation to extreme O2 levels in Drosophila melanogaster
Multiple mechanisms drive genomic adaptation to extreme O2 levels in Drosophila melanogaster
The genomic details of adaptation to extreme environments remain challenging to characterize. Using new methods to analyze flies experimentally evolved to survive extreme O2 conditions, the authors find a surprising level of synchronicity in selective sweeps, de novo mutations and adaptive recombination events.
- Arya Iranmehr
- , Tsering Stobdan
- & Gabriel G. Haddad
-
Article
| Open Access Complete sequences of Schizosaccharomyces pombe subtelomeres reveal multiple patterns of genome variation
Complete sequences of Schizosaccharomyces pombe subtelomeres reveal multiple patterns of genome variation
Sequencing and mapping of long repetitive regions can be challenging due to technical difficulties in sequencing and assembly of the sequence data. Here authors report the complete sequences of subtelomeric homologous (SH) regions of the fission yeast Schizosaccharomyces pombe to reveal highly polymorphic and hot spots for genome variation features.
- Yusuke Oizumi
- , Takuto Kaji
- & Junko Kanoh
-
Article
| Open Access Genomic signatures of recombination in a natural population of the bdelloid rotifer Adineta vaga
Genomic signatures of recombination in a natural population of the bdelloid rotifer Adineta vaga
Ancient, asexual lineages are rare as a lack of recombination is usually an evolutionary dead end. Here, authors compare complete genomes of 11 individual bdelloid rotifers that suggest evidence of regular genetic exchange between individuals in a species that was previously thought to be asexual.
- Olga A. Vakhrusheva
- , Elena A. Mnatsakanova
- & Alexey S. Kondrashov
-
Article
| Open Access Donkey genomes provide new insights into domestication and selection for coat color
Donkey genomes provide new insights into domestication and selection for coat color
A new donkey reference genome and comparisons with wild asses yields insights into the evolutionary history of donkey domestication and identifies a genetic variant that results in the non-Dun coat colours of domestic donkeys.
- Changfa Wang
- , Haijing Li
- & Jifeng Zhong
-
Article
| Open Access Evolution of the potassium channel gene Kcnj13 underlies colour pattern diversification in Danio fish
Evolution of the potassium channel gene Kcnj13 underlies colour pattern diversification in Danio fish
Fish show an amazing variety of colour patterns; however, the genetic basis for the evolution of this diversity is largely unexplored. Here, the authors identify the potassium channel gene Kcnj13 as evolved to contribute to differences in colour patterns between closely related Danio fish species.
- Marco Podobnik
- , Hans Georg Frohnhöfer
- & Uwe Irion
-
Article
| Open Access Rapid de novo evolution of lysis genes in single-stranded RNA phages
Rapid de novo evolution of lysis genes in single-stranded RNA phages
Leviviruses are phages with ssRNA genomes that encode a protein (Sgl) that induces host autolysis by interfering with bacterial cell wall synthesis. Identification of sgl genes is complicated by their small size and lack of sequence similarity. Here, Chamakura et al. use bioinformatic and experimental approaches to identify sgl genes in 244 leviviral genomes.
- Karthik R. Chamakura
- , Jennifer S. Tran
- & Ry Young
-
Article
| Open Access Evidences for a role of two Y-specific genes in sex determination in Populus deltoides
Evidences for a role of two Y-specific genes in sex determination in Populus deltoides
Dioecy has evolved independently from hermaphroditic ancestors in different plant lineages. Here, the authors assemble Populus deltoides male and female genomes, and show the putative roles of a femaleness gene and a maleness gene in sex determination, which suggests independent evolution in different poplar species.
- Liangjiao Xue
- , Huaitong Wu
- & Tongming Yin
-
Article
| Open Access Evolutionary and functional genomics of DNA methylation in maize domestication and improvement
Evolutionary and functional genomics of DNA methylation in maize domestication and improvement
Variation and evolution of DNA methylation during maize domestication remain largely unknown. Here, the authors generate genome and methylome sequencing data as well as HiChIP-based interactome data to investigate the adaptive and phenotypic consequences of methylation variations in maize.
- Gen Xu
- , Jing Lyu
- & Jinliang Yang
-
Article
| Open Access Chromosome-level genome assembly of a parent species of widely cultivated azaleas
Chromosome-level genome assembly of a parent species of widely cultivated azaleas
Azaleas are one of the most diverse ornamental plants and have cultural and economic importance. Here, the authors report a chromosome-scale genome assembly for the primary ancestor of the azalea cultivar Rhododendro simsi and identify transcription factors that may function in flower coloration at different stages.
- Fu-Sheng Yang
- , Shuai Nie
- & Jian-Feng Mao
-
Article
| Open Access Chromatin accessibility landscape and regulatory network of high-altitude hypoxia adaptation
Chromatin accessibility landscape and regulatory network of high-altitude hypoxia adaptation
Tibetan adaptation to the high-altitude environment represents a case of natural selection during recent human evolution. Here the authors investigated the chromatin and transcriptional landscape of umbilical endothelial cells from Tibetan and Han Chinese donors and provide genome-wide characterization of the hypoxia regulatory network associated high-altitude adaptation.
- Jingxue Xin
- , Hui Zhang
- & Bing Su
-
Article
| Open Access Factor analysis of ancient population genomic samples
Factor analysis of ancient population genomic samples
Principal component analysis is often used in studies of ancient DNA, but does not account for the age of the samples. Here, the authors present a factor analysis (FA) which corrects for this by including the effect of allele frequency drift over time.
- Olivier François
- & Flora Jay
-
Article
| Open Access Senescence and entrenchment in evolution of amino acid sites
Senescence and entrenchment in evolution of amino acid sites
Amino acid propensities at sites change over evolutionary time, due to epistatically interacting sites or environmental changes. Here, the authors develop an approach to distinguish between these, and model the fitness dynamics of each, then annotate indicative sites in vertebrate and insect genomes.
- A. V. Stolyarova
- , E. Nabieva
- & G. A. Bazykin
-
Article
| Open Access Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolution
Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolution
Multicellularity requires complex coordinated gene expression. Fukushima and Pollock find that gene expression in different organs is likely to constrain future patterns of gene expression evolution, particularly following gene duplication.
- Kenji Fukushima
- & David D. Pollock
-
Article
| Open Access Origin and cross-species transmission of bat coronaviruses in China
Origin and cross-species transmission of bat coronaviruses in China
Bats are a likely reservoir of zoonotic coronaviruses (CoVs). Here, analyzing bat CoV sequences in China, the authors find that alpha-CoVs have switched hosts more frequently than betaCoVs, identify a bat family and genus that are highly involved in host-switching, and define hotspots of CoV evolutionary diversity.
- Alice Latinne
- , Ben Hu
- & Peter Daszak
-
Article
| Open Access Ancient genomes in South Patagonia reveal population movements associated with technological shifts and geography
Ancient genomes in South Patagonia reveal population movements associated with technological shifts and geography
How Indigenous populations in the southern tip of South America have changed over time has been unclear. Here the authors generate genome-wide data for 20 ancient individuals and examine how past migrations and admixture events correlate to geography and shifts in the archaeological record.
- Nathan Nakatsuka
- , Pierre Luisi
- & David Reich
-
Article
| Open Access Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes
Transposable elements contribute to cell and species-specific chromatin looping and gene regulation in mammalian genomes
A fraction of mammalian CTCF binding sites fall within transposable elements (TEs) but their contribution to the evolution of 3D chromatin structure is unknown. Here the authors investigate the effect of TE-driven CTCF binding site expansions on chromatin looping in humans and mice, and provide evidence that TEs contribute to cell-specific and species-specific chromatin looping diversity and variable gene regulation in mammalian genomes.
- Adam G. Diehl
- , Ningxin Ouyang
- & Alan P. Boyle
-
Article
| Open Access Extreme genetic signatures of local adaptation during Lotus japonicus colonization of Japan
Extreme genetic signatures of local adaptation during Lotus japonicus colonization of Japan
Local adaptation contributes to plant colonization across extreme environmental gradients. Here, the authors reconstruct the colonization history of Lotus japonicus in Japan and identify extreme genetic signatures of local adaptation to a cold climate using genome resequencing and common garden experiments.
- Niraj Shah
- , Tomomi Wakabayashi
- & Stig Uggerhøj Andersen
-
Article
| Open Access Unraveling cis and trans regulatory evolution during cotton domestication
Unraveling cis and trans regulatory evolution during cotton domestication
Relatively little is known about the complexity of regulatory evolution accompanying polyploid crop domestication. Here, using reciprocal hybrids between wild and domesticated allotetraploid cotton lines, the authors catalog cis and trans regulatory variants and show their equivalent effects on cotton fiber domestication.
- Ying Bao
- , Guanjing Hu
- & Jonathan F. Wendel
-
Article
| Open Access Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits
Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits
Rare structural variants may account for a significant fraction of variation in complex traits. Here the authors analyse 14 Drosophila melanogaster genomes and find that structural variants are common, found in functionally important genes, and associated with QTLs.
- Mahul Chakraborty
- , J. J. Emerson
- & Anthony D. Long
-
Article
| Open Access A streamlined and predominantly diploid genome in the tiny marine green alga Chloropicon primus
A streamlined and predominantly diploid genome in the tiny marine green alga Chloropicon primus
The Chloropicophyceae represent an important group of green algae in tropical oceans, but there is only limited genomic resource available. Here, the authors present the genome sequence of Chloropicon primus, revealing a diploid structure and the presence of a propionate detoxification pathway.
- Claude Lemieux
- , Monique Turmel
- & Jean-François Pombert
-
Article
| Open Access Harmonious genetic combinations rewire regulatory networks and flip gene essentiality
Harmonious genetic combinations rewire regulatory networks and flip gene essentiality
Studying how genetic variants in different genes interact and their combinatorial output is experimentally and analytically challenging. Here, the authors quantify the effects of more than 5000 mutation pairs in the yeast GAL regulatory system, finding that many combinations can be predicted with statistical models.
- Aaron M. New
- & Ben Lehner
-
Article
| Open Access Meta-analytic evidence that sexual selection improves population fitness
Meta-analytic evidence that sexual selection improves population fitness
Sexual selection has the potential to either increase or decrease absolute fitness. Here, Cally et al. perform a meta-analysis of 65 experimental evolution studies and find that sexual selection on males tends to increase fitness, especially in females evolving under stressful conditions.
- Justin G. Cally
- , Devi Stuart-Fox
- & Luke Holman
-
Article
| Open Access Defining the genetic and evolutionary architecture of alternative splicing in response to infection
Defining the genetic and evolutionary architecture of alternative splicing in response to infection
Genetic ancestry might influence immunological response to infection at different regulatory levels. Here, the authors use RNA-Seq to investigate the variability of alternative splicing patterns in resting and stimulated monocytes of African- and European-descent.
- Maxime Rotival
- , Hélène Quach
- & Lluis Quintana-Murci
-
Article
| Open Access Tracking the origin of two genetic components associated with transposable element bursts in domesticated rice
Tracking the origin of two genetic components associated with transposable element bursts in domesticated rice
Transposable element (TE) bursts shape genome evolution but their origin remains unclear. Here, the authors show that a burst is restricted to only a few domesticated rice accessions and is associated with the acquisition of two TE variants, Ping16A and Ping16A_Stow, not the loss of TE silencing.
- Jinfeng Chen
- , Lu Lu
- & Susan R. Wessler
-
Article
| Open Access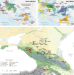 Ancient human genome-wide data from a 3000-year interval in the Caucasus corresponds with eco-geographic regions
Ancient human genome-wide data from a 3000-year interval in the Caucasus corresponds with eco-geographic regions
The Caucasus mountain range has impacted on the culture and genetics of the wider region. Here, the authors generate genome-wide SNP data for 45 Eneolithic and Bronze Age individuals across the Caucasus, and find distinct genetic clusters between mountain and steppe zones as well as occasional gene-flow.
- Chuan-Chao Wang
- , Sabine Reinhold
- & Wolfgang Haak
 The genetic architecture underlying prey-dependent performance in a microbial predator
The genetic architecture underlying prey-dependent performance in a microbial predator