
Abstract
The mechanisms by which the apolipoprotein E ε4 (APOEε4) allele influences the pathophysiological progression of Alzheimer’s disease (AD) are poorly understood. Here we tested the association of APOEε4 carriership and amyloid-β (Aβ) burden with longitudinal tau pathology. We longitudinally assessed 94 individuals across the aging and AD spectrum who underwent clinical assessments, APOE genotyping, magnetic resonance imaging, positron emission tomography (PET) for Aβ ([18F]AZD4694) and tau ([18F]MK-6240) at baseline, as well as a 2-year follow-up tau-PET scan. We found that APOEε4 carriership potentiates Aβ effects on longitudinal tau accumulation over 2 years. The APOEε4-potentiated Aβ effects on tau-PET burden were mediated by longitudinal plasma phosphorylated tau at threonine 217 (p-tau217+) increase. This longitudinal tau accumulation as measured by PET was accompanied by brain atrophy and clinical decline. Our results suggest that the APOEε4 allele plays a key role in Aβ downstream effects on the aggregation of phosphorylated tau in the living human brain.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
Data availability
The data from the TRIAD study used in the present work is not publicly available as the information could compromise the participants’ privacy. Therefore, the raw and analyzed data will be made available from the corresponding author (T.A.P.) upon reasonable request from a qualified academic investigator for the sole purpose of replicating the procedures and results presented in this article. Arrangements for data sharing for replication of the findings in the TRIAD dataset are subject to standard data-sharing agreements and further information can be found in the study’s website (https://triad.tnl-mcgill.com/).
Code availability
The codes used for the data analyses in the present work will be made available from the corresponding author (T.A.P.) upon request.
References
Polanco, J. C. et al. Amyloid-β and tau complexity—towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 14, 22–39 (2018).
Knopman, D. S. et al. Alzheimer disease. Nat. Rev. Dis. Primers 7, 33 (2021).
Jack, C. R. Jr. et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018).
Farrer, L. A. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997).
Genin, E. et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907 (2011).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993).
Zhao, N., Liu, C. C., Qiao, W. & Bu, G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 83, 347–357 (2018).
Holtzman, D. M., Herz, J. & Bu, G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006312 (2012).
Lim, Y. Y. et al. APOE ε4 moderates amyloid-related memory decline in preclinical Alzheimer’s disease. Neurobiol. Aging 36, 1239–1244 (2015).
Kantarci, K. et al. APOE modifies the association between Aβ load and cognition in cognitively normal older adults. Neurology 78, 232–240 (2012).
Mormino, E. C. et al. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology 82, 1760–1767 (2014).
Shi, Y. et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017).
Litvinchuk, A. et al. Apolipoprotein E4 reduction with antisense oligonucleotides decreases neurodegeneration in a tauopathy model. Ann. Neurol. 89, 952–966 (2021).
Wang, C. et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109, 1657–1674 (2021).
Therriault, J. et al. APOEε4 potentiates the relationship between amyloid-β and tau pathologies. Mol. Psychiatry https://doi.org/10.1038/s41380-020-0688-6 (2021).
Farfel, J. M., Yu, L., De Jager, P. L., Schneider, J. A. & Bennett, D. A. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 37, 19–25 (2016).
Dincer, A. et al. APOE ε4 genotype, amyloid-β, and sex interact to predict tau in regions of high APOE mRNA expression. Sci. Transl. Med. 14, eabl7646 (2022).
Koutsodendris, N. et al. Neuronal APOE4 removal protects against tau-mediated gliosis, neurodegeneration and myelin deficits. Nat. Aging 3, 275–296 (2023).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991).
Braak, H. & Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357 (1997).
Therriault, J. et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat. Aging 2, 526–535 (2022).
Nelson, P. T. et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381 (2012).
Pascoal, T. A. et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 143, 2818–2830 (2020).
Jack, C. R. Jr. et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216 (2013).
Montal, V. et al. Network tau spreading is vulnerable to the expression gradients of APOE and glutamatergic-related genes. Sci. Transl. Med. 14, eabn7273 (2022).
Young, C. B. et al. APOE effects on regional tau in preclinical Alzheimer’s disease. Mol. Neurodegener. 18, 1 (2023).
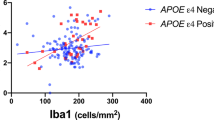
Ferrari-Souza, J. P. et al. APOEε4 associates with microglial activation independently of Aβ plaques and tau tangles. Sci. Adv. 9, eade1474 (2023).
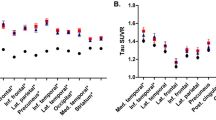
Therriault, J. et al. Association of apolipoprotein E ε4 with medial temporal tau independent of amyloid-β. JAMA Neurol. 77, 470–479 (2020).
La Joie, R. et al. Association of APOE4 and clinical variability in Alzheimer disease with the pattern of tau- and amyloid-PET. Neurology 96, e650–e661 (2021).
Groot, C. et al. Phospho-tau with subthreshold tau-PET predicts increased tau accumulation rates in amyloid-positive individuals. Brain https://doi.org/10.1093/brain/awac329 (2023).
Mattsson-Carlgren, N. et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive tau PET in Alzheimer’s disease. Sci. Adv. 6, eaaz2387 (2020).
Barthelemy, N. R. et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 26, 398–407 (2020).
Reimand, J. et al. Association of amyloid-β CSF/PET discordance and tau load 5 years later. Neurology 95, e2648–e2657 (2020).
Mila-Aloma, M. et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med. 28, 1797–1801 (2022).
Ashton, N. J. et al. Differential roles of Aβ42/40, p-tau231 and p-tau217 for Alzheimer’s trial selection and disease monitoring. Nat. Med. 28, 2555–2562 (2022).
Therriault, J. et al. Association of phosphorylated tau biomarkers with amyloid positron emission tomography vs tau positron emission tomography. JAMA Neurol. 80, 188–199 (2022).
Querfurth, H. W. & LaFerla, F. M. Alzheimer’s disease. N. Engl. J. Med. 362, 329–344 (2010).
Karikari, T. K. et al. Blood phospho-tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nat. Rev. Neurol. 18, 400–418 (2022).
Wang, C. et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657 (2018).
Brecht, W. J. et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 24, 2527–2534 (2004).
Mattsson-Carlgren, N. et al. Soluble P-tau217 reflects amyloid and tau pathology and mediates the association of amyloid with tau. EMBO Mol. Med. 13, e14022 (2021).
Pichet Binette, A. et al. Amyloid-associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer’s disease. Nat. Commun. 13, 6635 (2022).
Petersen, R. C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256, 183–194 (2004).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Montagne, A. et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581, 71–76 (2020).
Triana-Baltzer, G. et al. Development and validation of a high-sensitivity assay for measuring p217+tau in plasma. Alzheimers Dement. 13, e12204 (2021).
Saykin, A. J. et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimers Dement. 11, 792–814 (2015).
Ferrari-Souza, J. P. et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 27, 4781–4789 (2022).
Pascoal, T. A. et al. In vivo quantification of neurofibrillary tangles with [(18)F]MK-6240. Alzheimers Res. Ther. 10, 74 (2018).
Cselenyi, Z. et al. Clinical validation of 18F-AZD4694, an amyloid-β-specific PET radioligand. J. Nucl. Med. 53, 415–424 (2012).
Jack, C. R. Jr. et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 141, 1517–1528 (2018).
Pascoal, T. A. et al. Longitudinal 18F-MK-6240 tau tangles accumulation follows Braak stages. Brain 44, 3517–3528 (2021).
Diedrichsen, J., Balsters, J. H., Flavell, J., Cussans, E. & Ramnani, N. A probabilistic MR atlas of the human cerebellum. Neuroimage 46, 39–46 (2009).
Jack, C. R. Jr. et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 13, 205–216 (2017).
Therriault, J. et al. Determining amyloid-β positivity using (18)F-AZD4694 PET imaging. J. Nucl. Med. 62, 247–252 (2021).
Tukey, J.W. Exploratory Data Analysis (Addison-Wesley, 1977).
Moscoso, A. et al. CSF biomarkers and plasma p-tau181 as predictors of longitudinal tau accumulation: implications for clinical trial design. Alzheimers Dement. 18, 2614–2626 (2022).
Therriault, J. et al. Frequency of biologically defined Alzheimer disease in relation to age, sex, APOE ε4, and cognitive impairment. Neurology 96, e975–e985 (2021).
Worsley, K. J., Taylor, J. E., Tomaiuolo, F. & Lerch, J. Unified univariate and multivariate random field theory. Neuroimage 23, S189–S195 (2004).
Lerch, J., Hammill, C., van Eede, M. & Cassel, D. RMINC: Statistical Tools for Medical Imaging NetCDF (MINC) Files (2017).
Acknowledgements
We acknowledge all study participants and staff at the McGill Center for Studies in Aging. We thank Cerveau Technologies for the use of [18F]MK-6240. We also thank D. Jolly, A. Kostikov, M. Samoila-Lactatus, K. Ross, M. Boudjemeline and S. Li for assisting in the radiochemistry production, as well as R. Strauss, E. Strauss, G. Gagne, C. Mayhew, T. Vinet-Celluci, K. Wan, S. Sbeiti, M. Jin Joung, M. Olmand, R. Nazar, H.-H. Hsiao, R. Bouhachi and A. Aliaga for helping with the acquisition of the data. We thank the following funding agencies for their support: National Institutes of Health (grants R01AG075336 and R01AG073267 to T.A.P. and R01AG068398 to K.B.); Alzheimer’s Association (grants AARFD-22-974627 to B.B.; AACSF-20-648075 to T.A.P.; NIRG-12-92090 and NIRP-12-259245 to P.R.-N.; AARF-21-850325 to T.K.K.; AARFD-22-923814 to P.C.L.F.; AARGD-21-850670 to E.R.Z.; and ADSF-21-831376-C, ADSF-21-831381-C and ADSF-21-831377-C to H.Z.); Brain Canada Foundation (CFI Project 34874 and 33397 to P.R.-N.); CAPES (grant 88887.336490/2019-00 to B.B.); CNPq (grant 200691/2021-0 to J.P.F.-S. and 312306/2021-0 to E.R.Z.); Fonds de Recherche du Québec–Santé (Chercheur Boursier, grant 2020-VICO-279314 to P.R.-N.); CIHR-CCNA Canadian Consortium of Neurodegeneration in Aging (grants MOP-11-51-31; RFN 152985, 159815 and 162303 to P.R.-N.); Weston Brain Institute (grants 8400707, 8401154 and 8401103 to P.R.-N.); Colin Adair Charitable Foundation (grant to P.R.-N.); Swedish Research Council (grant 2021-03244 to T.K.K.; 2018-02532 to H.Z.; and 2017-00915 and 2022-00732 to K.B.); Wallenberg Scholar (grant 2022-01018 to H.Z.); BrightFocus Foundation (grant A2020812F to T.K.K.); Swedish Alzheimer Foundation (Alzheimerfonden; grant AF-930627 to T.K.K.; and AF-930351, AF-939721 and AF-968270 to K.B.); Swedish Brain Foundation (Hjärnfonden; grant FO2020-0240 to T.K.K.); Agneta Prytz-Folkes & Gösta Folkes Foundation (grant 2020-00124 to T.K.K.); European Union’s Horizon Europe research and innovation program (grant 101053962 to H.Z.); Swedish State Support for Clinical Research (grant ALFGBG-71320 to H.Z.); Alzheimer Drug Discovery Foundation (grant 201809-2016862 to H.Z. and RDAPB-201809-2016615 to K.B.); Bluefield Project, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (grant FO2022-0270 to H.Z.); European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie (grant 860197 (MIRIADE) to H.Z.); the European Union Joint Program–Neurodegenerative Disease Research (grant JPND2021-00694 to H.Z. and JPND2019-466-236 to K.B.); The UK Dementia Research Institute at UCL (grant UKDRI-1003 to H.Z.); National Academy of Neuropsychology (grant ALZ-NAN-22-928381 to E.R.Z.); Fundação de Amparo a pesquisa do Rio Grande do Sul (grant 21/2551-0000673-0 to E.R.Z.); Instituto Serrapilheira (grant Serra-1912-31365 to E.R.Z.); Hjärnfonden (grants FO2017-0243 and ALZ2022-0006 to K.B.); the Swedish state under the agreement between the Swedish government and the County Councils, the ALF agreement (grants ALFGBG-715986 and ALFGBG-965240 to K.B.); the Alzheimer’s Association 2021 Zenith Award (grant ZEN-21-848495 to K.B.); and the Alzheimer’s Association 2022–2025 grant (SG-23-1038904 QC to K.B.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.P.F.-S. and T.A.P. conceived the study. J.P.F.-S. and T.A.P. prepared the figures, tables and drafted the paper with input from B.B., P.C.L.F., A.L.B., G.P., F.Z.L., D.T.L., J.T., C.T., C.S., Y.-T.W., M.C., S.S., A.C.M., M.V., G.B., M.S.K., J.S., N.R., V.P., N.M.P., A.C., O.L.L., W.E.K., J.-P.S., S.G., D.O.S., G.T.-B., Z.S.S., H.C.K., T.K.K., V.L.V., D.L.T., N.J.A., H.Z., K.B., E.R.Z. and P.R.-N. J.P.F.-S., B.B., P.C.L.F., G.P., F.Z.L., D.T.L., J.T., C.T., C.S., Y.-T.W., M.C., S.S., A.C.M., M.V., G.B., M.S.K., J.S., N.R., V.P., N.M.P., A.C., O.L.L., W.E.K., J.-P.S., S.G., D.O.S., V.L.V., E.R.Z., P.R.-N. and T.A.P. contributed to the acquisition, processing and analysis of imaging data. J.P.F.-S., A.L.B., G.T.-B., Z.S.S., H.C.K., T.K.K., N.J.A., H.Z. and K.B. contributed to the analysis of the fluid biomarker data. T.A.P. supervised this work. D.L.T. assisted in the statistical analysis. All authors performed a critical review of the paper for intellectual content and approved the final paper draft.
Corresponding author
Ethics declarations
Competing interests
S.G. has served as a scientific advisor to Cerveau Technologies. N.J.A. has given lectures in symposia sponsored by Lilly and Quanterix. G.T.-B., Z.S.S. and H.C.K. are employees of Janssen R&D and receive salary and stock from its parent company, Johnson & Johnson. H.Z. has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Passage Bio, Pinteon Therapeutics, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen and Roche and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. K.B. has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Prothena, Roche Diagnostics and Siemens Healthineers and is a co-founder of BBS, which is a part of the GU Ventures Incubator Program. E.R.Z. serves on the scientific advisory board of Next Innovative Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks Alifiya Kapasi and Christina Young for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Flowchart of included participants.
aOut of the 13 APOEε2 carriers who were excluded, twelve had the ε2/ε3 genotype (8 CU older adults, 2 with MCI, and 2 with AD dementia), and one had the ε2/ε4 genotype (individual with MCI). Abbreviations: AD = Alzheimer’s disease; ADAD = autosomal dominant Alzheimer’s disease; APOE = apolipoprotein E; Aβ = amyloid-β; CU = cognitively unimpaired; EOAD = early-onset Alzheimer’s disease; MCI = mild cognitive impairment; MRI = magnetic resonance imaging; PET = positron emission tomography; TRIAD = Translational Biomarkers in Aging and Dementia.
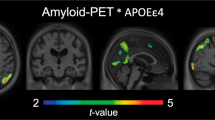
Extended Data Fig. 2 Sensitivity analyses testing the association of APOEε4 carriership and Aβ burden with tau accumulation including APOEε2 carriers.
(a) Violin plot of temporal meta-ROI tau-PET SUVR change across groups. The horizontal line inside each box depicts the median, and box ends represent the 25th and 75th percentiles, with whiskers extending to 1.5 × interquartile range. The horizontal dashed line represents the threshold for tau accumulation. Groups were compared using two-sided analysis of covariance with Tukey’s multiple comparisons test. (b) The figure shows the ORs from logistic regression on being classified as tau accumulator. Only the concomitant presence of Aβ+ and APOEε4 carriership was associated with higher odds of being classified as tau accumulator (Aβ- APOEε4 noncarrier: reference group; Aβ- APOEε4 carrier: OR = 1.7, 95% CI 0.2 to 9.9, P = 0.582; Aβ+ APOEε4 noncarrier: OR = 1.7, 95% CI 0.3 to 9.6, P = 0.559; and Aβ+ APOEε4 carrier: OR = 21.4, 95% CI 3.7 to 159.8, P = 0.001; Supplementary Fig. 1b). (c) The scatter-plot displays the association between global Aβ-PET SUVR and temporal meta-ROI tau-PET SUVR change in APOEε4 noncarriers (blue) and carriers (red). The error bands indicate the 95% CI. Density plots along the x and y axes provide the data distribution. The β estimate and P value were computed from a linear regression model assessing the interaction of APOEε4 status and global Aβ-PET burden on longitudinal tau-PET. Regressions were two-sided and adjusted for age, sex, diagnosis, and baseline tau-PET burden. The interaction model also accounted for APOEε4 status and global Aβ-PET burden main effects. Analyses were conducted including individuals bearing the ε2 allele of the APOE gene who were excluded from the main analyses (70 CU older adults, 28 with MCI, and 9 with AD dementia). Abbreviations: AD = Alzheimer’s disease; APOEε4 = apolipoprotein E ε4; Aβ = amyloid-β; CI = confidence interval; CU = cognitively unimpaired; MCI = mild cognitive impairment; OR = odds ratio; PET = positron emission tomography; ROI = region of interest; SUVR = standardized uptake value ratio.
Extended Data Fig. 3 Association of APOEε4 carriership and Aβ burden with tau accumulation in the subgroup of non-demented participants.
(a) Violin plot of temporal meta-ROI tau-PET SUVR change across groups. The horizontal line inside each box depicts the median, and box ends represent the 25th and 75th percentiles, with whiskers extending to 1.5 × interquartile range. The horizontal dashed line represents the threshold for tau accumulation. Groups were compared using two-sided analysis of covariance with Tukey’s multiple comparisons test. (b) The figure shows the ORs from logistic regression on being classified as tau accumulator. Only the concomitant presence of Aβ+ and APOEε4 carriership was associated with higher odds of being classified as tau accumulator (Aβ- APOEε4 noncarrier: reference group; Aβ- APOEε4 carrier: OR = 1.5, 95% CI 0.2 to 9.2, P = 0.696; Aβ+ APOEε4 noncarrier: OR = 0.9, 95% CI 0.1 to 7.8, P = 0.910; and Aβ+ APOEε4 carrier: OR = 18.8, 95% CI 2.0 to 228.7, P = 0.013; Supplementary Fig. 1c). (c) The scatter-plot displays the association between global Aβ-PET SUVR and temporal meta-ROI tau-PET SUVR change in APOEε4 noncarriers (blue) and carriers (red). The error bands indicate the 95% CI. Density plots along the x and y axes provide the data distribution. The β estimate and P value were computed from a linear regression assessing the interaction of APOEε4 status and global Aβ-PET burden on longitudinal tau-PET. Regressions were two-sided and adjusted for age, sex, diagnosis, and baseline tau-PET burden. The interaction model also accounted for APOEε4 status and global Aβ-PET burden main effects. Analyses were conducted in the subgroup of non-demented participants (62 CU older adults and 25 with MCI). Abbreviations: APOEε4 = apolipoprotein E ε4; Aβ = amyloid-β; CI = confidence interval; CU = cognitively unimpaired; MCI = mild cognitive impairment; OR = odds ratio; PET = positron emission tomography; ROI = region of interest; SUVR = standardized uptake value ratio.
Extended Data Fig. 4 Plasma p-tau217+ change according to longitudinal tau accumulation status.
Violin plot of plasma p-tau217+ change across groups defined based on longitudinal tau accumulation status. The horizontal line inside each box depicts the median, and box ends represent the 25th and 75th percentiles, with whiskers extending to 1.5 × interquartile range. Groups were compared using two-sided analysis of covariance. The P value was computed from a model adjusted for age, sex, diagnosis, and baseline plasma p-tau217+ levels. Analyses were conducted on a subset of 65 individuals (Supplementary Table 4). Abbreviation: p-tau217+ = phosphorylated tau at threonine 217.
Supplementary information
Supplementary Information
Supplementary Tables 1–12 and Fig. 1.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ferrari-Souza, J.P., Bellaver, B., Ferreira, P.C.L. et al. APOEε4 potentiates amyloid β effects on longitudinal tau pathology. Nat Aging 3, 1210–1218 (2023). https://doi.org/10.1038/s43587-023-00490-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-023-00490-2
This article is cited by
-
Multifaceted roles of APOE in Alzheimer disease
Nature Reviews Neurology (2024)
