
Abstract
Energy-band gradient halide perovskites are highly desired candidates for fabricating high performance optoelectronic devices. Here, we demonstrate that a mixed halide perovskite single crystal undergoes phase segregation in the longitudinal direction under above-bandgap light illumination. As a result, a micron thick layer with vertically gradient halide composition and thus graded valence band edge is generated at the crystal surface. The resultant gradient structure can facilitate the hole extraction at its interface with a hole transport layer. The longitudinal phase segregation of mixed halide perovskite single crystal is likely driven by abundant defects at the surface. Moreover, the segregation rate is increased in air compared to nitrogen probably due to the combined effect of oxygen and moisture. These findings not only deepen the understanding of phase segregation mechanism in mixed halide perovskite, but also indicate a promising avenue of fabricating vertically energy-band gradient perovskite and enhancing the perovskite-based optoelectronic device performance.
Similar content being viewed by others
Introduction
Halide perovskites are a class of material with chemical formular of ABX3, in which X is typically Cl, Br, I or a mixture thereof. The band structure of halide perovskites largely depends on the halide composition in X site, which can be tuned continuously by changing the ratio of Cl−, Br−, and I−1,2,3. A gradually varying halide composition leads to the formation of a graded valence band configuration4,5. A longitudinally energy-band gradient perovskites with varying halide composition can be interfaced with a hole transport layer (HTL) and used for accelerating the hole extraction and enhancing the efficiency of optoelectronic devices like solar cells and detectors6,7,8. Alternatively, such structures can facilitate photoexcited carrier extraction from high bandgap material towards lower bandgap one and improve the emitting efficiency of devices such as scintillators9,10,11.
A perovskite structure with intrinsic halide gradient in the longitudinal direction can be obtained by growing, evaporating or spin-coating successive multilayer of planar perovskite12. It is also possible to fabricate longitudinally gradient perovskite structure based on post-treatment such as ion-exchange reaction, by carefully controlling the exchange duration and temperature12,13. Phase segregation of mixed halide perovskite also offers a promising avenue for fabricating gradient structure. Mixed halide perovskite such as APb(BrxI1-x)3 is known to undergo phase segregation under above-gap illumination or external bias, when the Br content is higher than a threshold (i.e.,15% for MAPb(BrxI1-x)3, MA = CH3NH3)14,15,16,17,18,19,20,21. Phase segregation deteriorates the stability of mixed halide perovskite-based devices such as solar cells22. Tremendous efforts have been made to understand its underlying mechanism and most of these studies were carried out in polycrystalline thin films. Grain boundaries play an important role in the phase segregation of mixed halide perovskite polycrystalline films, in which the iodide ion migration mainly proceeds in the lateral orientation towards the grain boundaries23,24,25. However, it remains unclear how the phase segregation progresses in a mixed halide perovskite sample with absence of grain boundaries such as a single crystal. Perovskite single crystal possesses abundant defects at the surface compared to the interior, which may trigger the segregation in the vertical direction. This offers a potential avenue towards generating longitudinally gradient structure with varying halide composition.
In this work, we explore such possibility by studying the phase segregation of MAPb(BrxI1-x)3 single crystals under above-bandgap light excitation. Our depth resolved compositional analysis reveals the formation of a micron thick surface layer with varying halide composition along the depth dimension, indicating the occurrence of a longitudinal phase segregation in the near-surface layer of the single crystal. The resultant gradient structure is composed of multiple phases with various halide compositions as confirmed by low temperature photoluminescence (PL) measurements. These are uncommon observations in mixed halide perovskites (so far, they have been seen only in cathodoluminescence measurements assisted with intricate statistical analysis)26. Our further investigation reveals that the vertical segregation of the mixed halide perovskite single crystal is likely driven by surface defects and its rate is altered by the atmosphere significantly. The obtained gradient structure facilitates the hole extraction when the low bandgap side is interfaced with a HTL, which can potentially lead to improved performance of mixed halide perovskite single crystal-based optoelectronic devices such as solar cells and detectors.
Results and Discussion
Synthesis and characterization of mixed halide single crystal
Two MAPb(BrxI1-x)3 single crystals with I-rich and Br-rich compositions are synthesized based on inverse temperature crystallization method27, by tuning the ratio of Br and I in precursor solutions. The synthesis detail is included in the Supplementary Methods. We first study the phase segregation of the I-rich crystal with Br content x of 0.29, which exhibits tetragonal phase and exposes top (100) facet. This crystal undergoes phase segregation under ultraviolet (UV) light illumination, generating two extra I-rich compositions, as shown in Supplementary Fig. 1.
In comparison, the Br-rich crystal possesses cubic phase which exhibits cuboid shape and exposes (100) facets. Figure 1a shows the X-ray diffraction (XRD) pattern of the Br-rich MAPb(BrxI1-x)3 single crystal, in which the (200) diffraction peak is located at 30.05°. This corresponds to a lattice parameter of 5.88 \({{{\text{\AA }}}}\) and a Br content x of 0.905 according to the reported lattice parameter-composition relationship19. The phase segregation of the Br-rich crystal with such a high Br content can potentially generate more phases with different compositions. For this reason, the mixed halide perovskite crystal with Br-rich composition is selected for further study.

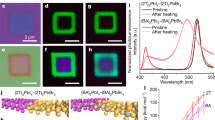
a XRD patterns of the sample before and after phase segregation. The inset shows the magnified spectrum around 29°. The bottom panel shows the reference powder XRD patterns for MAPbI3 and MAPbBr3. b UV-vis absorption spectra of the sample before and after phase segregation. c PL evolution during phase segregation, extracted from region 1 which is labelled in the inset confocal map of crystal surface. d Schematics showing a longitudinally gradient perovskite structure and the SEM-EDS analysis results of phase segregated sample, obtained using incident electrons of two energies. The energy deposition of 5 kV and 15 kV electrons obtained from simulation is also shown. e, f Evolution of I 3d and Br 3d XPS spectra with etch time. g The quantified percentage of two halide ions obtained from XPS spectra at different etch time.
Evidencing longitudinal phase segregation
We combine XRD, UV-visible absorption and PL measurements to investigate the phase segregation of this initially Br-rich MAPb(BrxI1-x)3 single crystal (x = 0.905). XRD pattern is recorded for the crystal before and after UV illumination (Fig. 1a). After light illumination, an extra diffraction peak appears at \(2\theta =29^\circ\) as shown in the inset figure, which likely corresponds to the I-rich phases generated due to phase segregation. Figure 1b shows the absorption spectra before and after UV illumination. As seen, the initial sample exhibits an absorption edge at wavelength of about 600 nm, and after illumination for a few minutes, an absorption edge emerges around 720 nm which corresponds to I-rich compositions, indicating the occurrence of phase segregation. Simultaneously, the absorption edge at 600 nm becomes less sharp than the initial sample which can be correlated with the defect formation due to the phase segregation.
We also capture a series of confocal PL maps after the crystal surface is subjected to 405 nm light illumination for different duration. The PL spectra of four separate regions 1, 2, 3 and 4 are extracted from the confocal maps as labelled in the inset of Figure 1c. Figure 1c reports the PL evolution of region 1 (for the PL spectra of other regions see Supplementary Fig. 2). Interestingly, all the regions exhibits similar PL evolution trend, in which the emission intensity of initial phase at around 560 nm decreases with illumination. Simultaneously, an emission feature emerges around 670 nm with its peak position redshifting and intensity increasing with illumination. These results suggest that phase segregation occurs predominantly in the longitudinal direction of the mixed halide perovskite single crystal.
It is worth to note that slight differences are also observed among PL spectra of different regions. (Supplementary Fig. 2) This is probably related with a mild horizontal phase segregation induced by the stochastic composition fluctuations pre-existing on the pristine mixed halide perovskite surface16. Similar observation was also made previously for MAPb(BrxI1−x)3 single crystal microplate28. Nevertheless, the observed PL difference between various regions is very small, and the halide ion migration in the vertical direction dominates the phase segregation process of single crystal. Note that near the crystal edge there is an exception and a more obvious horizontal segregation is observed, as shown by the PL spectra in Supplementary Fig. 3.
To further confirm the occurrence of longitudinal phase segregation, we study the composition of phase segregated sample by scanning electron microscope energy-dispersive X-ray spectroscopy (SEM-EDS) and depth resolved X-ray photoelectron spectroscopy (XPS) measurements. Figure 1d summarizes the composition of the overall sample surface obtained from SEM-EDS analysis using incident electron beam of two different energies (5 and 15 keV). As shown, a Br/I ratio of 9.5 is obtained with an incident electron energy of 15 keV. The Br/I ratio decreases to 2.0 when the incident electron energy is decreased to 5 keV. Since an electron beam with lower energy penetrates shallower into the target material and probes the information from a thinner surface layer, the results in Figure 1d suggest that the sample surface contains a higher content of I, and the inner regions are more abundant in Br. The SEM-EDS results confirm that segregation progresses in the longitudinal direction of the mixed halide perovskite single crystal and enables the formation of a structure with vertically gradient halide composition, as depicted in Figure 1d. To estimate the thickness of gradient structure, we simulate the deposited energy distribution of the incident electron beam in MAPb(BrxI1-x)3 (x = 0.905) using Monte Carlo program CASINO. As shown in Figure 1d, the incident electron beam with 5 and 15 keV deposits the energy within a layer of approximately 450 nm and 1170 nm respectively. This result indicates that the Br/I ratio of 450 nm thick near-surface layer is 2.0. With increasing depth, the iodide content decreases, and the overall Br/I ratio of 1170 nm thick near-surface layer approaches 9.5, which is the same as the initial crystal. Further, we perform cross sectional confocal PL analysis of the segregated sample after cleaving. (for the PL spectra see Supplementary Fig. 4) The results indicate that the gradient structure only distributes at near-surface region and the composition of the interior lattice remains to be the same as pristine phase. Based on the above analysis, it can be concluded that the longitudinal phase segregation of the mixed halide perovskite single crystal leads to the formation of a micron thick layer with vertically gradient halide composition at the crystal surface.
The depth distribution of various elements is further probed using XPS coupled with argon ion etching. XPS spectra are recorded every 300 s throughout a total etching time of 1500 s. Figure 1e, f reports the evolution of I 3d and Br 3d XPS spectra, which illustrates that the peak area of I 3d declines with etch time, while that of Br 3d increases very slightly. The quantified percentage of two halide ions is shown in Figure 1g. This result confirms that iodide ion is the preferentially mobile halide ion migrating from interior to surface during the longitudinal phase segregation, while the motion of bromide ion in the opposite direction is very mild.
Supplementary Fig. 5 reports the Pb 4 f, N1s and C 1 s XPS spectra. Intense peaks are observed at the side of Pb2+ feature and both C 1 s and N 1s intensity drops with etching time. These results indicate that the XPS measurement inevitably induces radiolysis of the perovskite crystal29,30,31, which involves multiple processes such as (1) reduction of lead ions and generation of Pb0 that corresponds to the observed intense peaks at the side of Pb2+ feature in Pb 4 f XPS; (2) formation of gaseous species containing nitrogen and carbon, which desorb easily under the high vacuum conditions. Due to the radiolysis, an accurate composition of the gradient structure cannot be obtained from XPS analysis.
Revealing the mechanism of longitudinal phase segregation
Mild vertical ion migration was also identified in an earlier study which employed mixed halide perovskite polycrystalline film26. It was proposed that the vertical ion migration originates from gradient carrier distribution along the depth of the surface 100 ~ 200 nm thick layer under UV excitation. The gradient carrier distribution leads to gradient lattice constant and drives the halide ions with larger radius (iodide ions) to migrate towards the surface. It is intriguing to investigate whether the gradient carrier distribution is the major driving force of vertical segregation in mixed halide perovskite single crystal. For this purpose, we use a two-photon laser (800 nm) to generate carriers in the entire thickness of the crystal in which no obvious gradient distribution of photogenerated carriers is induced along the depth. Figure 2a shows a series of PL spectra recorded under continuous two-photon excitation in air, in which the sample only displays an emission from the pristine phase initially and later another emission emerges which corresponds to the I-rich phases. One can evaluate the rate of phase segregation by fitting the variation of I-rich emission intensity against illumination time with \(I\left(t\right)=A-B{e}^{-{kt}}\)16. The obtained segregation rate \(k\) under two-photon excitation is 3.4 × 10−4 s−1 as shown in Fig. 2b. This value is close to what measured under single-photon excitation in air, as will be shown later, indicating the secondary role of gradient carrier distribution in phase segregation. Moreover, compared with single-photon excitation, the emission peak from the pristine phase under two-photon excitation is redshifted. The shape of the I-rich emission feature under two-photon excitation is also different from that of UV excitation. These observations can be correlated with the different penetration and detection depths of two excitation sources. In the case of UV excitation, the detected photons come from the surface; while the photons in two-photon PL spectra come from a thicker layer which contains phases with multiple iodide compositions and a significant photon recycling occurs before the photon detection. Based on the above analysis, it can be inferred that without inducing gradient carrier distribution, two-photon excitation also triggers vertical phase segregation and results in the formation of a vertically gradient structure. The vertical segregation of mixed halide perovskite single crystal is mainly driven by other factors rather than photogenerated carrier distribution.

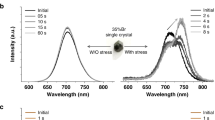
a PL evolution under two-photon excitation using 800 nm laser. b Variation of the I-rich emission intensity with respect to the illumination time under two-photon excitation. c TRPL of top and bottom crystal surface under single-photon excitation using 405 nm laser; d Intensity of I-rich emission feature with illumination time, extracted from the PL evolution of top and bottom surface under single-photon excitation. The experimental data and the fitting are shown in solid circles and lines respectively.
It is possible that surface defects induce electric field in the vertical direction and drive the migration of iodide ions, triggering the halide redistribution in the vertical direction. This speculation accords well with the general understanding of halide segregation process which underscores the important role of perovskite surfaces in halide segregation process of any perovskite materials32,33,34,35. Since the top and bottom surface of the same crystal have different surface defect density, we compare the phase segregation rate of these two crystal surfaces to illustrate the role of surface defects. Figure 2c shows the time-resolved PL (TRPL) spectra measured from two surfaces which can be well fitted with bi-exponential function \({{{\rm{I}}}}\left({{{\rm{t}}}}\right)={{{{\rm{A}}}}}_{1}{{{{\rm{e}}}}}^{-{{{\rm{t}}}}/{{{{\rm{\tau }}}}}_{1}}+{{{{\rm{A}}}}}_{2}{{{{\rm{e}}}}}^{-{{{\rm{t}}}}/{{{{\rm{\tau }}}}}_{2}}\). (for the fitting parameters see Supplementary Table 1) The effective decay lifetime can be calculated according to \({\tau }_{{eff}}=({\sum }_{i=1}^{2}{A}_{i}{{\tau }_{i}}^{2})/({\sum }_{i=1}^{2}{A}_{i}{\tau }_{i})\). The obtained effective lifetime of top surface is 767.3 ns, which is nine times longer compared to the bottom surface (85.6 ns), suggesting that the bottom crystal surface has a much higher surface defect density than the top surface. We monitor the PL evolution of top and bottom surface under 405 nm excitation. Both exhibit the decrease of pristine phase emission and increase of I-rich emission. Figure 2d plots the intensity of I-rich emission feature with illumination time for top and bottom surfaces. By fitting, the obtained segregation rate of the bottom surface is about eight times larger than that of the top surface. The strikingly higher segregation rate of more defective surface confirms the vital role that surface defects play in vertical phase segregation in the mixed halide perovskite single crystal.
We further investigate the effect of atmosphere on longitudinal phase segregation by monitoring the PL evolution of two identical samples in air and N2 respectively under UV excitation (405 nm). The humidity and oxygen levels of the air are 80.0% and 20.9% respectively. Note that the samples in this set of experiments are mounted inside a cuvette filled with N2 or air, and a home-built spectroscopy system is used for PL measurement. The intensity of equipped UV illumination source is 0.36 mW cm−2 which is larger than that of confocal microscope used in Figure 1c and therefore a later stage of phase segregation is captured under this condition. Figure 3a, b reports the PL evolution of the crystal in atmosphere of air and N2 respectively, both of which show decreasing emission of the pristine phase and emergence of I-rich emission feature. With further illumination, the intensity of I-rich feature increases and its peak position redshifts. Figure 3c plots the variation of peak intensity with illumination time for both atmospheres. The obtained segregation rate increases from 7.0 × 10−6 s−1 in N2 to 5.0 × 10−4 s−1 in air, by nearly a factor of 70.

a, b PL evolution in air and N2. c, d Variance of emission intensity, position, and FWHM (full width at half maxima) of the PL feature around 690 nm with illumination time. The dots in (c) are experimental data extracted from PL spectra, and solid lines on top of the dots are from fitting. The fluctuation of PL position before reaching the plateau is highlighted by the blue circle in (d).
The enhancement of phase segregation rate in air is likely due to the combined effect of oxygen and water. On one hand, the water intercalation into the perovskite lattice can reduce the activation energies for vacancy-mediated ion migration36, which favors the phase segregation. Water molecule can also favor the trapping of migrated halide ions at vacancy sites of halide on perovskite surface37,38. On the other hand, I- can be oxidized to form I2 by oxygen in the presence of water39, which potentially accelerates the phase segregation process. The latter can be understood by referring to a recently proposed model of Ross A. Kerner et al.5. This model suggests that the phase segregation is triggered by preferential oxidation of iodide ions due to the presence of photogenerated holes. We infer that the oxygen and water molecules absorbed to the crystal surface can assist the oxidation of iodide ions, resulting in a faster phase segregation.
Note that the segregation rate of our crystal in air is 100 times smaller than what reported for phase segregation in polycrystalline film under similar conditions16. The obviously different rates points to different segregation mechanisms in single crystal and polycrystalline film. In polycrystalline film, phase segregation mainly occurs in the horizontal direction because the grain boundaries not only provide a reducing environment for oxidation product of iodide ions (i.e. I2) to get reduced and re-enter5, but also induce an electric field to drive the migration of iodide ions35,40. Due to the grain boundaries, the average activation energy of ion migration is low and the horizontal segregation rate in polycrystalline film is high32. In contrast, a mixed halide perovskite single crystal is free of grain boundaries and the segregation of a single crystal is dominated by the ion migration along the longitudinal dimension which has a relatively low migration rate.
Identifying multiple intermediate phases in the gradient structure
In the above study, we also observe that the full width at half maximum (FWHM) of the I-rich emission first decreases with illumination and then remains unchanged after 150 s for samples in both atmospheres, as illustrated in Fig. 3d. This is related with formation of multiple phases with different halide compositions when the phase segregation progresses.
Initially upon the illumination, multiple intermediate phases with various iodide contents are formed, all of which emit and together compose the PL spectra, leading to a large FWHM. With continuous supplement of iodide ions, the intermediate phases eventually transform into more stable I-rich phases, causing the narrowing of FWHM which finally reaches a plateau. Phase segregation occurs step by step in N2, while the oxygen and moisture in air leads to faster formation of stable I-rich phases. As a result, a wider FWHM is observed in N2 than air in the initial stage. Besides, due to the coexistence of multiple intermediate phases of different energy band, energy funneling should occur towards lower bandgap regions, as reported before41. It is likely that efficient energy funneling between adjacent domains also contributes to the narrowing of FWHM with illumination time. To confirm the presence of energy funneling among I-rich phases, we measure the TRPL spectra in several selected detection ranges. Supplementary Fig. 6 shows the TRPL of three selected detection energy windows which are centered at 650, 680 and 720 nm with a width of 30 nm. (for the fitting parameters see Supplementary Table 2) The obtained effective lifetime increases from 181.9, 345.2 to 596.9 ns when the central detection wavelength changes from 650, 680 to 720 nm. This evolution trend of decay lifetime confirms the occurrence of energy funneling among various I-rich compositions.
Figure 3d also reports the peak position variation of I-rich emission feature with illumination time in air and N2. The step-by-step occurrence of phase segregation in N2 causes the PL position to undergo a fluctuation before reaching the plateau, as highlighted by the blue circle. This is in contrast with the smooth redshift observed in air. Besides, the PL position of the I-rich phases eventually stabilizes at a less reddish wavelength in air (706 nm) than the one in N2 (710 nm), despite of more significant phase segregation. The less reddish wavelength in air may be related to the expulsion of volatile molecular iodine species formed due to iodide ion oxidation42.
To identify the intermediate phases formed during segregation, we conduct low temperature PL study. At lower temperatures, the carrier-phonon coupling can be reduced, and PL peaks are narrower, which allow better identification of multiple emission peaks that are otherwise convoluted into one PL emission feature at room temperature. Moreover, due to the retarded ion diffusion at lower temperatures, transformation from one intermediate phase to another can be slowed down or even prohibited, enabling the recognition of metastable phases.
It is worth to mention that the high vacuum conditions used in low temperature PL measurements accelerate the phase segregation. As a result, the I-rich emission of the first PL measurement is already pronounced, and the peak position and intensity barely changes with further illumination (see the PL evolution at different temperatures in Supplementary Fig. 7). The vacuum effect is validated by varying the chamber pressure from 50 to 0.05 mbar and measuring the PL spectra at two different locations of the same crystal. As seen in Supplementary Fig. 8, at lower pressure the I-rich emission feature exhibits an intensity increase and a peak redshift, which confirms that the high vacuum accelerates the phase segregation. The promoted segregation in high vacuum is probably related with lattice shrinkage and strain formation, as shown by a control experiment performed on a single-halide (MAPbBr3) single crystal. Supplementary Fig. 9 presents the XRD patterns of this crystal before and after four hours storage in vacuum. As shown, after vacuum storage the diffraction peaks of MAPbBr3 shift towards higher angle which points to lattice shrinkage. Moreover, after vacuum storage the diffraction peaks show an increase of FWHM, indicating the generation of strain. The strain arises from inhomogeneous shrinkage along different crystalline planes, which was reported for other types of perovskites before43.
Figure 4a shows the contour map of PL emission in a temperature range of 10 ~ 260 K under 405 nm excitation. As expected, the PL intensity increases significantly with decreasing temperature, due to less carrier-phonon coupling and lower probability of exciton dissociation. Besides, a Br-rich emission feature is seen around 620 nm in low temperature PL spectra, apart from the 690 nm I-rich emission feature. This Br-rich emission is however very mild in room temperature PL measurements, indicating that these Br-rich phases are metastable and can only be stabilized at low temperatures. With increasing temperatures, the iodide ion migration is accelerated, which enables the Br-rich phase to transform efficiently into I-rich phases. Figure 4b, c display the PL spectra at two representative temperatures of 25 K and 70 K. (for the PL spectra at other temperatures see Supplementary Fig. 10) Gazing on the emission spectral feature around 690 nm, we find it starts displaying three distinct peaks when the temperature falls below 70 K. These peaks become broader and merge into one at higher temperatures. The emission feature around 690 nm can thus be fitted with three Gaussian functions, which are labelled as peak 1, 2 and 3. The emission feature around 620 nm can be fitted by two Gaussian functions, which are labelled as peak 4 and 5. We infer that multiple emission peaks observed in low temperature PL spectra stem from intermediate phases with different halide compositions that are formed during the phase segregation. The composition of the intermediate phases can be estimated by referring to the bandgap-composition relationship of MAPb(BrxI1-x)3 \({E}_{{g;I}/{Br}}\,(x)={E}_{{g;I}}(1-x)+{E}_{{g;Br}}x-{bx}(1-x)\), where \({E}_{{g;Br}}=2.38\) eV, \({E}_{{g;I}}=1.58\) eV, b = 0.33 eV19. According to the positions of emission peak 1 ~ 5, the compositions of these intermediate phases can be estimated to be MAPb(Br0.64I0.36)3, MAPb(Br0.53I0.47)3, MAPb(Br0.41I0.59)3, MAPb(Br0.33I0.67)3 and MAPb(Br0.29I0.71)3. It is worth to mention that the emission peak position observed in PL measurement depends on the Stokes shift, halide composition, temperature, size and depth of corresponding phase, and the halide compositions of intermediate phases obtained from the emission peak position are only a rough estimation.

a Contour map showing the PL emission intensity in the temperature range of 10 ~ 230 K. b, c PL spectra at two representative temperatures of 25 K and 70 K and multi-peak fitting. The black line illustrates the raw profiles directly from the experiment and the yellow dashed lines on top of black lines are constructed from the deconvoluted profiles by summing up all the individual components. The close shape of black and yellow lines indicates good fit of these PL spectra. d Portion of emission peak 1, 2 and 3 at different temperature obtained by calculating the ratio of individual peak area over the full area of I-rich emission feature.
We further investigate the portion of different I-rich phases at different temperature by calculating the ratio of individual peak area of peak 1, 2 and 3 over the full area of I-rich emission feature. As summarized in Fig. 4d, the portion of lower energy peak 1 increases with increasing temperature, while the portion of peak 2 decreases, and that of peak 3 remains relatively stable. A similar trend is observed for the two peaks in Br-rich emission feature, where the portion of peak 4 turns larger while that of peak 5 becomes smaller with increasing temperature. This is reasonable considering that more iodide ions are required to enter the lattice and form the phase with higher content of iodide, posing a higher energy barrier for the transformation which can only be overcome at higher temperatures.
Potential application of longitudinal phase segregation
To demonstrate the potential application of obtained gradient structure, we assemble a perovskite/HTL interface by coating a mixed halide perovskite (MAPb(BrxI1-x)3 with x = 0.905) single crystal with a HTL layer (Me-4PACz) on top. Supplementary Fig. 11 shows the TRPL spectra of perovskite crystal coated with HTL in comparison to that of bare perovskite crystal. (for the fitting parameters and the effective lifetime see Supplementary Table 3) The obtained effective decay lifetime of perovskite crystal coated with HTL (419.8 ns) is shorter than the bare crystal (767.4 ns), which indicates the hole extraction from perovskite towards HTL at their interface. Besides, it can also be seen that the initial decay of perovskite with HTL is slower than the bare crystal, which is likely due to the passivation of perovskite surface traps after coating the Me-4PACz, as has been reported earlier44.
The crystal coated with HTL is then illuminated with 420 nm light continuously. TRPL spectra are captured every 300 s from the interface. As seen in Fig. 5a, the decay becomes faster with illumination. It is very likely that a gradually varying halide composition along the depth is generated due to phase segregation under illumination. This leads to the formation of a graded valence band configuration. As a result, the hole extraction towards HTL is accelerated, which causes the faster decay of emission. The enhanced hole extraction can lead to improved performance of mixed halide perovskite single crystal-based optoelectronic devices such as solar cell and detector.

a Evolution of TRPL spectra measured at the perovskite/HTL interface under continuous 420 nm illumination. The inset shows the TRPL measurement configuration. b Variation of I-rich emission intensity with illumination time in three illumination cycles. In each cycle, the crystal is subjected to continuous UV illumination for 300 s after which a PL spectrum is captured; the light is then switched off and the sample remains in dark for 15 min. Experimental data and fitting curves are shown in solid circles and lines respectively.
We further study the reversibility of the phase segregation in mixed halide perovskite single crystals and evaluate the applicability in real device. To do so, we record the PL evolution in three sequential illumination cycles. In each cycle, the crystal is subjected to continuous UV illumination for 300 s and the PL spectrum is captured subsequently; the light is then switched off and the sample remains in dark for 15 mins. The results show that the intensity of I-rich emission nearly diminishes after dark storage, indicating the reversal of PL after removal of illumination. This accords well with previous studies in which the segregated structure reversed back to the mixed halide composition upon the removal of UV excitation due to entropy-driven ion diffusion23,45. However, we also find that the segregation rate increases significantly with more cycles. Figure 5b shows the variation of I-rich emission intensity in three cycles, and the segregation rate obtained from fitting is shown for each cycle. As seen, the phase segregation rate of the third cycle is 16 times larger compared with the first cycle. This result indicates that from a microscopic view the crystal lattice structure does not reverse completely during storage in dark. It is likely that some defects, such as halide vacancies induced in the previous phase segregation do not fully migrate back to their origin positions in dark conditions and serve as easy channels for the recurrence of phase segregation. Irreversibility of phase segregation was also reported earlier under some conditions such as electron beam excitations, which destruct the lattice and hinder the reversal process42. These results suggest that repetitive illumination can shorten the required time for establishing a gradient structure, which is beneficial for the applications in real devices.
Conclusions
In summary, we unveil the occurrence of phase segregation along the depth dimension of mixed perovskite single crystal under UV excitation. We identify the presence of multiple intermediate phases through low temperature PL studies in the resultant structure, refreshing the typical view of only two defined stoichiometric phases in previous studies and providing solid evidence for existence of multiple compositions. The longitudinal phase segregation is likely driven by the defects at the crystal surface and its rate is influenced by atmosphere. The longitudinal phase segregation results in a surface layer with gradient halide composition, which can form a graded valence band and enhance the hole extraction at the interface with adjacent hole transport layer in a mixed perovskite single crystal-based optoelectronic device.
Methods
Chemicals
Methylammonium bromide (MABr, 99%) and methylammonium iodide (MAI, 99%) from Aladdin. Lead bromide (PbBr2, 99%) and N,N-Dimethylformamide (DMF, 99.9%) from Macklin. \(\gamma\)-Butyrolactone (\(\gamma\)-GBL, 99%) from GLPBIO. [4-(3,6-dimethyl-9H-carbazol-9-yl)butyl]phosphonic acid (Me-4PACz, 99%) from TCL. All chemicals are used without further purification.
Synthesis of MAPb(BrxI1-x)3 (x = 0.905) single crystal
0.112 g of MABr (99%), 1.43 g of MAI (99%), 3.67 g of PbBr2 (99.9%) are dissolved in a mixed solution of 7 mL DMF and 3 mL γ-butylene and stirred at a speed of 1000 r.p.m. at 60°C for 20 mins. A 1 mol/L mixed-halide solution is obtained after filtering using PTFE, 0.22 μm.
1.5 mL precursor solution is transferred to a 3 mL vial wrapped with aluminium foil in glove box. It is then kept in the oil bath of 75 °C for 5 hours, and heated from 75 to 110 °C at a rate of 5 °C/h for crystal nucleation, and kept for 5 h for crystal growth. MAPb(BrxI1-x)3 (x = 0.905) single crystals can be obtained by repeating this process for several cycles as required. The samples are finally taken out of the vial with tweezers and gently tapped with tissues to wipe off the residual precursors from surface of single crystals.
Structural, compositional, and morphological analysis
XRD patterns for single crystals are obtained using a PANalytical Empyrean. SEM images and EDS spectra are recorded with a Hitachi TM4000 Tabletop SEM operating at voltage from 5 ~ 15 kV. XPS spectra are obtained by thermo-VG Scientific (ESCALAB 250) system using a monochromatized Al Kα (for XPS mode) equipped with an Argon ion gun under a pressure of 5.0 × 10−7 Pa.
PL and absorption spectroscopy
PL measurements of the single crystals are performed in a reflection mode using a 1 mm diameter optical fiber with continuous 405 nm excitation and detected by a BiaoQi-QYAS series PL spectrometer. Absorption spectra are recorded by a BiaoQi optical spectrum analyzer for which the measurement range is 350 ~ 1100 nm. Confocal PL maps are acquired at room temperature by using a Leica SP8X Confocal Laser Scanning Microscope. Low temperature PL study is carried out by using a ARS CS202AE-DMX-1AL with helium cooling. The two-photon PL spectra are recorded by a CCD camera (PIXIS: 100B, Princeton Instruments) coupled with a monochromator (HRS-300-MS, Princeton Instruments), pumped at 800 nm for two-photon excitation by using a CARBIDE laser system (1030 nm, 230 fs, Light Conversion) and a TOPAS optical parametric amplifier.
Data availability
The datasets generated during the current study are available from the corresponding author upon reasonable request. Supplementary information is available in the online version of the paper.
References
Kulkarni, S. A. et al. Band-gap tuning of lead halide perovskites using a sequential deposition process. J. Mater. Chem. A 2, 9221–9225 (2014).
Akkerman, Q. A. et al. Tuning the Optical Properties of Cesium Lead Halide Perovskite Nanocrystals by Anion Exchange Reactions. J. Am. Chem. Soc. 137, 10276–10281 (2015).
Noh, J. H., Im, S. H., Heo, J. H., Mandal, T. N. & Seok, S. I. Chemical Management for Colorful, Efficient, and Stable Inorganic–Organic Hybrid Nanostructured Solar Cells. Nano Lett. 13, 1764–1769 (2013).
Endres, J. et al. Valence and Conduction Band Densities of States of Metal Halide Perovskites: A Combined Experimental–Theoretical Study. J. Phys. Chem. Lett. 7, 2722–2729 (2016).
Kerner, R. A., Xu, Z., Larson, B. W. & Rand, B. P. The role of halide oxidation in perovskite halide phase separation. Joule 5, 2273–2295 (2021).
Qiao, H. W. et al. A Gradient Heterostructure Based on Tolerance Factor in High-Performance Perovskite Solar Cells with 0.84 Fill Factor. Adv. Mater. 31, 1804217 (2019).
Wang, X. et al. Solution-Processed Halide Perovskite Single Crystals with Intrinsic Compositional Gradients for X-ray Detection. Chem. Mater. 32, 4973–4983 (2020).
Cha, M. et al. Enhancing Perovskite Solar Cell Performance by Interface Engineering Using CH3NH3PbBr0.9I2.1 Quantum Dots. J. Am. Chem. Soc. 138, 8581–8587 (2016).
Shao, W. et al. Gradient heterostructure perovskite single crystals enable the improvement of radiative recombination for scintillator application. Phys. Chem. Chem. Phys. 22, 6970–6974 (2020).
Kim, M.-c et al. Observation of Enhanced Hole Extraction in Br Concentration Gradient Perovskite Materials. Nano Lett. 16, 5756–5763 (2016).
Gan, Z. et al. Efficient Energy Funnelling by Engineering the Bandgap of a Perovskite: Förster Resonance Energy Transfer or Charge Transfer? J. Phys. Chem. Lett. 11, 5963–5971 (2020).
Tian, W., Leng, J., Zhao, C. & Jin, S. Long-Distance Charge Carrier Funneling in Perovskite Nanowires Enabled by Built-in Halide Gradient. J. Am. Chem. Soc. 139, 579–582 (2017).
Elmelund, T., Scheidt, R. A., Seger, B. & Kamat, P. V. J. A. E. L. Bidirectional halide ion exchange in paired lead halide perovskite films with thermal activation. ACS Energy Lett. 4, 1961–1969 (2019).
Elmelund, T., Seger, B., Kuno, M. & Kamat, P. V. How Interplay between Photo and Thermal Activation Dictates Halide Ion Segregation in Mixed Halide Perovskites. ACS Energy Lett. 5, 56–63 (2020).
Chen, Z. A.-O., Brocks, G., Tao, S. A.-O. & Bobbert, P. A.-O. Unified theory for light-induced halide segregation in mixed halide perovskites. (2041–1723 (Electronic)).
Draguta, S. et al. Rationalizing the light-induced phase separation of mixed halide organic–inorganic perovskites. Nat. Commun. 8, 200 (2017).
Moradi, S. et al. High-throughput exploration of halide perovskite compositionally-graded films and degradation mechanisms. Commun. Mater. 3, 13 (2022).
Suchan, K. et al. Multi-Stage Phase-Segregation of Mixed Halide Perovskites under Illumination: A Quantitative Comparison of Experimental Observations and Thermodynamic Models. Adv. Funct. Mater. 33, 2206047 (2023).
Brennan, M. C., Draguta, S., Kamat, P. V. & Kuno, M. Light-Induced Anion Phase Segregation in Mixed Halide Perovskites. ACS Energy Lett. 3, 204–213 (2018).
Peng, S. et al. Kinetics and mechanism of light-induced phase separation in a mixed-halide perovskite. Matter 6, 2052–2065 (2023).
Wang, K. et al. How Ternary Cations and Binary Halogens Stabilize Trigonal FA1–x–yMAxCsyPbI3–zBrz Perovskites: From a Single Crystal Perspective. Chem. Mater. 34, 1179–1190 (2022).
Knight, A. J. et al. Halide Segregation in Mixed-Halide Perovskites: Influence of A-Site Cations. ACS Energy Lett. 6, 799–808 (2021).
Mao, W. et al. Light-induced reversal of ion segregation in mixed-halide perovskites. Nat. Mater. 20, 55–61 (2021).
Hoke, E. T. et al. Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chem. Sci. 6, 613–617 (2015).
Funk, H. et al. In Situ TEM Monitoring of Phase-Segregation in Inorganic Mixed Halide Perovskite. J. Phys. Chem. Lett. 11, 4945–4950 (2020).
Shirzadi, E. et al. Deconvolution of Light-Induced Ion Migration Phenomena by Statistical Analysis of Cathodoluminescence in Lead Halide-Based Perovskites. Adv. Sci. 9, 2103729 (2022).
Saidaminov, M. I. et al. High-quality bulk hybrid perovskite single crystals within minutes by inverse temperature crystallization. Nat. Commun. 6, 7586 (2015).
Mao, W. et al. Visualizing Phase Segregation in Mixed-Halide Perovskite Single Crystals. Angew. Chem. Int. Ed. 58, 2893–2898 (2019).
McGettrick, J. D. et al. Sources of Pb(0) artefacts during XPS analysis of lead halide perovskites. Mater. Lett. 251, 98–101 (2019).
Dang, Z., Luo, Y., Xu, Y., Gao, P. & Wang, X.-S. Transformation and degradation of metal halide perovskites induced by energetic electrons and their practical implications. Nano Futures 5, 032001 (2021).
Lin, W.-C. et al. In situ XPS investigation of the X-ray-triggered decomposition of perovskites in ultrahigh vacuum condition. npj Mater. Degrad. 5, 13 (2021).
Meggiolaro, D., Mosconi, E. & De Angelis, F. Formation of Surface Defects Dominates Ion Migration in Lead-Halide Perovskites. ACS Energy Lett. 4, 779–785 (2019).
Wu, X. et al. Insight into the Defect Chemistry and Ion Migration in Perovskite Fabricated by Hybrid Chemical Vapor Deposition. ACS Appl. Energy Mater. 6, 12198–12205 (2023).
Yoon, S. J., Kuno, M. & Kamat, P. V. Shift Happens. How Halide Ion Defects Influence Photoinduced Segregation in Mixed Halide Perovskites. ACS Energy Lett. 2, 1507–1514 (2017).
Belisle, R. A. et al. Impact of Surfaces on Photoinduced Halide Segregation in Mixed-Halide Perovskites. ACS Energy Lett. 3, 2694–2700 (2018).
Jong, U.-G. et al. Influence of water intercalation and hydration on chemical decomposition and ion transport in methylammonium lead halide perovskites. J. Mater. Chem. A 6, 1067–1074 (2018).
Sun, K. & Müller-Buschbaum, P. Shedding Light on the Moisture Stability of Halide Perovskite Thin Films. Energy Technol. 11, 2201475 (2023).
Yang, Z. et al. Multifunctional Phosphorus-Containing Lewis Acid and Base Passivation Enabling Efficient and Moisture-Stable Perovskite Solar Cells. Adv. Funct. Mater. 30, 1910710 (2020).
Cai, J., Shen, B., Li, Z., Chen, J. & He, C. Removal of elemental mercury by clays impregnated with KI and KBr. Chem. Eng. J. 241, 19–27 (2014).
Fang, S. et al. Stability in Photoinduced Instability in Mixed-Halide Perovskite Materials and Solar Cells. J. Phys. Chem. C. 125, 21370–21380 (2021).
Gan, Z. et al. Energy Funneling in Quasi-2D Ruddlesden–Popper Perovskites: Charge Transfer versus Resonant Energy Transfer. Adv. Photonics Res. 3, 2100283 (2022).
Li, Z. et al. Beyond the Phase Segregation: Probing the Irreversible Phase Reconstruction of Mixed-Halide Perovskites. Adv. Sci. 9, 2103948 (2022).
Guo, R. et al. Degradation mechanisms of perovskite solar cells under vacuum and one atmosphere of nitrogen. Nat. Energy 6, 977–986 (2021).
Yang, B., Cai, B., Zhou, T., Zheng, X. & Zhang, W.-H. Facile and sustainable interface modulation via a self-assembly phosphonate molecule for efficient and stable perovskite photovoltaics. Chem. Eng. J. 488, 150861 (2024).
Knight, A. J. et al. Electronic Traps and Phase Segregation in Lead Mixed-Halide Perovskite. ACS Energy Lett. 4, 75–84 (2019).
Acknowledgements
Natural Science Foundation of Guangdong Province, 2024A1515011004; National Natural Science Foundation of China, 62034009, 62104266; Shenzhen Science and Technology Innovation Commission (2022A006); Shenzhen Fundamental Research Program JCYJ20200109142425294. Zhiya Dang acknowledges Dr. Ahmed L. Abdelhady for the useful tips on perovskite single crystal synthesis. The authors thank to XPS measurement in the Surface and Structure Analysis Platform of Instrumental Analysis & Research Center, Sun Yat-sen University.
Author information
Authors and Affiliations
Contributions
C.Z. conceived the idea, synthesized the samples, conducted the experiments and interpreted the data. L.Y. carried out TRPL measurements and assisted in spectra analysis. L.F. and S.Q. performed two-photon PL measurements. L.T. conducted electron beam related simulation. L.Z., R.X. and G.P. supplemented part of the data analysis and provided useful suggestions. G.P. provided part of the financial support. D.Z. supervised the research project and wrote the manuscript. All the authors contributed to the general discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Hairen Tan and Jet-Sing Lee. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Z., Dang, Z., Luo, Y. et al. Energy-band gradient structure originated from longitudinal phase segregation of mixed halide perovskite single crystal. Commun Mater 5, 174 (2024). https://doi.org/10.1038/s43246-024-00588-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00588-z
