
Abstract
Identification of genomic and epigenomic determinants of drug resistance provides important insights for improving cancer treatment. Using agnostic genome-wide interrogation of messenger RNA and microRNA (miRNA) expression, DNA methylation, single-nucleotide polymorphisms, copy number alterations and single-nucleotide variants/indels in primary human acute lymphoblastic leukemia cells, we identified 463 genomic features associated with glucocorticoid resistance. Gene-level aggregation identified 118 overlapping genes, 15 of which were confirmed by genome-wide CRISPR screen. Collectively, this identified 30 of 38 (79%) known glucocorticoid-resistance genes/miRNAs and all 38 known resistance pathways, while revealing 14 genes not previously associated with glucocorticoid resistance. Single-cell RNA-sequencing and network-based transcriptomic modeling corroborated the top previously undiscovered gene, CELSR2. Manipulation of CELSR2 recapitulated glucocorticoid resistance in human leukemia cell lines and revealed a synergistic drug combination (prednisolone and venetoclax) that mitigated resistance in mouse xenograft models. These findings illustrate the power of an integrative genomic strategy for elucidating genes and pathways conferring drug resistance in cancer cells.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

Data availability
DNA methylation, gene expression and ChIP-seq data are available at the Gene Expression Omnibus (GEO) under accession no. GSE66708. MiRNA data can be found at GEO under accession no. GSE76849. Cell line RNA-seq data can be found at GEO under accession no. GSE115384. Validation cohort no. 1 RNA-seq data from 73 of the 320 patients in the independent second cohort can be found at GEO under accession no. GSE115525. Additional RNA-seq data from validation cohort no. 1 (n = 247) can be found at GEO under accession no. GSE124824. PAX5 CHIP-seq can be found at GEO under accession no. GSE115764. Cell line ATAC-seq data can be found at GEO under accession no. GSE129066. Genotype data can be found in dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000638.v1.p1. Source data have been provided in the form of unprocessed images for all western blots (Figs. 3, 4 and 7 and Extended Data Figs. 4, 7 and 9) and for graphs (Figs. 1, 3, 4, 5 and 8 and Extended Data Figs. 4, 5, 7 and 9) in the manuscript. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Code used to generate for the polygenomic analysis and the TAP analysis can be found on GitHub at https://github.com/evanslabSJCRH/Polygenomic-Analysis. The NetBID code can be found at https://github.com/jyyulab/NetBID. Any custom code generated for our analyses not specifically listed here or in the text may be requested from W.E.E. (William.Evans@stjude.org). All R packages or other software used is given in Methods for each relevant analysis.
References
Pieters, R. et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet 338, 399–403 (1991).
Pui, C. H. & Evans, W. E. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin. Hematol. 50, 185–196 (2013).
Pui, C. H. & Evans, W. E. Acute lymphoblastic leukemia. N. Engl. J. Med. 339, 605–615 (1998).
Pui, C. H. & Evans, W. E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354, 166–178 (2006).
Clavell, L. A. et al. Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N. Engl. J. Med. 315, 657–663 (1986).
Rhen, T. & Cidlowski, J. A. Antiinflammatory action of glucocorticoids-new mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723 (2005).
Dordelmann, M. et al. Prednisone response is the strongest predictor of treatment outcome in infant acute lymphoblastic leukemia. Blood 94, 1209–1217 (1999).
Boer, M. L. D. et al. Patient stratification based on prednisolone–vincristine–asparaginase resistance profiles in children with acute lymphoblastic leukemia. J. Clin. Oncol. 21, 3262–3268 (2003).
Kaspers, G. J. et al. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood 90, 2723–2729 (1997).
Schmidt, S. et al. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ. 11(Suppl 1), S45–S55 (2004).
Inaba, H. & Pui, C. H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 11, 1096–1106 (2010).
Bachmann, P. S. et al. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 67, 4482–4490 (2007).
Song, Q.-Q., Xie, W.-Y., Tang, Y.-J., Zhang, J. & Liu, J. Genetic variation in the glucocorticoid pathway involved in interindividual differences in the glucocorticoid treatment. Pharmacogenomics 18, 293–316 (2017).
Downing, J. R. et al. The pediatric cancer genome project. Nat. Genet. 44, 619–622 (2012).
Paugh, S. W. et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 47, 607–614 (2015).
Campana, D. Minimal residual disease in acute lymphoblastic leukemia. Semin. Hematol. 46, 100–106 (2009).
Cave, H. et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer—Childhood Leukemia Cooperative Group. N. Engl. J. Med. 339, 591–598 (1998).
Pui, C. H. et al. Clinical impact of minimal residual disease in children with different subtypes of acute lymphoblastic leukemia treated with response-adapted therapy. Leukemia 31, 333–339 (2017).
Pottier, N. et al. The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J. Natl Cancer Instit. 100, 1792–1803 (2008).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Rogatsky, I., Hittelman, A. B., Pearce, D. & Garabedian, M. J. Distinct glucocorticoid receptor transcriptional regulatory surfaces mediate the cytotoxic and cytostatic effects of glucocorticoids. Mol. Cell Biol. 19, 5036–5049 (1999).
Heidari, N., Miller, A. V., Hicks, M. A., Marking, C. B. & Harada, H. Glucocorticoid-mediated BIM induction and apoptosis are regulated by Runx2 and c-Jun in leukemia cells. Cell Death Dis. 3, e349 (2012).
Lochmann, T. L., Bouck, Y. M. & Faber, A. C. BCL-2 inhibition is a promising therapeutic strategy for small cell lung cancer. Oncoscience 5, 218–219 (2018).
Pham, L. V. et al. Strategic therapeutic targeting to overcome venetoclax resistance in aggressive B-cell lymphomas. Clin. Cancer Res. 24, 3967 (2018).
Du, X. et al. Hippo/Mst signaling couples metabolic state and function of CD8α+ dendritic cells for cytotoxic T-cell priming. Nature 558, 141–145 (2018).
Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018).
Jing, D. et al. Lymphocyte-specific chromatin accessibility pre-determines glucocorticoid resistance in acute lymphoblastic leukemia. Cancer Cell 34, 906–921 (2018). e908.
Jing, D. et al. Opposing regulation of BIM and BCL2 controls glucocorticoid-induced apoptosis of pediatric acute lymphoblastic leukemia cells. Blood 125, 273–283 (2015).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Sugimura, R. et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell 150, 351–365 (2012).
Presul, E., Schmidt, S., Kofler, R. & Helmberg, A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J. Mol. Endocrinol. 38, 79–90 (2007).
Pui, C. H., Ochs, J., Kalwinsky, D. K. & Costlow, M. E. Impact of treatment efficacy on the prognostic value of glucocorticoid receptor levels in childhood acute lymphoblastic leukemia. Leuk. Res. 8, 345–350 (1984).
Irving, J. A., Minto, L., Bailey, S. & Hall, A. G. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 65, 9712–9718 (2005).
Tremblay, C. S. et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia 30, 1993–2001 (2016).
Li, Y. et al. IL-7 receptor mutations and steroid resistance in pediatric T cell acute lymphoblastic leukemia: a genome sequencing study. PLoS Med. 13, e1002200 (2016).
Delgado-Martin, C. et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia 31, 2568–2576 (2017).
Oppermann, S. et al. Janus and PI3-kinases mediate glucocorticoid resistance in activated chronic leukemia cells. Oncotarget 7, 72608–72621 (2016).
Kruth, K. A. et al. Suppression of B-cell development genes is key to glucocorticoid efficacy in treatment of acute lymphoblastic leukemia. Blood 129, 3000–3008 (2017).
Wei, G. et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 10, 331–342 (2006).
Bonapace, L. et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Invest. 120, 1310–1323 (2010).
Piovan, E. et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 24, 766–776 (2013).
Nicholson, L. et al. Quantitative proteomic analysis reveals maturation as a mechanism underlying glucocorticoid resistance in B lineage ALL and re-sensitization by JNK inhibition. Br. J. Haematol. 171, 595–605 (2015).
Chan, L. N. et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 542, 479–483 (2017).
Jones, C. L. et al. Loss of TBL1XR1 disrupts glucocorticoid receptor recruitment to chromatin and results in glucocorticoid resistance in a B-lymphoblastic leukemia model. J. Biol. Chem. 289, 20502–20515 (2014).
Jones, C. L. et al. MAPK signaling cascades mediate distinct glucocorticoid resistance mechanisms in pediatric leukemia. Blood 126, 2202–2212 (2015).
Hosono, N. et al. Glutathione S-transferase M1 inhibits dexamethasone-induced apoptosis in association with the suppression of Bim through dual mechanisms in a lymphoblastic leukemia cell line. Cancer Sci. 101, 767–773 (2010).
Kotani, A. et al. miR-128b is a potent glucocorticoid sensitizer in MLL-AF4 acute lymphocytic leukemia cells and exerts cooperative effects with miR-221. Blood 114, 4169–4178 (2009).
Han, B. W. et al. A set of miRNAs that involve in the pathways of drug resistance and leukemic stem-cell differentiation is associated with the risk of relapse and glucocorticoid response in childhood ALL. Hum. Mol. Genet. 20, 4903–4915 (2011).
Zhao, J. J. et al. Targeting the miR-221-222/PUMA/BAK/BAX pathway abrogates dexamethasone resistance in multiple myeloma. Cancer Res. 75, 4384–4397 (2015).
Spijkers-Hagelstein, J. A., Mimoso Pinhancos, S., Schneider, P., Pieters, R. & Stam, R. W. Src kinase-induced phosphorylation of annexin A2 mediates glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia 27, 1063–1071 (2013).
Spijkers-Hagelstein, J. A. et al. Elevated S100A8/S100A9 expression causes glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia 26, 1255–1265 (2012).
Aries, I. M. et al. EMP1, a novel poor prognostic factor in pediatric leukemia regulates prednisolone resistance, cell proliferation, migration and adhesion. Leukemia 28, 1828–1837 (2014).
Yang, J. J. et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood 120, 4197–4204 (2012).
Meyers, J. A., Taverna, J., Chaves, J., Makkinje, A. & Lerner, A. Phosphodiesterase 4 inhibitors augment levels of glucocorticoid receptor in B cell chronic lymphocytic leukemia but not in normal circulating hematopoietic cells. Clin. Cancer Res. 13, 4920–4927 (2007).
Zhou, M. et al. Targeting of the deubiquitinase USP9X attenuates B-cell acute lymphoblastic leukemia cell survival and overcomes glucocorticoid resistance. Biochem. Biophys. Res. Commun. 459, 333–339 (2015).
Rocha, J. C. et al. Pharmacogenetics of outcome in children with acute lymphoblastic leukemia. Blood 105, 4752–4758 (2005).
Mullighan, C. G. et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471, 235–239 (2011).
Malyukova, A. et al. FBXW7 regulates glucocorticoid response in T-cell acute lymphoblastic leukaemia by targeting the glucocorticoid receptor for degradation. Leukemia 27, 1053–1062 (2013).
Park, H. W. et al. Alternative Wnt signaling activates YAP/TAZ. Cell 162, 780–794 (2015).
Cortijo, C., Gouzi, M., Tissir, F. & Grapin-Botton, A. Planar cell polarity controls pancreatic beta cell differentiation and glucose homeostasis. Cell Rep. 2, 1593–1606 (2012).
Cabral, A. L., Hays, A. N., Housley, P. R., Brentani, M. M. & Martins, V. R. Repression of glucocorticoid receptor gene transcription by c-Jun. Mol. Cell Endocrinol. 175, 67–79 (2001).
Holleman, A. et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. NJEM 351, 533–542 (2004).
Coustan-Smith, E. et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood 117, 6267–6276 (2011).
Cheok, M. H. et al. Treatment-specific changes in gene expression discriminate in vivo drug response in human leukemia cells. Nat. Genet. 34, 85–90 (2003).
Yeoh, E. J. et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1, 133–143 (2002).
Kuan, P. F., Wang, S., Zhou, X. & Chu, H. A statistical framework for Illumina DNA methylation arrays. Bioinformatics 26, 2849–2855 (2010).
French, D. et al. Acquired variation outweighs inherited variation in whole genome analysis of methotrexate polyglutamate accumulation in leukemia. Blood 113, 4512–4520 (2009).
Paugh, S. W. et al. MicroRNAs form triplexes with double stranded DNA at sequence-specific binding sites; a eukaryotic mechanism via which microRNAs could directly alter gene expression. PLoS Comput. Biol. 12, e1004744 (2016).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Liu, Y. et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211 (2017).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Li, W. et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281 (2015).
Cheng, C. & Parzen, E. Unified estimators of smooth quantile and quantile density functions. J. Stat. Plan. Infer. 59, 291–307 (1997).
De Vore, R. A. The Approximation of Continuous Functions by Positive Linear Operators (Springer-Verlag, 1972).
Cheng, C. in Optimality, Vol. 49, Lecture Notes—Monograph Series (ed. Rojo, J.) 51–76 (Institute of Mathematical Statistics, 2006).
Maaten, L. V. D. Accelerating t-SNE using tree-based algorithms. J. Machine Learn. Res. 115, 3221–3245 (2014).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Ward, J. H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244 (1963).
Savic, D. et al. Distinct gene regulatory programs define the inhibitory effects of liver X receptors and PPARG on cancer cell proliferation. Genome Med. 8, 74 (2016).
Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016).
Khaw, S. L. et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 128, 1382–1395 (2016).
Acknowledgements
We thank the patients and families who participated in these institutional review board-approved studies. We also thank the technical staff in our labs (H. Williams, N. Atkinson, D. Maxwell, J. Hunt, B. Smart, Y. Wang, A. John and T. Lin), D. Bucci at Ohio State University, the Hartwell Center for Bioinformatics & Biotechnology at St. Jude Children’s Research Hospital and other National Cancer Institute-funded Cancer Center Shared resources that supported much of the research reported herein. We thank in particular the staff of the Animal Resources Center at St. Jude Children’s Research Hospital; T. Rogers, the veterinarian involved in our study; and M. Payton for her help in our animal studies. We also thank J. Meijerink at Princess Maxima Center for advising us on previously published mechanisms of glucocorticoid resistance. Research reported in this publication was supported in part by funds from the NIH (grant nos. R01 CA36401 (to W.E.E.), P50 GM115279 (to M.V.R., J.J.Y., C.G.M. and W.E.E.), U01 GM92666 (to M.V.R. and W.E.E.)), a St. Jude Comprehensive Cancer Center grant (no. CA21765) from the National Cancer Institute, and the American Lebanese Syrian Associated Charities. St. Jude Children’s Research Hospital received a donation from Abbvie Pharmaceuticals to support the Family Commons, a treatment and research-free space for patients and families. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
R.J.A., S.W.P. and W.E.E. conceived the study. R.J.A., E.J.B., K.R.C. and W.E.E. provided the methodology. R.J.A. and E.J.B. did the investigations. R.J.A., R.C., L.S., J.L., D.P., S.W.P., C.C., J.Y., J.C.P., D.S. and Y.G. performed the formal analysis. R.J.A., S.W.P., J.R.M., W.Y. and C.S. carried out the data curation. R.J.A. and W.E.E. wrote the original draft of the manuscript. R.J.A., S.W.P., R.C., L.S., J.L., D.C.F., C.E.L., E.J.B., W.Y., J.R.M., J.A.B, J.C.P, J.D.D., K.R.C., D.P., C.J.C., S.N., A.K., S.E.K., E.L.-L., B.D., C.S., Y.G., K.H., K.G.R., S.P., S.M.K., W.S., E.M.P., M.R.L., H.I., C.G.M., S.J., C.-H.P., C.C., D.S., J.Y., C.G., J.J.Y., M.V.R. and W.E.E. wrote, reviewed and edited the manuscript. W.E.E., J.J.Y. and M.V.R. were responsible for acquiring funds. E.J.B., K.R.C., S.J., C.H.P., J.J.Y., M.V.R. and W.E.E. supervised the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
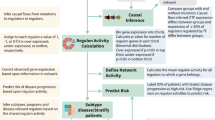
Extended Data Fig. 1 Polygenomic analysis workflow.
a, Flowchart depicting cohorts, genomic assays and detailed analysis pipeline for polygenomic analyses of multiple feature types (mRNA, miRNA, DNA methylation, SNVs, CNVs and WES mutations) as determinants of prednisolone sensitivity in patients diagnosed with acute lymphoblastic leukemia (“lm” = linear model). b, Table describing age, race, gender and molecular subtype of discovery cohort (n = 225 patients) from polygenomic analysis. The P-values represent differences between the discovery cohort enrolled on the two clinical trials (Fisher’s Exact Test p-value; Total 15 and Total 16).
Extended Data Fig. 2 Validation of gene expression signature, relation to treatment response and WES variant connectivity.
a, Connectivity between polygenomic signatures for mutation (n = 227 mutations) and mRNA expression (n = 254 mRNA probes; Fisher’s Exact Test clustering p-values and linear model p-value for connectivity). b, Characteristics of WES mutations with linear model p-value < 0.05 vs. LC50. (SIFTcat Del = Deleterious and Tol = Tolerated). c, RNA sequencing of ALL cells from St. Jude Total XVI patients (n = 73 patients; validation cohort #1; Fisher’s Exact Test clustering p-value) clustered with gene expression signature from discovery cohort analysis. d, Publicly available DCOG/COALL patient cohort (n = 145 patients; validation cohort #2; Fisher’s Exact Test clustering p-value) clustering with gene expression signature from patient discovery cohort. e, Clustering of gene expression vs. LC50. Red denotes genes correlated with LC50 or minimal residual disease (MRD) in positive direction. Blue denotes genes correlated in negative direction with LC50 or MRD. Clustering performed to show concordance of genes discriminating LC50 or MRD. f, Boxplot denoting Prednisolone LC50 in patients from discovery cohort with the major ALL molecular subtypes. Red circles denote prednisolone resistant patients, green denotes sensitive patients, and black denotes intermediate sensitivity. Upper line is the upper quartile (75%) middle line is the median and lower line is lower quartile (25%) boundary for Prednisolone LC50.
Extended Data Fig. 3 Gene level integration of genomic variants related to prednisolone resistance.
Each panel depicts -log10 p-values for the association of the indicated genomic feature with prednisolone LC50, and the aggregated gene-level linear model p-value based on all genomic features is shown for each gene at the top right. Red triangles represent mRNA probes within the gene body, orange diamonds depict copy number variants, blue squares are DNA methylation probes, grey circles SNVs, and purple circles miRNAs within 50 kb upstream or downstream of gene region (n = 203 patients). a, SMARCA4, a component of the SWI/SNF complex, has been previously linked to glucocorticoid resistance in pediatric ALL19. b, NLRP3 encodes NALP3, an inflammasome component that activates caspase 1, and has been previously associated with ALL resistance to glucocorticoids15. c, PTTG1IP encodes the pituitary tumor-transforming gene 1 protein-interacting protein that interacts with the proto-oncogene PTTG1 (also known as securin). d, CELSR2 is a G-protein coupled receptor involved in non-canonical Wnt signaling. PTTG1IP and CELSR2 are novel genes from the current study associated with glucocorticoid resistance.
Extended Data Fig. 4 CELSR2 knockdown blunts glucocorticoid responsiveness of 697 cells and increases sensitivity to venetoclax.
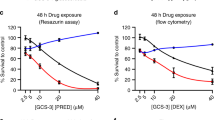
a, Volcano plot for untreated CELSR2 knockdown ALL cell lines vs. non-target control in 697 cell line (n = 3 independent experiments; linear model p-value). Left side of plot depicts genes with reduced expression in CELSR2 knockdown cells and genes to the right had increased in expression in CELSR2 knockdown cells. b, Volcano plot of gene expression after 24 h of prednisolone treatment of CELSR2 knockdown vs. non-target control ALL cells (697; n = 3 independent experiments; linear model p-value). c, Dose-response plot (mean ± S.D.; n = 3 independent experiments) of two shRNA constructs vs non-targeting control and un-transduced NALM-6 leukemia cell line. d, CELSR2 (n = 3 independent experiments) e, NR3C1 (n = 4 independent experiments) f, BCL2 (n = 5 independent experiments) g, BIM (n = 4 independent experiments) and h, Bim/Bcl2 protein expression (mean ± S.D; n = 4 independent experiments; two-tailed t-test p-values; * = p < 0.05,** = p < 0.01, *** = p < 0.001, **** = p < 0.0001) in NALM-6 cells comparing controls (NTC; solid bars) to CELSR2-knockdown (shCELSR2) either prior to prednisolone treatment (0HR) or after 24 hr prednisolone treatment (24HR). i, The 75 most highly upregulated (top) or downregulated (bottom) genes after 24 h treatment with 10 µM prednisolone. Blue and green bars depict mRNA expression (mean ± S.D.; n = 3 independent experiments) in 697 cells transfected with non-target control vector and gold bars depict cells expressing shRNA for CELSR2 knockdown.
Extended Data Fig. 5 Venetoclax and prednisolone synergize in primary ALL with low CELSR2 expression and CELSR2 knockdown in cell lines disregulation of Bim/Bcl2 axis.
a, Response surface model plot of cytotoxicity from prednisolone plus venetoclax at concentrations indicated for the 697 leukemia cell line transduced with non-targeting control vector. b, Response surface model plot for the 697 leukemia cell line transduced with CELSR2 shRNA knockdown vector (for a and b individual points represent n = 3 independent experiments performed in technical duplicate; response surface model two-tailed t-test p-value). The alpha (α) value indicates antagonism < 0 or synergy > 0 with greater synergy from higher value. P-value describes overall model fit. Individual plots of prednisolone effect (mean ± S.D.; n = 3 independent experiments) c, NALM-6 and d, 697 leukemia cell lines at one concentration of venetoclax (mean ± S.D.; n = 3 independent experiments). Black lines are non-targeting control cells and red lines are CELSR2 knockdown cells, dashed lines indicate predicted additivity curve fit based on single drug treatments; data left of the dashed lines represent additivity/synergy. Solid lines represent fit of measured values. e, Venetoclax sensitivity of independent cohort of patients (n = 96 ALL patients) grouped based on prednisolone sensitivity (LC50) f, Bcl2 expression associated with sensitivity to venetoclax (n = 81 ALL patients) g, Primary ALL cells from patients (n = 6 patient samples) and human leukemia cell lines assessed for additivity/synergy with prednisolone and venetoclax (for all box plots horizontal bars depict medians and boxes represent 25th and 75th percentiles, whiskers represent ± 1.5x IQR; linear model p-values). h, mRNA expression (n = 1 experiment run in technical triplicate) of CELSR2 in patient samples assessed for synergy.
Extended Data Fig. 6 NetBID identifies regulatory nodes of prednisolone resistance.
a, Enrichment of previously reported resistance genes (n = 40 genes and miRNAs; Wilcoxon two-tailed p-value) in NetBID results. b, Heatmap of top 48 NetBID-predicted drivers (‘symbol’_’regulon size’) are ranked by integrated NetBID p-value. Left: color-coded by z-score and labeled by p-value of NetBID results in TOTXVI, TOTXV, and combination (Comb); Right: differential expression of each driver itself, color-coded by z-score and labeled by signed fold-change in TOTXVI, TOTXVI and combination (Comb; (Stouffer’s combined Bayesian generalized linear model “NetBID” p-value; n = 203 patients). c, CELSR2 regulon from B-ALLi (n = 399 genes). Legends of node and edge follow Fig. 6c. d, Enrichment of NetBID-inferred CELSR2 regulon (n = 399 genes) in differentially expressed genes of CELSR2 knockdown vs. NTC in Nalm-6 human ALL cell lines (n = 222 genes; Wilcoxon two-tailed p-value) upon prednisolone treatment for 24 hr (top) Blue lines inside the box indicate the down-regulation of CELSR2 itself, labeled p-value and signed fold-change. e, Enrichment of previously reported resistance genes (n = 40 genes and miRNAs; Wilcoxon two-tailed p-value) in differentially expressed genes of CELSR2 knockdown vs. NTC in NALM-6 ALL cell lines without prednisolone treatment.
Extended Data Fig. 7 CELSR2 mRNA expression is related to PAX5 expression in primary ALL cells.
a, Subnetwork (top 50 interactions ranked by mutual information) of PAX5 and CELSR2 from B-ALLi (n = 185 patients). Legends of node and edge follow Fig. 6c, except that nodes in green are those in top 48 drivers (Fig. 6b). b, CELSR2 expression positively correlates with PAX5 expression in primary acute lymphoblastic leukemia cells (n = 203 patients; black line represents regression fit associated with linear model p-value and Rsq). c, Open chromatin regions defined by ATAC-seq (n = 2 independent experiments) in three sensitive and three resistant human leukemia cell lines and H3K27 acetylation from ENCODE in upstream 5′ region of CELSR2. ENCODE binding site in GM12878 lymphoid cells for PAX5 and CHIP-seq peaks from NALM-6 cells for PAX5 binding are indicated at bottom of the plot. d, PAX5 (**** = 3.5 × 10−5) e, CELSR2 (*** = 3.0 × 10−4) f, NR3C1(**** = 3.2 × 10−5) protein expression (mean ± S.D.) in NALM-6 leukemia cell lines stably expressing shRNA knockdown constructs targeting PAX5 (for d-f n = 5 independent experiments; two-tailed t-test p-values).
Extended Data Fig. 8 Single cell transcriptomics defines distinct expression signatures in primary B-ALL cells.
a, Clustering of bone marrow cells from a prednisolone sensitive patient (n = 2,427 control cells; n = 924 treated cells) based on top 1000 most highly expressed genes b, Identification of distinct cell populations in a prednisolone sensitive patient CD19 + B-cells (red), CD3E + T-cells (blue), ALAS2 + Erythrocytes (purple) and CD14 + Macrophages (green) c, Control vs. treatment for all cell clusters in prednisolone sensitive patient (red = control, blue = treated) d, Clustering of bone marrow cells from a prednisolone resistant patient (n = 686 control cells; n = 759 treated cells) based on top 1000 most highly expressed genes e, Identification of distinct cell populations in a prednisolone resistant patient CD19 + (red) and CD3E + T-cells (blue) f, Control (C) vs. treatment (T) for all cell clusters in prednisolone resistant patient (red = control, blue = treated).
Extended Data Fig. 9 Chromatin status in glucocorticoid sensitive and resistant human ALL cell lines, and perturbation of non-canonical WNT signaling by reduction of CELSR2 expression.
a, ATAC-seq for six human leukemia cell lines, three prednisolone sensitive and three resistant cell lines depicting open chromatin in the region upstream of NR3C1 (n = 2 independent experiments). b, H3K27Ac data from ENCODE (black box) showing lymphocyte regulatory region in GM12878 cell line (pink) c, RefSeq NR3C1 transcripts d, ENCODE transcription factor binding sites for PAX5, NR3C1, TEAD4 and non-canonical Wnt effectors (NFATC1 and AP-1 [JUN and FOS]) e, Representative western blot and f, Barplot (mean ± S.D.; n = 3 independent experiments; two tailed t-test p values) depicting total cellular protein expression of signaling components from planar cell polarity and Ca2+/NFAT non-canonical Wnt signaling protein CELSR2 knockdown vs. control cells with or without 10 µM prednisolone treatment for 24 hr. g, Representative western blot and h, Barplot (mean ± S.D.; n = 3 independent experiments; two-tailed t-test p-values) depicting cytoplasmic protein expression of signaling components from planar cell polarity and Ca2+/NFAT non-canonical Wnt signaling protein CELSR2 knockdown vs. control cells with or without 10 µM prednisolone treatment for 24 hr.
Supplementary information
Supplementary Information
Supplementary Table 5 and Notes.
Source data
Rights and permissions
About this article

Cite this article
Autry, R.J., Paugh, S.W., Carter, R. et al. Integrative genomic analyses reveal mechanisms of glucocorticoid resistance in acute lymphoblastic leukemia. Nat Cancer 1, 329–344 (2020). https://doi.org/10.1038/s43018-020-0037-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-020-0037-3
This article is cited by
-
CRISPR screening in hematology research: from bulk to single-cell level
Journal of Hematology & Oncology (2023)
-
Dasatinib overcomes glucocorticoid resistance in B-cell acute lymphoblastic leukemia
Nature Communications (2023)
-
NetBID2 provides comprehensive hidden driver analysis
Nature Communications (2023)
-
Pharmacotypes across the genomic landscape of pediatric acute lymphoblastic leukemia and impact on treatment response
Nature Medicine (2023)
-
An alternative CYB5A transcript is expressed in aneuploid ALL and enriched in relapse
BMC Genomic Data (2022)
