
Abstract
The contributing genetic factors of vertigo remain poorly characterized, particularly in individuals of non-European ancestries. Here we show the genetic landscape of vertigo in an Asian population-based cohort. In a two-stage genome-wide association study (Ncase = 6199; Ncontrol = 54,587), we identify vertigo-associated genomic loci in DROSHA and ZNF91/LINC01224, with the latter replicating the findings in European ancestries. Gene-based association testing corroborates these findings. Interestingly, both genes are enriched in cerebellum, a key structure receiving sensory input from the vestibular system. Subjects carrying risk alleles from lead SNPs of DROSHA and ZNF91 incur a 1.74-fold risk of vertigo than those without. Moreover, composite clinical-polygenic risk scores allow differentiation between patients and controls, yielding an area under receiver operating characteristic curve of 0.69. This study identified novel genomic loci for vertigo in an Asian population-based cohort, which may help identifying high risk subjects and provide mechanistic insight in understanding the pathogenesis of vertigo.
Similar content being viewed by others
Introduction
Vertigo is a severe subtype of dizziness characterized by an illusion of movement such as a sense of rotation, linear displacement, or tilt. The vestibular nerves afferent from the inner ear structures such as otolith organs and the semicircular canals maintain a balanced tonic rate of firing into the brainstem vestibular nuclei. Any asymmetrical involvement of the baseline activity in any location in the peripheral or central vestibular pathways may lead to vertigo. The lifetime prevalence of significant dizziness ranges between 17 and 30%, and for vertigo attributed to peripheral or central vestibular diseases, 3–10%1,2. As a common presenting complaint in primary care clinics or emergency departments, vertigo not only causes great suffering to the affected subjects but also leads to a considerable socioeconomic burden3 However, the underlying pathophysiology of vertigo has not been fully elucidated.
Studies have suggested familial aggregation in some common causes of vertigo such as benign paroxysmal positional vertigo (BPPV)4, vestibular migraine5, or Meniere’s disease6, but in certain types of vertigo such as vestibular neuritis, there is no apparent family history. In addition, there is significant inter-ethnic difference in the prevalence of certain types of vertigo, such as Meniere’s disease7. Little is known regarding how genetic factors may contribute to the predisposition of developing these individual types of vertigo. A genome-wide linkage scan in 20 multigenerational families demonstrated the linkage of familial benign recurrent vertigo to 22q12; however, remarkable heterogeneity was also noted8. A study including 131 patients with vestibular neuritis and 2609 controls of European ancestry showed genome-wide associations with vestibular neuritis in 4 regions functionally relevant to virus hypothesis and insulin metabolism9; however, the case number is limited and the results await independent replications. A recent large-scale genome-wide meta-analysis combining data from Iceland, the UK, the US, and Finland for the first time identified six variants to be associated with vertigo, which provided the genetic basis of the disease and implicated potential pathogenesis10. However, whether these results could be extrapolated to subjects of non-European ancestry remains unclear.
To identify the genetic landscape of vertigo in Asians, we used a genome-wide association study (GWAS) for participants recruited from the Taiwan Biobank11. By identifying susceptible genes for self-reported vertigo in Han Chinese residing in Taiwan, this study provides novel mechanistic insights for a better understanding of the inter-ethnic similarities and differences of the pathophysiology of vertigo.
Results
Baseline characteristics of the subjects
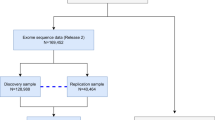
After excluding subjects with kinship relation and comorbid disorders with vertigo, including self-reported migraine, inflammatory bowel disease, peptic ulcer, and gastroesophageal reflux disorder, 3735 subjects with vertigo and 31,736 controls were enrolled in the discovery cohort, and 2464 subjects with vertigo and 22,851 controls were enrolled in the replication cohort. In total, 6199 subjects with vertigo and 54,587 controls were eligible for combined analysis (see Supplementary Fig. 1). Demographic characteristics of participants are shown in Supplementary Table 1. The female-to-male ratio was higher, and the age was older in the subjects with vertigo than in the controls.
Quality control
After applying stringent quality control (QC) criteria, we obtained 10,546,339 variants with an average call rate of 99.25% ± 0.9%. After correction for PC1–PC10, sex, age, and unbalanced case-control ratios (with Scalable and Accurate Implementation of GEneralized mixed model (SAIGE12), quantile–quantile plot of results from logistic regression P value showed that the value of the genomic inflation factor lambda calculated based on the 50th percentile (median) was 1.009 for the discovery cohort and 0.99 for the replication cohort (Supplementary Fig. 2). The polygenicity in our cohort is 0.7726.
GWAS results
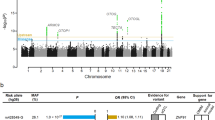
The two-stage genome-wide analysis using imputation data identified one replicable genomic locus with multiple SNPs reaching GWAS significance within or near Drosha Ribonuclease III (DROSHA) and Chromosome 5 Open Reading Frame 22 (C5orf22) at both discovery and replication cohorts after correction for age, sex, PC1–PC10, and case-control imbalance (with SAIGE12) (Fig. 1 and Table 1). The lead SNP in the combined analysis was rs6859527 (odds ratio (OR), 0.704; 95% confidence interval (CI), 0.666‒0.744; P = 2.90 × 10−33). In the discovery cohort, there were two additional significant loci at the Zinc Finger Protein 91 (ZNF91) and Sodium Voltage-Gated Channel Alpha Subunit 5 (SCN5A), which, however, did not reach GWAS significance in the replication cohort. Nevertheless, in the combined analysis, multiple SNPs within or near ZNF91 were significantly associated with vertigo (lead SNP rs295402 (OR 1.173, 95% CI 1.130‒1.218; P = 2.03 × 10−15). The lead SNP (rs7645178) in SCN5A in the combined analysis was of only suggestive GWAS significance (OR 1.101, 95% CI 1.059‒1.145; P = 1.74 × 10−6). In the combined analysis, there was also a significant locus at Brain Abundant Membrane Attached Signal Protein 1 (BASP1) (lead SNP rs1234168883; OR 1.428, 95% CI 1.268‒1.609; P = 1.31 × 10−8). However, the association of BASP1 with vertigo diminished after conditional analysis on the lead SNP in DROSHA. The regional association plots of these loci are demonstrated in Fig. 2. We further calculated the attributable risk of lead SNPs in these significant loci (i.e., rs6859527, rs6450850, rs295402). We found that in comparison with subjects who did not carry any of the risk alleles, the odds ratio of having vertigo in subjects carrying five risk alleles was 1.495 (95% CI 1.249‒1.789; P = 1.16 × 10−5) and that in subjects carrying six risk alleles was 1.740 (95% CI 1.442‒2.101; P = 7.93 × 10−9). Because rs6859527 and rs6450850 are in high LD (r = 1), which may affect the burden when calculating the number of risk alleles, we also analyzed the results with only the two SNPs without LD (i.e., rs6450850 and rs295402). Subjects who carried three or four risk alleles of these two SNPs had a higher risk of having vertigo compared to those without any risk alleles (OR: 1.442 (P = 0.0001) and 1.740 (P < 0.0001), respectively).

The horizontal axis shows the chromosomal position, and the vertical axis shows the significance of tested markers. The threshold for genome-wide significance (P < 5 × 10−8) is indicated by a gray dash line.

Each dot represents a single-nucleotide polymorphism (SNP) derived from the fine-mapped imputation data. The horizontal axis gives the genomic coordinate, and the vertical axis the significance level (−log10 P value). The top SNP for each locus is marked with a purple diamond (CRCh38/hg19). SNPs are colored based on their correlation (r2) with the labeled lead SNP according to the legend. The solid blue line shows the recombination rate from 1000 Genomes EAS data (right vertical axis). The gray dashed line corresponds to P = 5 × 10−8. Figures were obtained from LocusZoom.
Gene-based association testing
Gene-based association testing used the mean association signal from all SNPs within each gene, accounting for LD. Gene-based analysis as computed by MAGAMA using summary statistics of our GWAS identified two loci, which was driven by the lead SNPs of ZNF91 and DROSHA. The Q–Q plot of the gene-based test computed by MAGMA13 is shown in Supplementary Fig. 3.
Cross-population meta-analysis
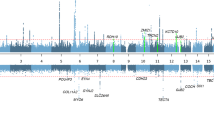
We conducted a cross-population meta-analysis for the European GWAS10 and ours using METAL14. The Manhattan plot of the meta-analysis is shown in Fig. 3. The lead loci of the European GWAS and ours remained the most significant loci in the meta-analysis. While not all the lead SNPs in the European study were replicable in our GWAS, all these variants except rs7130190, which was unavailable in our dataset, were significantly associated with vertigo (Table 2). Moreover, the SNP-based heritability differed between the European and Taiwanese cohorts, and the transethnic genetic-effect correlations were significantly different from 1, showing that although the phenotypes have clear genetic overlap, the per allele effect sizes differ significantly between the two populations (Supplementary Table 2).

Manhattan plot of the meta-analysis of the European and Taiwanese vertigo GWAS.
Association between PRS and vertigo risk
We calculated three sets of PRS, including one derived from genome-wide data and another two sets composed of the lead SNPs of the significant loci in the current study and the previous European GWAS. By combining the PRS with the 10 principal components and the clinical demographics, including age, sex, and family history of vertigo, the AUC for discriminating patients from controls was 69.00% (95% CI = 67.95–70.04%) using the LDPred model. The PRS-family history (FH) based on a logistic model (PRS-FHlog) had a similar performance (AUC 69.09%, 95% CI: 68.04–70.13%). The estimated prevalence ratio was 1.48 based on the comparison of subjects with 5-th (the highest) versus those with 1-th (the lowest) PRS quintile. This suggested that individuals with the highest PRS quintile were 1.48 times as likely as those with the lowest PRS quintile to have vertigo in the Taiwanese population. Similar to the performance of genome-wide PRS, the model using the PRS derived from the three lead SNPs identified in this study (i.e., rs6450850, rs6859527, and rs295402) combined with clinical variables had an AUC of 69.24% (95% CI = 68.19–70.29%). In contrast, the model using the PRS derived from the six lead SNPs identified from the European GWAS combined with clinical variables had an AUC of 66.53% (95%CI = 65.47–67.58%). Moreover, to improve the cross-ethnicity prediction accuracy of PRS, we applied PRS-CSx “meta” function to borrow information from the European GWAS on vertigo10. By combining the PRS obtained from PRS-CSx with the 10 principal components and the clinical demographics, including age, sex, and family history of vertigo, the AUC for discriminating patients from controls was 69.58%.
SNP heritability and genetic correlation between vertigo and migraine
We estimated the SNP-based heritability (h2) of vertigo at 3.68% (SE 0.8%) on the observed scale and 10.08% (SE 2.18%) on the liability scale. We estimated the genetic covariance between vertigo and migraine on the liability scale was 3.64% (SE 2.01%) and the genetic correlation of vertigo and migraine was 26.03% (SE 13.99%, P = 0.063).
eQTL and enrichment analysis
eQTL analysis using the GTEx online platform15 (accessed on December 1, 2023) found that the lead SNP rs295402 in ZNF91 has significant eQTLs on LINC01224, CTB-176F20.3, ZNF91. However, rs6859527 and rs6450850 in DROSHA did not have significant eQTLs. To gain more biological insights from the vertigo-associated genes from multi-ethnic data, we also included the results of the meta-analysis of the current study and the European GWAS. The eQTLs of the lead SNPs and genes of the meta-analysis are summarized in Supplementary Table 3. Interestingly, the lead variants in ZNF91, TECTA, and OTOP1/TMEM128 also had significant eQTLs on the genes themselves. Taking the missense variant rs612969 in TECTA for example, single tissue analysis found that the eQTL of rs612969 was most significant in thyroid, cerebellum, and caudate nucleus; multi-tissue eQTLs using cross-tissue meta-analysis also showed that rs612969 in these tissues had high posterior probability of significant eQTL (m-value ≥ 0.99) (GTEx Analysis Release V8; dbGaP Accession phs000424.v8.p2). Furthermore, we explored the eQTLs of the combined European and Taiwanese GWAS data on GTEx v8 and BRAINEAC brain tissues using the FUMA platform. A total of 408 significant entries were identified on C2orf72, OTOGL, TECTA, TMEM128, and ZNF91, respectively, with more than two-third of them being significant on cerebellum or cerebellar hemispheres (Supplementary Data 1).
MAGMA gene-property analyses implemented in FUMA combining the significant genes in Taiwanese and European cohorts using the summary statistics of the meta-analysis did not demonstrate significantly enriched tissues. However, bulk tissue gene expression analysis on GTEx demonstrated that DROSHA, ZNF91, and TECTA are enriched in the brain, particularly in the cerebellum and cerebellar hemisphere (Fig. 4 and Supplementary Fig. 4). In addition, the OTOG, OTOGL, and TECTA are enriched in pituitary gland while ARMC9 and OPOT1 are not specifically enriched in brain tissues (Supplementary Fig. 4). As GTEx did not include data from vestibules, we also explored the gene expression of the significant genes in the vestibules using data from umgear.org and SHIELD. Most of the genes except Znf91 are expressed in vestibular sensory epithelium or vestibular sensory neurons in mice (see Supplementary Table 3 for details).

The bulk tissue gene expression data was obtained from the GTEx platform. Note that both DROSHA, ZNF91 and TECTA were most abundantly expressed in cerebellum and cerebellar hemisphere. TPM stands for transcript per million.
Discussion
This study demonstrated the genetic landscape of vertigo in the general population in Han Chinese residing in Taiwan. To the best of our knowledge, this is also the first that identified replicable genomic loci associated with vertigo in Asians. We identified DROSHA and ZNF91 to be significantly associated with vertigo, and another two loci SCN5A and OTOGL with suggestive GWAS significance. Notably, ZNF91 and OTOGL were recently discovered to be associated with vertigo in an European genome-wide meta-analysis10, suggesting a shared genetic susceptibility to vertigo across ethnicities. Supporting this, our cross-population meta-analysis confirmed that all the previously reported genes in the European study were significantly associated with vertigo. The composite prediction model combining PRS derived from the three lead SNPs and clinical features could differentiate patients from controls with nearly 70% accuracy, not inferior to that of composite scores from genome-wide PRS and clinical features, suggesting that SNPs other than the lead SNPs did not substantially contribute to the risk of vertigo in the general population, which was also supported by the finding of the low SNP-based heritability. In contrast, subjects carrying all the 6 risk alleles of the lead SNPs had a 1.74-fold risk to have vertigo than the noncarriers.
The implicated genes from the lead SNPs may provide novel mechanistic insights to better understand the pathophysiology of the common types of vertigo. Gene expression analysis demonstrated that both DROSHA and ZNF91, as well as TECTA identified in European populations, were enriched in the cerebellum and cerebellar hemispheres, the key structures responsible for the maintenance of equilibrium and coordination of movements. Vertigo is a common manifestation of cerebellar dysfunction and activation of cerebellum on functional neuroimaging has been consistently identified in subjects with vertigo16. However, how these genes may affect cerebellar function is unclear. In addition, most of the vertigo-associated genes in this and previous studies were expressed in the vestibules, which may also provide mechanistic explanations of vertigo.
DROSHA gene encodes a ribonuclease III double-stranded RNA-specific ribonuclease which participates in the initial step of microRNA (miRNA) biosynthesis. Drosha cleaves the stem-loop structure from the primary miRNA (pri-miRNA) in the nucleus, yielding the precursor miRNA (pre-miRNA), which is then exported to the cytoplasm and processed by the cytoplasmic Dicer to generate mature miRNAs. In patients with idiopathic sudden sensorineural hearing loss with some of them having vertigo, Dicer is downregulated and correlated with Drosha17. In a study that examined miRNA expression in inner ear fluid (i.e., endolymph and perilymph) extracted from human temporal bones in patients with BPPV, some inner ear fluid-specific miRNAs were identified18. As the homeostasis of inner ear fluid is associated with vestibular symptoms, these miRNAs might participate in regulating vestibular functions. In fact, in a rat study for unilateral vestibular deafferentation, miRNAs 218a-5p, 219a-5p, and 221-3p were found to regulate vestibular compensation19. MicroRNA-219a-5p was also found to modulate the expression of CaMKIIγ and protein kinase C to facilitate vestibular compensation in acute vertigo20. In addition, the miR-183 family gene cluster including miR-183, miR-96, and miR-182 was related to vestibular function21. In a study using next-generation sequencing, some miRNAs were found highly expressed in cochlear and vestibular sensory epithelium, with miR-182 being the most highly expressed one22. How the Drosha protein may interact with these vestibule-associated microRNAs remains to be explored. On the other hand, one of the most common type of vertigo—vestibular migraine could also be functionally relevant to Drosha, as some circulating miRNAs have been found to be dysregulated in migraine23,24. Further studies are needed to evaluate how the miRNA biosynthesis regulated by Drosha, particularly in the cerebellum or vestibular organs, contributes to the pathogenesis of vertigo.
ZNF91 exerts its function as a transcription factor by binding and repressing SINE-VNTR-Alu (SVA) retrotransposons elements25. ZNF91, together with multiple zinc finger proteins, is involved in a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway: hsa05168 Herpes simplex virus 1 (HSV-1) infection26. ZFN91 was found to be a transcription repressor of the human FcγRIIB gene promoter27. FcγRIIB, as one of the gamma receptors for the Fc portion of IgG, plays an important role in regulating antiviral immune responses. A recent study suggested that a polymorphism in FCGRIIB may be associated with the host ability to suppress HSV-1 reactivation by increasing the expression of the IgG3 subclass28. Interestingly, reactivation of latent HSV-1 infection has been considered a culprit of vestibular neuritis29,30, an important cause of acute vertigo. One GWAS of vestibular neuritis also implicated the potential involvement of the host factor for HSV-1 replication by demonstrating an association of nuclear receptor subfamily 3 group C member 2 (NR3C2) with vestibular neuritis9, while NR3C2 has been shown to have antiviral activity against HSV-131. Hence, it is plausible that the significant association between ZNF91 and vertigo in our study might be in part attributed to its potential modulatory effect against HSV-1 infection.
Although cross-population fine mapping in GWAS presents several challenges, primarily due to the genetic diversity found across different populations such as variations in allele frequencies and patterns of linkage disequilibrium, our meta-analysis demonstrated that the missense variant rs612969 identified in European study remained the most significant variant in TECTA combining the data from different ethnicities. In addition to the proposed function in inner ears, particularly the effect on age-related hearing impairment inferred by the European study10, our analysis suggests that TECTA may also contribute to vertigo pathogenesis by affecting cerebellum or caudate nucleus, as indicated by the significant eQTL of the missense variant rs612969 on TECTA in the cerebellum and caudate nucleus as well as the significant enrichment of TECTA in cerebellum.
The loci with suggestive significance of association with vertigo might also be functionally relevant to the pathogenesis of vertigo. SCN5A encodes the sodium channel NaV1.5 that has an important role in controlling heart rate. The mutations of SCN5A have been linked to various forms of cardiac arrhythmia and cardiomyopathy32, which are potential causes of dizziness or vertigo. Another possibility that SCN5A may be involved in the pathogenesis of vertigo is that NaV1.5 has been identified in vestibular ganglion33,34 and the dysfunction of the channel might lead to vestibular symptoms. OTOGL (Otogelin Like) encodes proteins responsible for inner ear function and the mutations in OTOGL have been found to lead to sensorineural hearing loss with or without vestibular hyporeflexia35,36. In the European GWAS10, the association of OTOGL with vertigo is mainly driven by BPPV, suggesting that OTOGL may mediate vertigo via inner ear dysfunction. Nevertheless, these links were purely speculative, and the function of these vertigo-associated genes may not be specific to vertigo but may related to dizziness in a broad sense.
Although our findings provide genetic evidence to support prevailing theories of vertigo pathogenesis, there were limitations in our study. First, the self-reported vertigo in this study is broadly defined and may include multiple potential etiologies. Employing the International Classification of Diseases (ICD)-defined phenotype as a sensitivity analysis could help alleviate concerns about phenotype definition. Unfortunately, the Taiwan Biobank did not provide corresponding ICD codes for the participants and applying the linkage for the anonymized data between the Taiwan Biobank database and the Taiwan’s National Health Insurance Research Database (NHIRD) is complicated and could not be done within a reasonable timeframe. In the current study, the various functions of the identified genes suggest that these variants might be associated with some of the most common forms of vertigo with different pathogenic mechanisms or nonspecifically with dizziness. Further studies are needed to investigate whether these genes are associated with individual vestibular disorders with ICD-based phenotyping. Nevertheless, despite the heterogeneity of the etiologies and susceptible genes of vertigo, our PRS models demonstrated that subjects who have higher PRS are more likely to have vertigo than those with lower PRS, suggesting that the susceptibility to vertigo in individuals is intricately modulated by at least selected susceptible genes. However, the PRS study is heavily influenced by the phenotype definition. Because the phenotype defined in our study may be different from that defined based on ICD codes, it might not be optimal to compare the performance of PRS based on self-reported vertigo with that based on ICD code-defined vertigo for predicting self-report vertigo in our cohort. The performance of these PRS could be interpreted with caution. Furthermore, while we have successfully replicated part of the findings of the European GWAS, not all the risk variants were replicable in our study. As allele frequencies and linkage disequilibrium patterns differ between ethnicities, future multi-ethnic studies are needed to explore shared and ethnicity-specific variants associated with vertigo. In addition, future studies are needed to elucidate the association of these susceptible genes with specific phenotypes to better understanding for the pathogenesis of vertigo.
Methods
Inclusion and ethics
This study was approved by the Institutional Review Boards of Taipei Veterans General Hospital (2013-11-001AC) and Academia Sinica, Taiwan (AS-IRB02-10836). The study was conducted according to the principles expressed in the Declaration of Helsinki. All collected information was de-identified before statistical data analysis. The corresponding authors had full access to all the data in the study and had final responsibility for the decision to submit for publication. All ethical regulations relevant to human research participants were followed.
Study participants and data collection
This study was a two-stage case-control GWAS, including a discovery cohort and a replication cohort, followed by a combined analysis of both cohorts to examine the significance of the validated single-nucleotide polymorphisms (SNPs). All participants were unrelated and of Han Chinese ancestry recruited from the Taiwan Biobank. The Taiwan Biobank is a general population-based public health research resource established by the National Center for Genomics Medicine, Academia Sinica, Taiwan, which recruited cancer-free adults aged between 30 and 70 years11. Taiwan Biobank is the largest publicly available genetic database of individuals with East Asian ancestry that provides adequate coverage of genetic diversity across all Han Chinese11. Participants had to fill out a structured questionnaire that included questions on personal information and medical history. The structured questionnaire contained two questions to specifically to inquire the personal or family history of vertigo. The questions were (1) Have you ever experienced vertigo in your life? and (2) Do your family has history of vertigo? Participants reporting history of vertigo were defined as cases. Participants without self-reported vertigo but with family history of vertigo were excluded from both cases and controls. Participants without self-reported vertigo nor family history of vertigo were used as potential controls and subjected to the following quality control procedures.
The initial sample used in this study contains a total of 103,332 individuals who had been genotyped with TWB2 array. Participants with no phenotype data (n = 11,080), participants with gender mismatch (n = 2) and genotype call rate <0.95 (n = 0) were excluded. The rest participants were entered into the following analysis.
High-quality SNPs for kinship estimation and computing principal components and genomic relation matrix (GRM) were extracted by the following criteria: (1) SNP IDs, chromosomes, physical positions, minor alleles and major alleles were all identical in both datasets; (2) call rate >0.99; (3) minor allele frequency (MAF) > 0.01; (4) Hardy–Weinberg equilibrium (HWE) P value < 0.001; (5) choose independent SNPs: (5a) consider a window of 50 SNPs, (5b) calculate LD between each pair of SNPs in the window, remove one of a pair of SNPs if the LD is greater than 0.2, and (5c) shift the window 10 SNPs forward and repeat the procedure. After extraction, 438,288 SNPs were used in the Genetic relatedness calculations via the PLINK --genome command. After excluding participants with kinship relations within three generations, 86,868 participants (Pi_hat >0.125) were eligible for analysis. We homogenized the controls by removing individuals that have comorbid disorders with vertigo (n = 26,082). Comorbid disorders are defined by a data-driven method using the Partitioning Around Medoids (PAM) algorithm in the cluster package of R (version 3.6, https://arxiv.org/abs/1810.05691). The participants were then divided into a discovery cohort and replication cohort depending on the time of recruitment.
Single-nucleotide polymorphism (SNP) genotyping and imputation
All study subjects received whole-genome SNP genotyping using the Axiom Genome-Wide Array TWB2 chip11 at the National Center for Genomics Medicine, Academia Sinica. The imputation was carried out by the Taiwan biobank. Well-imputed SNPs (information score >0.8) out of the 16,211,759 variants on chr1-chr22 were retained, followed by systematic quality control (QC). The QC criteria were applied to exclude SNPs if they (a) had a total call rate <95% in cases and controls combined, (b) had a minor allele frequency (MAF) of <0.1% in cases and controls combined, (c) showed significant (P < 4 × 10−9) deviation from Hardy–Weinberg equilibrium in controls, and (d) a significant difference in their genotype call rates in case and control data (P < 1 × 10−10). For sample filtering, we excluded arrays with generated genotypes for fewer than 95% of loci. Finally, 10,546,339 imputation loci were yielded.
Statistics and reproducibility
The genomic inflation factor (GIF) was calculated, and principal component analysis (PCA) was conducted to identify the top 10 principal components (PCs) via Eigenstrat and 438,288 SNPs in typing datasets. We calculated the distribution of expected P values under the null hypothesis and genomic inflation value (λ). Logistic regression was used to evaluate the association of SNPs with vertigo by adjusting age, sex, and the top 10 PCs (PC1–PC10), and an additive genetic model was conducted using PLINK37 (version 1.9). Refined association tests were performed using R’s SAIGE package (version 0.35) for adjustment of the imbalanced case-control ratio. P < 5 × 10−8 was designated as GWAS significance, and P < 5 × 10−5 was designated as suggestive GWAS significance. Manhattan and quantile–quantile (Q–Q) plots were graphed using the R package. The positional gene mapping and fine mapping of significant loci were generated using LocusZoom38 and Probabilistic Identification of Causal SNPs (PICS2)39. The proportion of variance explained by a given SNP was calculated using Nagelkerke pseudo R2.
Gene-based analysis
We further performed the MAGMA gene-based association analysis40 implemented in FUMA13, which calculates a gene test-statistic (P value) based on all SNPs located within genes, using imputation data. The input SNPs were mapped to 19,270 protein coding genes. Genome-wide significance was defined at P = 0.05/19,270 = 2.595 × 10−6.
Univariate LD-score regression
Linkage Disequilibrium Score Regression (LDSC v1.0.1) was used to estimate the proportion of a true polygenic signal opposed to confounding biases, and to calculate SNP-based heritability41. We estimated LD Scores from 1446 samples in the Taiwan Biobank whole-genome sequencing data using an unbiased estimator of r2 with 1-cM windows, singletons excluded and no r2 cutoff. Heritability estimates were converted to the liability scale assuming a population prevalence of vertigo of 5%. In addition, as vertigo is a common comorbidity or variant of migraine, i.e., vestibular migraine, we evaluated the genetic correlation of vertigo with migraine using a previously established cohort consisting of 3173 Taiwanese migraine patients and 24,528 healthy controls42.
Attributable risk of lead SNPs in significant loci
To evaluate the combined effects of the susceptible genes, we calculated the odds ratio of vertigo in participants stratified by the numbers of risk alleles from three leads SNPs of the susceptible loci.
Cross-population meta-analysis
To investigate the potential variants associated with vertigo across populations, we performed meta-analysis of the European GWAS and ours. There are 5,121,713 SNPs in common between Skuladottir et al.10 and our dataset. A fixed-effects model based on inverse-variance weighting, as implemented in METAL14, was used to combine summary statistics from the two studies. Genetic loci which have previously been reported to be associated with vertigo in the European ancestry10 were tested for association with vertigo in both our combined analysis. and the cross-population meta-analysis. To gain insight on how the genetic architecture of vertigo varies between populations, we also performed a transethnic genetic-correlation estimate from summary statistics using Popcorn43.
Polygenic risk score model
As some types of vertigo are suggested to be hereditary, the polygenic risk scores (PRS) formed from a set of vertigo-associated genetic variants may serve as a useful predictor of vertigo risk. To evaluate the possibility of identifying the subjects with high risks to develop vertigo, we built prediction models by including genome-wide PRS and clinical demographics. We used the discovery cohort to conduct GWAS for generating the estimates of regression coefficients, its standard error and associated p value (\({\hat{\beta }}_{j}\), \({\hat{\sigma }}_{j}\) and \({p}_{j}\)) for each SNP j based on the univariate logistic regression analysis. With SNPs that passed the QC in GWAS analysis, the SNPs with MAF less than 1% were excluded, which is a common criterion for PRS analysis. To calculate PRS, we have used the standard clumping and thresholding (C + T) method44, but its generated PRS did not improve the prediction model significantly (with P value > 0.05) compared to the models adjusting for age, sex, and with or without family history of vertigo for prediction analysis. Therefore, we used another method that has been applied widely in several studies, the LDPred method45. LDpred is a Bayesian shrinkage approach that estimates a posterior mean effect size for each genetic variant based on an infinitesimal prior or a point-normal distribution for sparsity assumption. The fraction of the causal variants with nonzero effect size serves as a tuning parameter \({{\rm{\rho }}}\) because it is generally unknown in practice and it varies among different traits/diseases. The parameter space for \({{\rm{\rho }}}\) was considered as {1, 0.3, 0.1, 0.03, 0.01, 0.003, 0.001}, where \({{\rm{\rho }}}=1\) is for the infinitesimal model.
The optimal PRS model was chosen from the value of \({{\rm{\rho }}}\) that maximized the prediction performance, area under the ROC curve (AUC), evaluated in the replication cohort. The optimal PRS was included in a logistic regression model adjusting for age, sex, with or without family history of vertigo for prediction analysis. Moreover, to stratify the subjects from the low- to high-risk groups by their PRS, we transformed the PRS into 5 quintile variables. The prevalence ratio of vertigo for individuals with the highest PRS quintile and the lowest was estimated. In addition to the PRS by LDpred method, we also calculated the PRS derived from the lead SNPs in this study and the previously reported SNPs associated with vertigo10.
As including family history (FH) may affect the prediction accuracy of PRS, we also adopted another method, PRS-FH, using a logistic model to see if it could enhance the clinical utility of PRS46. Moreover, because PRS may attenuate cross-population predictive performance, we employed PRS-CSx47, which is an extension of PRS-CS48 that uses a global and a local shrinkage parameter for SNP effect size distribution to build PRS using GWAS summary statistics for a single population. To improve the cross-ethnicity prediction accuracy of PRS, PRS-CSx further assumes shared genetic causal effects across populations and borrows information among them.
To improve the prediction performance of PRS for the Taiwanese population, we applied PRS-CSx “meta” function to borrow information from the European GWAS on vertigo. PRS scores were derived by the meta-analysis of the GWAS summary statistics from the smaller sample size of our study and from the larger sample size of the European study, respectively.
Gene expression, tissue enrichment analysis, and pathway analysis
The tissue-specific expression of genes in individual tissues was explored with based on the expression data from the Genotype-Tissue Expression (GTEx) project15. We also performed tissue expression analysis based on the MAGMA gene property in FUMA13, which tested for positive relationships between tissue-specific gene expression in 30 general tissue types and 54 specific tissue types in the GTEx v7 & v8 RNA-seq data. In addition, to investigate the expression of the vertigo-associated genes in vestibules, we also explore their expression data on gEAR platform (https://umgear.org) and SHIELD49, which offer RNA-seq data for inner ear tissues.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS summary statistics have been provided to the NHGRI-EBI GWAS Catalog and the study accession number is GCST90432100. Anonymized data from TWB is available for application by contacting biobank_rs@ibms.sinica.edu.tw. In addition, analyzed data related to this study, intended for reasonable use, can be shared by request by contacting the corresponding authors.
References
Murdin, L. & Schilder, A. G. Epidemiology of balance symptoms and disorders in the community: a systematic review. Otol. Neurotol. 36, 387–392 (2015).
Neuhauser, H. K. Epidemiology of vertigo. Curr. Opin. Neurol. 20, 40–46 (2007).
Kovacs, E., Wang, X. & Grill, E. Economic burden of vertigo: a systematic review. Health Econ. Rev. 9, 37 (2019).
Gizzi, M. S., Peddareddygari, L. R. & Grewal, R. P. A familial form of benign paroxysmal positional vertigo maps to chromosome 15. Int. J. Neurosci. 125, 593–596 (2015).
Bahmad, F. Jr. et al. Locus for familial migrainous vertigo disease maps to chromosome 5q35. Ann. Otol. Rhinol. Laryngol. 118, 670–676 (2009).
Gallego-Martinez, A. & Lopez-Escamez, J. A. Genetic architecture of Meniere’s disease. Hear Res. 397, 107872 (2019).
Ohmen, J. D. et al. Genetic evidence for an ethnic diversity in the susceptibility to Ménière’s disease. Otol. Neurotol. 34, 1336–1341 (2013).
Lee, H. et al. A genome-wide linkage scan of familial benign recurrent vertigo: linkage to 22q12 with evidence of heterogeneity. Hum. Mol. Genet 15, 251–258 (2006).
Rujescu, D. et al. Genome-wide association study in vestibular neuritis: involvement of the host factor for HSV-1 replication. Front Neurol. 9, 591 (2018).
Skuladottir, A. T. et al. A genome-wide meta-analysis uncovers six sequence variants conferring risk of vertigo. Commun. Biol. 4, 1148 (2021).
Wei, C. Y. et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom. Med. 6, 10 (2021).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Varangot-Reille, C., Herranz-Gomez, A., de la Nava, J., Suso-Martí, L. & Cuenca-Martínez, F. The experience of vertigo: a systematic review of neuroimaging studies. Brain Imaging Behav. 16, 2797–2808 (2022).
Kim, S., Lee, J. H. & Nam, S. I. Dicer is down-regulated and correlated with drosha in idiopathic sudden sensorineural hearing loss. J. Korean Med. Sci. 30, 1183–1188 (2015).
Rohde, M. et al. MicroRNA profile of human endo-/perilymph. J. Neurol. 265, 26–28 (2018).
Chang, M. Y. et al. MicroRNAs 218a-5p, 219a-5p, and 221-3p regulate vestibular compensation. Sci. Rep. 7, 8701 (2017).
Huang, R. & Bi, G. MicroRNA-219a-5p-mediated inhibition of CaMKIIγ facilitates vestibular compensation in acute vertigo by promoting protein kinase C expression. Ann. N. Y Acad. Sci. 1475, 78–88 (2020).
Mahmoudian-Sani, M. R. et al. MicroRNAs: effective elements in ear-related diseases and hearing loss. Eur. Arch. Otorhinolaryngol. 274, 2373–2380 (2017).
Rudnicki, A. et al. Next-generation sequencing of small RNAs from inner ear sensory epithelium identifies microRNAs and defines regulatory pathways. BMC Genomics 15, 484 (2014).
Cheng, C. Y. et al. Elevated circulating endothelial-specific microRNAs in migraine patients: a pilot study. Cephalalgia 38, 1585–1591 (2018).
Andersen, H. H., Duroux, M. & Gazerani, P. Serum MicroRNA signatures in migraineurs during attacks and in pain-free periods. Mol. Neurobiol. 53, 1494–1500 (2016).
Jacobs, F. M. et al. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516, 242–245 (2014).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Nishimura, T. et al. Characterization of the human FcγRIIB gene promoter: human zinc-finger proteins (ZNF140 and ZNF91) that bind to different regions function as transcription repressors. Int. Immunol. 13, 1075–1084 (2001).
Costa, A. S. et al. Relation between FCGRIIB rs1050501 and HSV-1 specific IgG antibodies in Alzheimer’s disease. J. Transl. Med. 18, 325 (2020).
Himmelein, S. et al. Differential involvement during latent herpes simplex virus 1 infection of the superior and inferior divisions of the vestibular ganglia: implications for vestibular neuritis. J. Virol. 91, e00331–17 (2017).
Roehm, P. C. et al. Cultured vestibular ganglion neurons demonstrate latent HSV1 reactivation. Laryngoscope 121, 2268–2275 (2011).
Haas, J. G., Weber, J., Gonzalez, O., Zimmer, R. & Griffiths, S. J. Antiviral activity of the mineralocorticoid receptor NR3C2 against Herpes simplex virus Type 1 (HSV-1) infection. Sci. Rep. 8, 15876 (2018).
Wilde, A. A. M. & Amin, A. S. Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 4, 569–579 (2018).
Liu, X. P. et al. Sodium channel diversity in the vestibular ganglion: NaV1.5, NaV1.8, and tetrodotoxin-sensitive currents. J. Neurophysiol. 115, 2536–2555 (2016).
Wooltorton, J. R. et al. Developmental changes in two voltage-dependent sodium currents in utricular hair cells. J. Neurophysiol. 97, 1684–1704 (2007).
Pan, C. et al. Novel heterozygous mutations in the otogelin-like (OTOGL) gene in a child with bilateral mild nonsyndromic sensorineural hearing loss. Gene 808, 146000 (2022).
Yariz, K. O. et al. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am. J. Hum. Genet. 91, 872–882 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Boughton, A. P. et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics 37, 3017–3018 (2021).
Taylor, K. E., Ansel, K. M., Marson, A., Criswell, L. A. & Farh, K. K.-H. PICS2: next-generation fine mapping via probabilistic identification of causal SNPs. Bioinformatics 37, 3004–3007 (2021).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Chen, S. P. et al. Genome-wide analyses identify novel risk loci for cluster headache in Han Chinese residing in Taiwan. J. Headache Pain. 23, 147 (2022).
Brown, B. C., Ye, C. J., Price, A. L. & Zaitlen, N. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99, 76–88 (2016).
Purcell, S. M. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009).
Vilhjálmsson, B. J. et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015).
Hujoel, M. L. A., Loh, P. R., Neale, B. M. & Price, A. L. Incorporating family history of disease improves polygenic risk scores in diverse populations. Cell Genom. 2, 100152 (2022).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Shen, J., Scheffer, D. I., Kwan, K. Y. & Corey, D. P. SHIELD: an integrative gene expression database for inner ear research. Database 2015, bav071 (2015).
Acknowledgements
This research has been conducted using the Taiwan Biobank. We thank all study participants for their generous contribution and the Taiwan Biobank for making the data available. This work was supported by the Brain Research Center, National Yang Ming Chiao Tung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan (SPC and SJW); Institute of Biomedical Sciences, Academia Sinica (CSF); the National Science and Technology Council, Taiwan [NSTC 108-2314-B-010 -022 -MY3, 110-2326-B-A49A-501 -MY3 & 112-2314-B-A49 -037 -MY3 (SPC); 108-2321-B-010-014-MY2, 108-2321-B-010-001-, 108-2314-B-010-023-MY3, 111-2321-B-A49 -004 - 113-2811-B-A49A-012- & 111-2314-B-A49 -090 -MY3 (SJW); and 108-2314-B-001-007, 109-2314-B-001 -006 -MY2 & 112-2314-B-001-010 (CSF)]; Ministry of Health and Welfare, Taiwan [MOHW107-TDU-B-211-123001 and MOHW 108-TDU-B-211-133001] (SJW); Taipei Veterans General Hospital, Taiwan [VGH-109-C-090 & V109E-005-1 (SJW); V111C-158, V112C-053 & V112D67-001-MY3-1 (SPC)]. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
The corresponding authors, Dr. Wang and Dr. Fann, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. S.P.C., C.S.-J.F., and S.J.W. conceived and designed the study. S.P.C., C.L.H., Y.H.L., L.L.P., T.H.C., Y.F.W., H.C.C., Y.M.C., C.S.-J.F., and S.J.W. were involved in the acquisition, analysis, and interpretation of data. S.P.C., C.L.H., and T.H.C. drafted the manuscript. All the authors provided critical revision of the manuscript for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: George Inglis and Dario Ummarino. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, SP., Hsu, CL., Chen, TH. et al. A genome-wide association study identifies novel loci of vertigo in an Asian population-based cohort. Commun Biol 7, 1034 (2024). https://doi.org/10.1038/s42003-024-06603-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06603-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
