
Abstract
This study aimed to create an innovative acidic nano catalyst capable of producing pyrimido[4,5-b]quinolines under environmentally friendly conditions. To achieve this objective, 1,3-benzenedisulfonyl amide (BDSA) was immobilized onto the surface of magnetic graphene oxide (GO/Fe3O4@PTRMS@BDSA@SO3H), and its surface was acidified using chlorosulfonic acid. The synthesized catalyst's structure was thoroughly examined and verified through various analyses, including FTIR, EDX, elemental mapping, FESEM, XRD, TGA, and DSC. This novel nano catalyst exhibited exceptional activity and selectivity in synthesizing pyrimido[4,5-b]quinoline derivatives under solvent-free conditions, at low temperatures, and with high efficiency. Its catalytic effectiveness stemmed from features such as easy and eco-friendly synthesis methods, abundant accessible catalytic sites, a high surface area, remarkable selectivity, and facile separation from the reaction medium. Additionally, the catalyst proved to be cost-effective, safe, scalable, and reusable for up to four times.
Similar content being viewed by others

Introduction
Heterogeneous catalysts are crucial for advancing sustainable and eco-friendly chemical processes, contributing significantly to environmental protection and cost reduction. Recently, there has been a heightened focus on adopting green approaches in synthetic methods1,2,3. Nano catalysts, known for their efficiency and sustainability, play a vital role in various transformations. In contemporary times, carbonaceous nano catalysts are increasingly employed in numerous reactions to synthesize bioactive scaffolds through environmentally friendly pathways in the chemical industry. Consequently, the ongoing research aims to develop nano catalysts using greener methods, with the goal of enhancing synthesis procedures and improving efficiency, yield, and atom-economy4,5,6.
Graphene oxide (GO), a carbon allotrope, exhibits numerous unique features, including a large surface area, high chemical performance, non-toxicity, thermal and chemical stability, electrical properties, strength, and reusability7,8,9,10. There exist multiple techniques for producing graphene, which can be divided broadly into two primary classifications: (a) direct methods, including mechanical exfoliation, liquid-phase chemical/electrochemical exfoliation, carbon nanotube unzipping, chemical vapor deposition, and epitaxial growth; and (b) indirect approaches that entail the synthesis of graphene from oxidized and exfoliated graphite on a significant scale11,12,13,14. GO has attracted considerable interest due to its remarkable properties and potential uses, but it also presents significant risks that must be carefully evaluated. Research indicates that GO can cause oxidative stress, inflammation, and cellular damage, potentially resulting in cytotoxic effects. Its ability to cross biological membranes raises further concerns about its impact on organs and tissues. Inhalation of GO nanoparticles may lead to respiratory problems, while ingestion or skin contact could result in additional health issues. Additionally, the environmental impact of GO, particularly its durability and behavior in ecosystems, is still largely unknown. As research progresses, it is essential to understand and mitigate the risks associated with graphene oxide to ensure its safe and sustainable application in various fields15,16.
Nanoparticles (NPs) are technically defined as particles with dimensions less than 100 nm, possessing unique properties not typically found in bulk samples of the same material. Depending on their general shape, nanoparticles can be categorized as 0D, 1D, 2D, or 3D. The main composition of nanoparticles is intricate, consisting of the surface layer, the shell layer, and the core—the central part of the NP, commonly referred to as the NP itself. These particles are classified into various groups based on their properties, shape, and size, including fullerenes, metal nanoparticles, ceramic nanoparticles, and polymer nanoparticles. These substances hold remarkable importance across diverse fields owing to their exceptional attributes, highlighted by their high surface area-to-volume ratio, distinctiveness. sub-micron dimensions, and sophisticated targeting mechanisms17,18,19,20.
Metal nanoparticles (MNPs) find growing applications globally in biomedicine and associated domains. Heterogeneous catalysts, especially those based on MNP-graphene composites, have shown distinct advantages over homogeneous catalysts regarding their straightforward separation from reaction mixtures, mild reaction conditions, elimination of cumbersome procedures, and notably, the capacity for regeneration and reuse without requiring prior activation21,22,23. Graphene undergoes oxidation to form graphene oxide (GO), showcasing a variety of oxygenated chemical moieties such as –COOH, –OH, carbonyl, and epoxy groups on its surface24,25,26. These functional groups enable the attachment of diverse organic functionalities and metal nanoparticles (MNPs). Hence, GO stands out as a highly effective catalyst with promising applications in the future. These investigations delve into how these interactions impact the electronic configuration of supported MNP catalysts, consequently influencing their catalytic efficiency and long-term stability27,28,29.
Multicomponent reactions (MCRs) provide a rapid and straightforward pathway to a diverse array of organic compounds. The products are formed by the simultaneous reaction of three or more reactants, incorporating all raw materials. The efficiency of these reactions significantly improves as they eliminate the need for intermediate separation. The advantages of MCRs include straightforward conditions, minimized byproducts, facile separation, energy conservation, and shortened reaction times30,31,32,33.
Quinoline-containing compounds display a range of biological activities, encompassing DNA binding and serving as DNA interstitial carriers. Pyrimidines, quinolines, and their derivatives play crucial roles as components in bioactive and pharmaceutical synthetic compounds. Pyrimidine-containing compounds have been employed as Abl kinase inhibitors, antimicrobial agents, and anti-hepatitis A substances. The quinoline moiety is a prevalent component in the structures of various bioactive compounds with antitumor, antihistaminic properties and DNA binding34,35,36.
Moreover, heterocycles containing the pyrimido quinoline scaffold find applications in anti-malarial, antibacterial, anti-allergic, anti-cancer, antimicrobial, and anti-inflammatory activities. Due to their significant biological effects, extensive endeavors have been dedicated to the production of pyrimido[4,5-b]quinolines through a single-step multicomponent reaction (MCRs) employing dimedone, arylaldehyde and 6-aminopyrimidine-2,4-dione with various catalysts. Nonetheless, there is a desire to explore alternative synthetic methods37,38. Various methods have been reported for the synthesis of pyrimido[4,5-b]quinoline derivatives, including a three-component reaction involving aldehyde, 6-aminopyrimidine-2,4-dione, and 1,3-diketone in the presence of catalysts such as Cs2.3H0.7PW10Mo2O4039, sulfonated DABCO enclosed in Agar40, Nano ZnO41, N,N-diethyl-N-sulfoethanaminium ([Et3N–SO3H][Cl]) chloride42, nanocrystal MgO43, and nano [Fe3O4@SiO2@BDIL]44, as well as [C4(DABCO)2]·2OH45.
Therefore, we have tried to prepare a sulfonated heterogeneous catalyst based on magnetized graphene oxide and modified with 1,3-disulfonylamide ligand as nitrogen-containing agent (GO/Fe3O4@PTRMS@BDSA). Then, the catalyst was validated through various instrumental analyses and employed in the synthesis of pyrimido[4,5-b]quinoline derivatives under solvent-free conditions, resulting in the rapid production of high-yield products.
Experimental
General
The chemicals utilized in this study were obtained from Merck company and used without additional purification. FT-IR spectroscopy was conducted employing a Perkin Elmer GX FT-IR spectrometer, covering the range of 4000–400 cm−1. 1H NMR and 13C NMR spectra were obtained in DMSO-d6 solvent using Bruker BioSpin GmbH 300 MHz FT NMR spectrometers. Using the BUCHI 510 device, the melting point of the products was determined. The structure of the novel LDH@PTRMS@BDSA catalyst was thoroughly characterized through a series of analyses, including FTIR, EDX, MAPPING, XRD, FESEM, TGA, and DSC. Using Philips PW1730, XRD patterns were taken in the angular range of 10 to 90 degrees (2θ). FESEM analysis was executed employing the FE-SEM TESCAN MIRA3 instrument. The synthesized catalyst underwent EDX analysis utilizing the EDAX-EDS equipment. TGA was performed on a TGA-DTA instrument in a nitrogen atmosphere, heating at a rate of 10 °C min−1 from 40 to 600 °C, and DSC measurements were taken with a DSC devise. The advancement of reactions and the purity of materials were investigated utilizing thin layer chromatography (TLC) and silica gel plates.
Instructions for the production of graphene oxide (GO)
GO was synthesized from natural graphite using a modified Hummers method. At first, in an ice bath, 50 mL of 98% sulfuric acid was introduced into a 250 mL flask, followed by adding 0.5 g of graphite. After 15 min, 2 g of potassium permanganate was slowly added over a period of 2 h. After three hours, 150 mL of distilled water was added cautiously, after 30 min, 10 mL of 30% hydrogen peroxide was gradually added. The resulting blend was subjected to washing with a 5% HCl solution followed by distilled water, using repeated centrifugation, and finally, it is dried in an oven at 60 °C for 24 h.
Instructions for the production of GO/Fe3O4
40 mg of graphene oxide was dissolved in 40 mL of distilled water, and the mixture was sonicated via bath sonication at room temperature for 30 min. A solution containing 0.21 mmol of FeCl2·4H2O and 0.4 mmol of FeCl3·6H2O was dissolved in 50 mL of deionized water, then added to the previous solution and heated to 85 °C. It was then treated with 30% ammonia until the pH of the solution reached 10. After stirring for 45 min, the resulting mixture was separated by centrifugation, washed with distilled water, and dried in an oven at 60 °C for 24 h.
Instructions for the production of GO/Fe3O4@PTRMS
200 mg of GO/Fe3O4 was added to a flask containing 30 ml of toluene and dispersed via ultrasonication for 30 min. Following this step, 15 mmol of PTRMS (3-chloropropyltrimethoxysilane) was added, and the resulting reaction mixture was stirred under reflux conditions for 24 h. Upon the reaction was finished, the magnetic nanosheets were separated using a strong external magnet, washed sequentially with solvents of toluene and ethanol, and subsequently is dried in a 60 °C oven for 24 h46.
Synthesis Procedure for 1,3-benzenedisulfonyl chloride
1,3-Benzenedisulfonic acid disodium salt (18 mmol) and PCl5 (16.5 mmol) as the chlorinating agent were added to a 250 mL flask. The mixture was fixed at 65 °C and stirred at this temperature for two hours. Upon, at the conclusion of the reaction, chloroform (100 mL) and dry ice (100 g) were added to the container, the organic layer was then separated using a decanter funnel..
Protocol for the synthesis of 1,3-benzenedisulfonyl amide (BDSA)
1 g of 1,3-benzenedisulfonyl chloride and 5 mL of ammonium chloride (30%) were added to a 50 mL flask and it was refluxed for 12 h. After the reaction concluded, the flask's opening was sealed with parafilm, and the reaction container was cooled to 0 °C to prompt the formation of desired crystals. These crystals were subsequently collected and dried at room temperature47.
Preparation of magnetic graphene oxide functionalized with the 1,3-benzenedisulfonylamide ligand (GO/Fe3O4@PTRMS@BDSA)
In a 100 mL flask, 0.5 g of GO/Fe3O4@PTRMS and 40 ml of toluene solvent were combined and subjected to ultrasonic waves for 10 min. Subsequently, 0.8 g of BDSA ligand was introduced into the reaction container, which was subjected for 24 h under reflux conditions. Upon finishing the reaction, the resulting magnetic nanoplates were collected with an external magnet and washed three times with toluene (3 × 3 ml) and twice with ethanol (2 × 3 ml). Eventually, the material was dried in an oven at 60 °C for 24 h, to complete the process.
Preparation of a sulfonic acid catalyst immobilized on magnetic graphene oxide functionalized with the BDSA ligand (GO/Fe3O4@PTRMS@BDSA@SO3H)
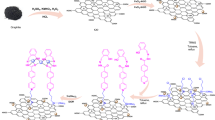
In a 50 ml flask set in an ice bath, 0.5 g of BDSA ligand-functionalized graphene oxide (GO/Fe3O4@PTRMS@BDSA) was poured in 10 ml of dichloromethane solvent. The mixture was sonicated for 5 min to ensure thorough dispersion. Following this, chlorosulfonic acid (3.5 mmol, equivalent to 0.233 ml) was slowly added drop by drop to the reaction mixture while maintaining the temperature of the reaction mixture at low temperature (0 °C). The resulting solution was then stirred with a magnetic stirrer for 12 h at room temperature. Upon finish of the reaction, the magnetic nanoparticles of GO/Fe3O4@PTRMS@BDSA@SO3H were isolated using an external magnet and washed five times with dichloromethane (5 × 3 mL). Finally, the material was dried in an oven at 60 °C for 24 h. The synthesis steps of the catalyst are depicted in Fig. 1.

The synthesis steps of the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst.
General method for the synthesis of pyrimido[4,5-b]quinoline derivatives in the presence of GO/Fe3O4@PTRMS@BDSA@SO3H nanoparticles
In a test tube, the reaction mixture comprising 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (1 mmol), dimedone (1 mmol), aromatic aldehyde derivatives (1 mmol), and GO/Fe3O4@PTRMS@BDSA@SO3H nanoparticles (0.05 g) was stirred using a magnetic stirrer under solvent-free conditions at 70 °C for a suitable duration. The reaction progress was monitored using TLC (normal hexane/ethyl acetate: 8/4). Once the reaction concluded and the desired compound formed, the reaction mixture was cooled to room temperature. To recover the catalyst, 3 mL of hot ethanol was introduced to the reaction mixture and stirred for 2 min. As the catalyst (GO/Fe3O4@PTRMS@BDSA@SO3H) remained insoluble, it was separated by centrifugation, washed with chloroform, and then dried in an oven at 60 °C. The solvent of the reaction mixture was evaporated, and the products were nebulized with ethanol, and the compounds were synthesized with high yield. FTIR, 1HNMR and 13CNMR spectra were used to identify the products and the melting point of all products was taken.
Results and discussion
Spectroscopic investigation to identify catalyst of sulfonic acid placed on magnetic graphene oxide coated with ligand of BDSA (GO/Fe3O4@PTRMS@BDSA@SO3H)
After the synthesis of the catalyst, various analyzes including FT-IR, EDX, MAPPING, FESEM, XRD, DSC, and TGA were studied to confirm and identify the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst. Figure 2 depicts spectrum of FT-IR (a) GO, (b) GO/Fe3O4, (c) magnetized graphene oxide functionalized with 3-chloropropyltrimethoxysilane (GO/Fe3O4@PTRMS), (d) ligand of 1,3-benzenedisulfonylamide (BDSA), (e) the BDSA ligand placed on graphene oxide (GO/Fe3O4@PTRMS@BDSA) and (f) GO/Fe3O4@PTRMS@BDSA@SO3H catalyst. In part (a), graphene oxide is characterized by distinct spectral features: a broad absorption peak at 3406 cm−1 corresponds to the stretching vibration of -OH groups on the graphene surface, accompanied by a peak at 1230 cm−1 corresponding to the bending vibration of –O–H groups. Additionally, the broad peak beginning at 2500 cm−1 signifies the presence of –O–H groups from COOH on the graphene. Notably, the absorption at 1726 cm−1 indicates stretching vibration of C=O acidic groups, while the absorption peak at 1366 cm−1 suggests the bending vibration of carboxyl groups, and the absorption peak at 1098 cm−1 indicates the C–O group. Furthermore, the vibration of the aromatic carbons of the graphite skeleton, which remains unoxidized, is observed in the 1628 region. In contrast, part (b) representing GO@Fe3O4 hybrid nanosheets exhibits a distinct spectrum from the preceding stage, characterized by a significant reduction in intensity of peaks corresponding to O–H and COOH groups at 3300–3550 cm−1 and 1728 cm−1. The presence of Fe NPs is verified by the emergence of peaks corresponding to Fe–O at 594 cm−1 and 620 cm−146. Part (c), concerning the functionalization of magnetic graphene oxide surface with 3-chloropropyltrimethoxysilane, the peak at 2935 cm−1 indicates the aliphatic stretching vibrations of CH groups from the 3-chloropropyltrimethoxysilane attached to the GO/Fe3O4 nanosheets. Part (d), associated with the stretching and bending vibrations of -NH groups indicated at 3371 cm−1, 3258 cm−1, and 1566 cm−1, respectively. Additionally, the stretching vibration of the S=O group is showed at 1324 cm−1 and 1144 cm−1. In part (e), the O–H stretching vibration on the graphene oxide surface is represented by the peak at 3428 cm−1, whereas the C–H stretching vibration is reflected in the peak at 2982 cm−1. Moreover, the stretching vibrations of sulfonyl groups are evident at 1323 cm−1 and 1145 cm−1, and the vibrational peaks of NH groups are located at 3371 cm−1 and 3257 cm−1, confirming the attachment of BDSA to GO/Fe3O4@TRMS. Lastly, part (f) pertains to the sulfonation of GO/Fe3O4@PTRMS@BDSA, where the observed broad peak spanning from 2500 to 3500 cm−1 is associated with the acidification of the catalyst, and the overlapped peak in the range of 1143–1324 cm−1 corresponds to the stretching vibrations of S=O groups.

FTIR spectrum of (a) GO, (b) GO/Fe3O4, (c) GO/Fe3O4@PTRMS, (d) ligand of 1,3-benzenedisulfonylamide (BDSA), (e) GO/Fe3O4@PTRMS@BDSA and (f) GO/Fe3O4@PTRMS@BDSA@SO3H.
The findings from the EDX analysis concerning the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst are shown in Fig. 3. The visual depiction verifies the existence of diverse elements within the catalyst, including carbon, oxygen, nitrogen, silicon, iron, and sulfur. Furthermore, the elemental mapping analysis of the mentioned catalyst, depicted in Fig. 4, corroborates the existence of these anticipated elements in the structure of the catalyst.

EDX analysis of GO/Fe3O4@PTRMS@BDSA@SO3H catalyst.

Elemental mapping analysis (MAPPING) of GO/Fe3O4@TRMS@BDSA@SO3H.
Table 1 presents the quantitative analysis of the constituent elements in the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst. The weight and atomic percentages of the elements are within acceptable ranges for the catalyst. Notably, sulfur has an atomic percentage of 5.29% and a weight percentage of 10.83%. This sulfur content is associated with the catalyst's acidification and the ligand on its surface.
The thermal analysis curves for the GO/Fe3O4@TRMS@BDSA@SO3H catalysis were done with a heating rate of 10 °C/min in temperature range of 40–530 °C (Fig. 5). Examination of the thermal resistance analysis graph reveals three distinct reduction stages.

TGA and DSC thermal analyzes of GO/Fe3O4@PTRMS@BDSA@SO3H catalyst.
The initial decrease is observed within the temperature range of 132 °C, which is attributed to the evaporation of solvents contained and water in the catalyst layers. Subsequently, next reduction is observed between 280 and 430 °C, indicative of the ligand decomposition within the structure. This indicates a reduction in weight of approximately 22%. Consequently, the ligand constitutes about 22% of the catalyst's weight. This means that for every gram of catalyst, the ligand weighs roughly 0.22 g. A subsequent mass reduction is observed around 430 °C, likely associated with catalyst degradation. The DSC curve corroborates the TGA curve and indicates the stability of the catalyst up to the temperature threshold of 280 °C.
The effectiveness of the composite is contingent upon the properties of the filler nanoparticles and their even distribution throughout the specimen, in turn, enhancing its chemical, physical, thermal, and electrochemical attributes. For the examination of the structure, morphology, and dimensions of the catalyst particles, FESEM analysis was employed. At lower magnification, SEM images in Fig. 6, part (a), delineate the contours and creases of graphene oxide nanosheets. In part (b), the appearance of white spherical dots indicates the presence of iron NPs, the gray particles signify the attachment of 3-chloropropyltrimethoxysilane to magnetic graphene46. Part (c) illustrates the deposition of acidic particles with a high and even dispersion across the catalyst surface.

FESEM images of (a1 and a2) GO, (b1 and b2) GO/Fe3O4@PTRMS and (c1 and c2) GO/Fe3O4@PTRMS@BDSA@SO3H.
XRD analysis was used to examine the structural and crystalline attributes of the synthesized catalyst (Fig. 7). Part (a) pertains to graphene oxide, where peaks at 30.93°, 10.58°, and 22.39° correspond to structure graphene. In part (b), representing magnetized graphene oxide, sharp peaks indicative of magnetite emerged at 34.97°, 41.53°, 50.46.8°, 50.46.8°, 67.29°, and 74.33°, alongside the previous peaks46. Moreover, part (c) displays peaks associated with both magnetized graphene and the sulfonation of the catalyst. The XRD difference of the two stages of GO/Fe3O4@PTRMS and GO/Fe3O4@PTRMS@BDSA@SO3H is the placement of the BDSA ligand on the GO/Fe3O4@PTRMS surface and the sulfonation of the GO/Fe3O4@PTRMS@BDSA surface, which changes the XRD patterns.

XRD of (a) GO, (b) GO/Fe3O4@PTRMS and (c) GO/Fe3O4@PTRMS@BDSA@SO3H.
The magnetic nature of the produced catalyst plays a crucial role in its easy retrieval and subsequent reuse. Consequently, through VSM analysis, the magnetic properties of the samples were evaluated. Figure 8 illustrates the VSM curves of (a) GO@Fe3O4, (b) GO@Fe3O4@PTRMS, (c) GO/Fe3O4@PTRMS@BDSA, and (d) the synthesized GO/Fe3O4@PTRMS@BDSA@SO3H nanocomposite. As depicted in the figure, the magnetic characteristic decreases with the layering configuration, signifying optimal layer placement. The magnetic value of the GO@Fe3O4 sample measures approximately 45.77, which decreases to about 24.26 upon the introduction of 3-chloropropyltrimethoxysilane on GO@Fe3O4, further declining to 15.24 with the subsequent addition of the ligand. Finally, upon sulfonation of the catalyst (GO/Fe3O4@PTRMS@BDSA@SO3H), the magnetic value reduces to 7.88, Enabling the extraction of nanoparticles from the reaction environment through the application of a magnetic field. Additionally, Fig. 9 depicting the separation of the nano catalyst via super magnetism, corroborating the VSM analysis and demonstrating the facile recovery of the nano catalyst.

VSM curves of (a) GO@Fe3O4, (b) GO@Fe3O4@PTRMS, (c) GO/Fe3O4@PTRMS@BDSA and (d) GO/Fe3O4@PTRMS@BDSA@SO3H.

Separation of GO/Fe3O4@PTRMS@BDSA@SO3H catalyst using an external magnet.
To determine the chlorosulfonic acid content loaded onto the catalyst surface, the reverse titration method was utilized. Initially, 0.5 g of the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst was added to 25 mL of 0.1 M sodium hydroxide solution. A few drops of phenolphthalein were then added, and the excess sodium hydroxide was titrated with 0.1 M hydrochloric acid. The volume of hydrochloric acid required was 8 ml. This volume was subtracted from the initial solution volume (25 mL) to calculate the volume (17 mL) necessary to neutralize the acidic sites on the catalyst surface. The amount of chlorosulfonic acid loaded onto the catalyst per 0.5 g was determined to be 1.7 mmol. The amount of acid on the catalyst's surface was calculated to be approximately 0.13 g.
Catalytic application of GO/Fe3O4@PTRMS@BDSA@SO3H in the preparation of pyrimido[4,5-b]quinoline derivatives
After validating the new nano catalyst through the aforementioned methods, its catalytic efficacy was assessed in the one-pot three-component synthesis of pyrimido[4,5-b]quinolines. Initially, dimedone (1.0 mmol), 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-diene (1.0 mmol), and 4-chlorobenzaldehyde (1.0 mmol) were chosen as model samples to optimize the synthesis conditions. The model reaction was conducted using various solvents such as water, ethanol, methanol, water/ethanol, acetonitrile, solvent-free, varying catalyst concentrations and at different temperatures. Results revealed that polar solvents like ethanol and water/ethanol positively impacted product yield,
However, the optimal yield and quickest reaction time were attained without solvent at 70 °C, employing 0.05 g of GO/Fe3O4@PTRMS@BDSA@SO3H nanoparticles. Moreover, comparison of reactions with and without the catalyst demonstrated a notable increase in product yield in the presence of the catalyst, whereas yields remained low without it after several hours. These findings are summarized in Table 2. Overall, GO/Fe3O4@PTRMS@BDSA@SO3H proved to be an efficient catalyst for synthesizing pyrimido[4,5-b]quinoline derivatives, offering both short reaction times and high efficiency.
Table 3 presents a comparison of the catalytic performance of GO/Fe3O4@PTRMS@BDSA@SO3H with its intermediates in the model reaction, which includes dimedone (1 mmol), malononitrile (1 mmol), 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (1.0 mmol), and 4-chlorobenzaldehyde (1.0 mmol). As shown in the table, using GO in the reaction resulted in an average yield of 44%. Magnetizing GO increased the yield to 58% and reduced the reaction time to 50 min. Activation of GO/Fe3O4 with PTRMS decreased the yield to 32%. Surface functionalization with the BDSA ligand did not significantly improve the yield, resulting in 39%. However, sulfonating the surface of GO/Fe3O4@PTRMS@BDSA dramatically increased the reaction yield to 93%, with the reaction time reduced to just 14 min, demonstrating the catalyst's high effectiveness.
Furthermore, to validate the efficacy of the novel catalyst and assess the method's versatility, a variety of pyrimido[4,5-b]quinoline derivatives were investigated under optimized conditions. This involved employing different benzaldehyde derivatives (1.0 mmol), dimedone (1.0 mmol), and 6-amino 1,3-dimethylpyrimidine-2,4(1H,3H)-DIN (1.0 mmol) with 50 mg of the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst at 70 °C in solvent-free condition. The outcomes are summarized in Table 4. The findings demonstrate the catalyst's high performance and short reaction time.
Turnover Number (TON) is defined as the number of moles or molecules of reactant A converted to product B per mole or molecule of catalyst. Turnover Frequency (TOF) indicates the number of moles or molecules of reactant A that one mole or molecule of catalyst can convert to product B per unit of time (second, minute, or hour). For heterogeneous catalysts, TOF and TON are expressed based on each active site or per g of catalyst because the exact number of catalyst molecules on the surface is unknown48,49. Table 4 lists the TOF and TON values for the products. The results indicate that a lower catalyst amount combined with higher efficiency results in a higher TON value. Additionally, if these conditions are met alongside a shorter reaction time, the TOF value increases. Therefore, higher TOF and TON values suggest a more efficient catalyst.
Remarkably, the effectiveness of the desired product was not significantly influenced by the presence of donor and acceptor groups on benzaldehyde. However, products synthesized with electron-accepting groups exhibited shorter reaction times compared to those with electron-donating groups, which required more time for synthesis.
To evaluate the catalyst's efficiency, a hot filtration test was conducted. The model reaction involved mixing dimedone (1.0 mmol), 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (1.0 mmol), and 4-chlorobenzaldehyde (1.0 mmol) with 50 mg of catalyst. This reaction was carried out at 70 °C without solvent. After 7 min, halfway through the total reaction time, ethanol was added to the reaction mixture to isolate the catalyst, resulting in a product yield of 69%. In a separate experiment, the catalyst was removed mid-reaction, and the process continued without it. The reaction achieved a product yield of 73% in 14 min, indicating that the catalyst played a significant role in advancing the reaction.
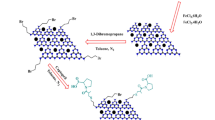
Figure 10 illustrates the suggested mechanism for synthesizing pyrimido[4,5-b]quinoline derivatives employing the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst. Initially, carbonyl benzaldehyde undergoes activation by the nano catalyst, rendering it susceptible to nucleophilic attack by dimedone, existing in its enolic form,

The proposed mechanism for the synthesis of pyrimido[4,5-b]quinoline derivatives in the vicinity of the catalyst of GO/Fe3O4@PTRMS@BDSA@SO3H.
leading to the creation of intermediate A. Subsequently, the removal of a water molecule gives rise to intermediate B, which further reacts with 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione, leading to the formation of intermediate C. Upon interaction with the catalyst, intermediate C undergoes intramolecular attack, forming intermediate D. Tautomerization and the elimination of a water molecule in proximity to the catalyst result in the synthesis of the product. The catalyst is then recovered and reused via a super magnet for subsequent cycles.
Catalyst recovery plays a pivotal role in assessing catalytic performance. After the model reaction, hot ethanol was added to the reaction mixture and stirred for 2 min. Subsequently, the mixture was dissolved, and using an external magnet, The separation of the catalyst was conducted from the reaction vessel. After undergoing multiple washes with ethanol and a single rinse with dichloromethane, the catalyst underwent drying in an oven set at 60 °C for 24 h. Subsequent reutilization in the model test revealed no significant alteration in its activity over a minimum of four consecutive cycles, underscoring its stability and resilience (Fig. 11).

Ability to recycle and reuse the GO/Fe3O4@PTRMS@BDSA@SO3H catalyst.
Figure 12-part a show that the FTIR spectrum of the recycled catalyst after four cycles is not significantly different from that of the synthesized catalyst. Additionally, the broad peak observed from 2500 to 3500 indicates acidification on the catalyst surface, while the broad peak from 1000 to 1400 indicates sulfur groups related to the acid and the sulfone group of the BDSI ligand on the catalyst surface. Additionally, Fig. 12 presents parts b and c, which show the XRD and VSM analyses of the recycled catalyst, respectively. The XRD analysis indicates that all the XRD peaks of the recycled catalyst match those of the non-recycled catalyst, except for a reduction in peak intensity. The VSM analysis estimates the magnetism of the recycled sample to be around 5, suggesting that some iron was lost during recycling. However, the catalyst still possessed sufficient magnetism to be easily separated from the reaction vessel using an external magnet. Part d of Fig. 12 shows the FESEM images of the recycled catalyst, which exhibit no significant changes compared to the original catalyst images. All these analyses demonstrate the durability and stability of the catalyst.

(a) FTIR spectrums of recycled catalyst after four times. (b) XRD spectrums of recycled catalyst after four times. (c) VSM of the recycled catalyst, and (d) FESEM of the recycled GO/Fe3O4@PTRMS@BDSA@SO3H catalyst surface.
The efficacy of the novel recoverable catalyst was assessed against previously reported catalysts for pyrimido[4,5-b]quinoline synthesis, and the findings are outlined in Table 5. As can be seen from the table, GO/Fe3O4@PTRMS@BDSA@SO3H catalyst as an efficient, reusable and green catalyst synthesized pyrimido[4,5-b]quinoline derivatives with high efficiency, lower temperature and short time.
Conclusion
In summary, the utilization of GO/Fe3O4@PTRMS@BDSA@SO3H as a novel solid acid magnetic nano catalyst facilitated the synthesis of pyrimido[4,5-b]quinoline derivatives under mild conditions. This catalyst underwent thorough validation through various analyses, such as FT-IR, EDX, elemental mapping, FESEM, XRD, TGA, and DSC analysis. The pyrimido[4,5-b]quinoline derivatives were synthesized via a highly efficient three-component reaction involving diverse benzaldehydes, dimedone, and 6-amino 1,3-dimethylpyrimidine-2,4(1H,3H)-DIN. Several advantages were observed in this study, including solvent-free conditions, straightforward purification, cost-effectiveness, high product yields, short reaction durations, simple workup procedures, and catalyst reusability. The solid acid nanomagnetic catalyst exhibited significant potential, demonstrating effective performance even after undergoing recycling for at least four cycles in the described synthetic protocol.
Data availability
Data is provided within the manuscript or supplementary information files.
file.
References
Toorbaf, M. & Moradi, L. Preparation of GO/SiO2/PEA as a new solid base catalyst for the green synthesis of some spirooxindole derivatives. RSC Adv. 11, 21840–21850 (2021).
Wang, X. S., Li, Q., Wu, J. R. & Zhang, M. M. Green method for the synthesis of benzo[f]pyrimido[4,5-b]quinoline derivatives catalyzed by iodine in aqueous media. Synth. Commun. 39, 3069–3080 (2009).
Al-Zubaidi, U. Z. I., Bahrami, K. & Khodamorady, M. Fe3O4@SiO2@CSH+VO3− as a novel recyclable heterogeneous catalyst with core–shell structure for oxidation of sulfides. Sci. Rep. 14, 1–13 (2024).
He, Z. H. et al. Photothermal catalytic CO2 oxidative dehydrogenation of propane to propylene over BiOX (X = Cl, Br, I) nanocatalysts. Green Chem. 24, 8270–8279 (2022).
Momeni, S. & Ghorbani-Vaghei, R. Synthesis, properties, and application of the new nanocatalyst of double layer hydroxides in the one-pot multicomponent synthesis of 2-amino-3-cyanopyridine derivatives. Sci. Rep. 13, 1–12 (2023).
Barzkar, A. & Beni, A. S. Fe3O4@C@MCM41-guanidine core–shell nanostructures as a powerful and recyclable nanocatalyst with high performance for synthesis of Knoevenagel reaction. Sci. Rep. 13, 1–13 (2023).
Hu, H., Du, S. & Xi, J. N-Doped holey graphene assembled on fibrous aluminum silicate for efficient carbocatalysis in fixed-bed systems. Green Chem. 24, 5255–5262 (2022).
Wu, H. et al. Graphene-oxide-catalyzed cross-dehydrogenative coupling of oxindoles with arenes and thiophenols. Adv. Synth. Catal. 362, 789–794 (2020).
Amiri-Zirtol, L. & Khabnadideh, S. A novel heterogeneous biocatalyst based on graphene oxide for synthesis of pyran derivatives. Sci. Rep. 14, 1–15 (2024).
Khabnadideh, S., Khorshidi, K. & Amiri-Zirtol, L. A novel heterogeneous acid–base nano-catalyst designed based on graphene oxide for synthesis of spiro-indoline-pyranochromene derivatives. BMC Chem. 17, 1–13 (2023).
Ioni, Y., Sapkov, I., Kirsanova, M. & Dimiev, A. M. Flame modified graphene oxide: Structure and sorption properties. Carbon N. Y. 212, 118122 (2023).
Saini, R., Naaz, F., Bashal, A. H., Pandit, A. H. & Farooq, U. Recent advances in nitrogen-doped graphene-based heterostructures and composites: Mechanism and active sites for electrochemical ORR and HER. Green Chem. 26, 57–102 (2024).
Helmi, M., Khoshdouni Farahani, Z., Hemmati, A. & Ghaemi, A. Facile synthesis of Persian gum–graphene oxide composite as a novel adsorbent for CO2 capture: Characterization and optimization. Sci. Rep. 14, 1–16 (2024).
Amiri-Zirtol, L. & Khabnadideh, S. GO treated with aminoethyl-piperazine as a reusable and eco-friendly organocatalyst for synthesis of some xanthen and pyran derivatives. ChemistrySelect 8, e202204007 (2023).
Singh, S. K. et al. Thrombus inducing property of atomically thin graphene oxide sheets. ACS Nano 5, 4987–4996 (2011).
Liu, S. et al. Antibacterial activity of graphite, graphite oxide, graphene oxide, and reduced graphene oxide: Membrane and oxidative stress. ACS Nano 5, 6971–6980 (2011).
Kouser, M., Chowhan, B., Sharma, N. & Gupta, M. Transformation of waste toner powder into valuable Fe2O3 nanoparticles for the preparation of recyclable Co(II)-NH2-SiO2@Fe2O3 and its applications in the synthesis of polyhydroquinoline and quinazoline derivatives. ACS Omega 7, 47619–47633 (2022).
Abdtawfeeq, T. H. et al. Ultrasound-assisted and one-pot synthesis of new Fe3O4 /Mo-MOF magnetic nano polymer as a strong antimicrobial agent and efficient nanocatalyst in the multicomponent synthesis of novel pyrano[2,3-d]pyrimidines derivatives. J. Inorg. Organomet. Polym. Mater. 33, 472–483 (2023).
Fattahi, B. & Dekamin, M. G. Fe3O4/SiO2 decorated trimesic acid-melamine nanocomposite: A reusable supramolecular organocatalyst for efficient multicomponent synthesis of imidazole derivatives. Sci. Rep. 13, 1–13 (2023).
Karimi, Z. & Rahbar-Kelishami, A. Preparation of highly efficient and eco-friendly alumina magnetic hybrid nanosorbent from red mud: Excellent adsorption capacity towards nitrate. J. Mol. Liq. 368, 120751 (2022).
Yang, A., Wang, Z. & Zhu, Y. Facile preparation and highly efficient sorption of magnetic composite graphene oxide/Fe3O4/GC for uranium removal. Sci. Rep. 11, 1–10 (2021).
Saffarian, H., Karimi, F., Yarie, M. & Zolfigol, M. A. Fe3O4@SiO2@(CH2)3-urea-quinoline sulfonic acid chloride: A novel catalyst for the synthesis of coumarin containing 1, 4 dihydropyridines. J. Mol. Struct. 1224, 129294 (2020).
Dardeer, H. M., Ibrahim, A. S., Gad, A. N. & Gaber, A. A. M. Bifunctional of Fe3O4@chitosan nanocomposite as a clarifying agent and cationic flocculant on different sugar solutions as a comprehensive semi industrial application. Sci. Rep. 14(1), 1–11 (2024).
Dai, G., Li, Q., Zang, D. & Wei, Y. A bifunctional molecular catalyst built up of l-proline grafted polyoxometalate for one-pot three-component green synthesis of heterocycles. Green Chem. 25, 6263–6269 (2023).
Parastar Gharehlar, M., Sheshmani, S., Nikmaram, F. R. & Doroudi, Z. Synergistic potential in spinel ferrite MFe2O4 (M = Co, Ni) nanoparticles-mediated graphene oxide: Structural aspects, photocatalytic, and kinetic studies. Sci. Rep. 14, 1–21 (2024).
Amiri-Zirtol, L., Amrollahi, M. A. & Mirjalili, B. F. GO-Fe3O4-Ti(IV) as an efficient magnetic catalyst for the synthesis of bis(indolyl)methanes and benzo[a]xanthen-11-one derivatives. Inorg. Nano-Metal Chem. 1–11 (2021).
Hemmati, S., Heravi, M. M., Karmakar, B. & Veisi, H. In situ decoration of Au NPs over polydopamine encapsulated GO/Fe3O4 nanoparticles as a recyclable nanocatalyst for the reduction of nitroarenes. Sci. Rep. 11, 1–14 (2021).
Guo, K., Baidak, A. & Yu, Z. Recent advances in green synthesis and modification of inorganic nanomaterials by ionizing and non-ionizing radiation. J. Mater. Chem. A 8, 23029–23058 (2020).
Amiri-zirtol, L. et al. A novel and efficient boron-containing magnetic catalyst based on graphene oxide (GO-Fe3O4-BFn) for synthesis of pyrazole and pyranopyrazole derivatives. J. Mol. Struct. 1307, 137868 (2024).
Moosavi-Zare, A. R. & Najafi, R. Multicomponent synthesis of pyrimido[4,5-b] quinolines over a carbocationic catalytic system. Sci. Rep. 13, 1–9 (2023).
Momeni, S. & Ghorbani-Vaghei, R. A facile one-pot synthesis of tetrahydrobenzo[b]pyrans and 2-amino-4H-chromenes under green conditions. RSC Adv. 14, 21608–21622 (2024).
Chen, R. et al. A three-component reaction to construct β-aminonitroso-α-diazocarbonyl compounds under metal-free conditions. Adv. Synth. Catal. 364, 1422–1426 (2022).
Momeni, S. & Ghorbani-Vaghei, R. An efficient, green and solvent-free protocol for one-pot synthesis of 1,4-dihydropyridine derivatives using a new recyclable heterogeneous catalyst. J. Mol. Struct. 1288, 135758 (2023).
Dinh Thanh, N. et al. Synthesis and structure of some substituted 2-amino-4-aryl-7-propargyloxy-4H-chromene-3-carbonitriles. Synth. Commun. 49, 102–117 (2019).
Jalili, F., Zarei, M., Zolfigol, M. A., Rostamnia, S. & Moosavi-Zare, A. R. SBA-15/PrN(CH2PO3H2)2 as a novel and efficient mesoporous solid acid catalyst with phosphorous acid tags and its application on the synthesis of new pyrimido[4,5-b]quinolones and pyrido[2,3-d]pyrimidines via anomeric based oxidation. Microporous Mesoporous Mater. 294, 109865 (2020).
Song, S., Dai, Y., Hong, Y., Li, X. & Yan, X. A simple continuous reaction for the synthesis of quinoline compounds. Green Chem. 24, 1714–1720 (2022).
Moosavi-Zare, A. R. & Najafi, R. Multicomponent synthesis of pyrimido[4,5-b] quinolines over a carbocationic catalytic system. Sci. Rep. 13, 16501 (2023).
Abdolmohammadi, S., Balalaie, S., Barari, M. & Rominger, F. Three-component green reaction of arylaldehydes, 6-amino-1,3-dimethyluracil and active methylene compounds catalyzed by Zr(HSO4)4 under solvent-free conditions. Comb. Chem. High Throughput Screen. 16, 150–159 (2013).
Khillare, K., Aher, D., Chavan, L. & Shankarwar, S. Cesium salt of 2-molybdo-10-tungstophosphoric acid as an efficient and reusable catalyst for the synthesis of uracil derivatives via a green route. RSC Adv. 11, 33980–33989 (2021).
Moghaddampour, I. M., Shirini, F. & Langarudi, M. S. N. Agar-entrapped sulfonated DABCO: Agelly acidic catalyst for the acceleration of one-pot synthesis of 1,2,4-triazoloquinazolinone and some pyrimidine derivatives. J. Mol. Struct. 1226, 129336 (2021).
Siddiqui, S. A. et al. Room temperature ionic liquid promoted improved and rapid synthesis of 2,4,5-triaryl imidazoles from aryl aldehydes and 1,2-diketones or α-hydroxyketone. Tetrahedron 61, 3539–3546 (2005).
Zare, A., Lotfifar, N. & Dianat, M. Preparation, characterization and application of nano-[Fe3O4@-SiO2@ R-NHMe2][H2PO4] as a novel magnetically recoverable catalyst for the synthesis of pyrimido [4, 5-b] quinolines. J. Mol. Struct. 1211, 128030 (2020).
Rad, A. M. & Mokhtary, M. Efficient one-pot synthesis of pyrido[2,3-d]pyrimidines catalyzed by nanocrystalline MgO in water. Int. Nano Lett. 5, 109–123 (2015).
Zare, A. & Barzegar, M. Dicationic ionic liquid grafted with silica-coated nano-Fe3O4 as a novel and efficient catalyst for the preparation of uracil-containing heterocycles. Res. Chem. Intermed. 46, 3727–3740 (2020).
Jolodar, O. G., Shirini, F. & Seddighi, M. Efficient synthesis of pyrano[2,3-d]pyrimidinone and pyrido[2,3-d]pyrimidine derivatives in presence of novel basic ionic liquid catalyst. Chinese J. Catal. 38, 1245–1251 (2017).
Momeni, S. & Ghorbani-Vaghei, R. Green synthesis of quinazoline derivatives using a novel recyclable nano-catalyst of magnetic modified graphene oxide supported with copper. Sci. Rep. 13, 1–17 (2023).
Momeni, S. & Ghorbani-Vaghei, R. Copper immobilized on modified LDHs as a novel efficient catalytic system for three-component synthesis of pyrano[2,3-d]pyrimidine and pyrazolo[4′,3′:5,6]pyrano[2,3-d]pyrimidine derivatives. ACS Omega 9, 10332–10342 (2024).
Kozuch, S. & Martin, J. M. L. ‘Turning over’ definitions in catalytic cycles. ACS Catal. 2, 2787–2794 (2012).
Ullah, H. et al. Chemical fixation of carbon dioxide catalyzed via cobalt (III) ONO pincer ligated complexes. Commun. Chem. 2(1), 1–9 (2019).
Shirini, F., Langarudi, M. S. N., Daneshvar, N., Jamasbi, N. & Irankhah-Khanghah, M. Preparation and characterization of [H2-DABCO][ClO4]2 as a new member of DABCO-based ionic liquids for the synthesis of pyrimido[4,5-b]-quinoline and pyrimido[4,5-d]pyrimidine derivatives. J. Mol. Struct. 1161, 366–382 (2018).
Moosavi-Zare, A. R., Goudarziafshar, H. & Bahrami, Z. Nano-[Cu-4C3NSP](Cl)2 as a new catalyst for the preparation of pyrimido[4,5-b]quinoline derivatives. Res. Chem. Intermed. 49, 507–523 (2023).
Shi, D. Q. et al. An Efficient synthesis of pyrimido[4,5-b]quinoline and indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine derivatives via multicomponent reactions in ionic liquid. J. Heterocycl. Chem. 45, 693–702 (2008).
Karami, Z. & Khodaei, M. M. Post-synthetic modification of IR-MOF-3 as acidic-basic heterogeneous catalyst for one-pot synthesis of pyrimido[4,5-b]quinolones. Res. Chem. Intermed. 48, 1773–1792 (2022).
Osanlou, F., Nemati, F. & Sabaqian, S. An eco-friendly and magnetized biopolymer cellulose-based heterogeneous acid catalyst for facile synthesis of functionalized pyrimido[4,5-b]quinolines and indeno fused pyrido[2,3-d]pyrimidines in water. Res. Chem. Intermed. 43, 2159–2174 (2017).
Zare, A., Dianat, M. & Eskandari, M. M. A novel organic–inorganic hybrid material: Production, characterization and catalytic performance for the reaction of arylaldehydes, dimedone and 6-amino-1,3-dimethyluracil. New J. Chem. 44, 4736–4743 (2020).
Sarmasti, N., Yousefi Seyf, J. & Khazaei, A. Synthesis and characterization of [Fe3O4@CQDs@Si(CH2)3NH2@CC@EDA@SO3H]+Cl− and Fe3O4@CQDs@Si(CH2)3NH2@CC@EDA@Cu nanocatalyts and their application in the synthesis of 5-amino-1,3-diphenyl-1H-pyrazole-4-carbonitrile and 1-(morpholino(phenyl)methyl)naphthalen-2-ol derivatives. Arab. J. Chem. 14, 103026 (2021).
Acknowledgements
We thank Bu-Ali Sina University, Center of Excellence Developmental of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) and for financial support.
Author information
Authors and Affiliations
Contributions
R.GH and S.M. conceived the experiments and provided experimental instructions, S.M. performed the experiments, analyzed the results and wrote the manuscript. The authors reviewed.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Momeni, S., Ghorbani-Vaghei, R. Facile synthesis of novel acidic modified magnetic graphene oxide and its application in the green synthesis of pyrimido[4,5-b]quinolines. Sci Rep 14, 21531 (2024). https://doi.org/10.1038/s41598-024-71461-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71461-9
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
