
Abstract
Cerebral cavernous malformations (CCMs) are low-flow vascular malformations in the brain associated with recurrent hemorrhage and seizures. The current treatment of CCMs relies solely on surgical intervention. Henceforth, alternative non-invasive therapies are urgently needed to help prevent subsequent hemorrhagic episodes. Long non-coding RNAs (lncRNAs) belong to the class of non-coding RNAs and are known to regulate gene transcription and involved in chromatin remodeling via various mechanism. Despite accumulating evidence demonstrating the role of lncRNAs in cerebrovascular disorders, their identification in CCMs pathology remains unknown. The objective of the current study was to identify lncRNAs associated with CCMs pathogenesis using patient cohorts having 10 CCM patients and 4 controls from brain. Executing next generation sequencing, we performed whole transcriptome sequencing (RNA-seq) analysis and identified 1,967 lncRNAs and 4,928 protein coding genes (PCGs) to be differentially expressed in CCMs patients. Among these, we selected top 6 differentially expressed lncRNAs each having significant correlative expression with more than 100 differentially expressed PCGs. The differential expression status of the top lncRNAs, SMIM25 and LBX2-AS1 in CCMs was further confirmed by qRT-PCR analysis. Additionally, gene set enrichment analysis of correlated PCGs revealed critical pathways related to vascular signaling and important biological processes relevant to CCMs pathophysiology. Here, by transcriptome-wide approach we demonstrate that lncRNAs are prevalent in CCMs disease and are likely to play critical roles in regulating important signaling pathways involved in the disease progression. We believe, that detailed future investigations on this set of identified lncRNAs can provide useful insights into the biology and, ultimately, contribute in preventing this debilitating disease.
Similar content being viewed by others


Introduction
Cerebral cavernous malformations (CCMs) are vascular lesions of the brain affecting approximately 0.5% of the human population1,2. They are associated with leaky endothelium and increased vascular permeability which often results in seizures, intracerebral hemorrhage and focal neurological deficits. CCMs can occur either sporadically with single lesion or as familial forms harboring multiple lesions. Familial CCMs are inherited in an autosomal dominant pattern with incomplete penetrance and variable expressivity3,4. Loss-of-function mutations are known in one of the three CCM genes: Krev interaction trapped 1 (CCM1/KRIT1), cerebral cavernous malformations 2 (CCM2) and programmed cell death 10 (PDCD10) predisposes to CCMs5,6. However, a growing body of evidence suggests that mutations of CCM genes are alone not fully sufficient to induce CCM lesion burden, therefore indicating the involvement of yet to be identified genetic factors for the disease progression7.
Noncoding RNAs represent RNA molecules that are not translated into proteins and are known to be involved in chromatin remodeling, post-transcriptional modifications, signal transduction and disease progression8,9,10. Based on their transcript size they are classified into two major categories: small non-coding RNAs of <200 nucleotides in length (microRNAs, snoRNAs, scaRNAs and piRNAs) and long non-coding RNAs (lncRNAs) of >200 nucleotides in length11. From our previous work, using RNA sequencing approach, we have shown that there are microRNAs and snoRNAs significantly deregulated in CCMs disease1,3. In this current study we focused on identifying and profiling long noncoding RNA (lncRNA) molecules in CCMs patients.
Long non-coding RNAs (lncRNAs) regulate gene expression by various mechanisms, such as chromatin level regulation, act as enhancer function, involvement in protein stability and modulation of transcription factor activity8,9,12. The role of lncRNAs in ischemic stroke and cerebrovascular pathologies has been well established13,14. Recently, Dykstra-Aiello et al. reported the correlation of lncRNAs identified in blood samples from stroke affected patients with the vascular risk factors15. Other studies too identified aberrantly expressed lncRNAs in focal ischemia, Hereditary Hemorrhagic Telangiectasia (HHT) and brain arteriovenous malformations (AVM) showing their clinical relevance in cerebrovascular malformations16,17,18. However, till date, the role of lncRNAs in CCMs disease has not been reported. Despite recent studies reporting the involvement of lncRNAs in animal models of stroke and cerebrovascular pathologies13,16, their role in CCMs disease remains elusive. We, therefore, investigated their association from surgically resected CCMs lesions. By using RNA-sequencing approach for profiling lncRNAs and their co-expressed protein coding genes (PCGs), this study will provide a new perspective on CCMs treatment strategies. Recent studies have investigated the functional relevance of lncRNAs using different computational approaches such as co-expression analysis and lncRNA-protein coding gene proximity analysis (cis or trans-regulation) strategies8,19,20,21. Since we used transcriptome or expression-based approach, we implemented lncRNA-mRNA co-expression analysis strategy to find functionally relevant lncRNAs deregulated in CCMs. Overall, this study is a unique effort to catalogue lncRNAs associated with CCMs disease and might open up new directions for better improved alternative therapies.
Results
Transcriptome profiling of CCM samples
CCMs originating from brainstem are rare and of particular interest due to their crucial relationship with the adjacent vascular and neural structures. Any mild or undetectable changes in these regions may result in higher bleeding rate associated with severe neurological deficits and morbidity22. Current treatment options for brainstem CCMs rely only on microsurgical interventions. Henceforth, alternative non-invasive treatment modalities are a prerequisite for preventing rebleeding in such eloquent locations.
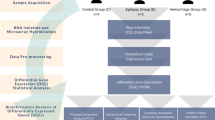
Following total RNA isolation from 10 brainstem CCMs (Table 1) and 4 temporal lobe epilepsy (TLE) controls (Table 2), samples were processed for library preparation and RNA sequencing (Fig. 1a). After the FASTQ files were adapter and quality trimmed, 70 to 92 million read sequences belonging to each sample were mapped to the human genome hg38, available on illumina’s iGenome site (http://support.illumina.com/sequencing/sequencing_software/igenome.html). The average number of reads entering the mapping process across all analyzed samples was 85.7 million. The average percentage of uniquely mapped reads was 85.4%, of reads mapped to multiple loci was 12.5%, and of unmapped reads was 1.8%.

Transcriptome profiles of CCM patients with differential expression patterns of LncRNAs and PCGs. (a) Heatmap showing differential expression of lncRNAs and PCGs between CCMs (n = 10) and control group (n = 4). (b) Kernal density graph showing coding potential probability of DE lncRNAs and DE PCGs. Probability or score is calculated using coding potential calculator (CPC). Green dotted lines divide coding and noncoding CPC scores. (c,d) Volcano plots shows up- (red) and down-regulated (blue) lncRNAs (c) and PCGs (d) respectively. Key significantly differentially expressed transcripts are highlighted with pink and known CCM related PCGs are highlighted with yellow. Vertical dotted lines represent log-fold change cut-off ±1.5 (right and left) and values above horizontal dotted lines represent transcripts with FDR <0.05 cut-off. (e) Expression status of CCM genes (grey bar) along with top up- (red bar) and down-regulated (blue bar) DE lncRNAs and PCGs. (f) Boxplots showing differentially expressed transcripts (lncRNAs and PCGs) on three previously known CCM susceptibility locus.
Differentially expressed lncRNAs and protein coding genes (PCGs) in CCMs
By comparing expression patterns between CCMs (n = 10) and control groups (n = 4) from RNA-seq, differentially expressed (DE) up- and down-regulated lncRNAs and protein coding genes (PCGs) were profiled. Among them, 1,967 lncRNAs and 4,928 PCGs were significantly differentially expressed (DE) between CCMs and control comparison groups. There were 1,475 and 2,290 up-regulated lncRNAs and PCGs; 492 and 2,638 down-regulated lncRNAs and PCGs, respectively. These differentially expressed (DE) transcripts were filtered using corrected p-value (p value adj <0.01) and an absolute log-fold change greater than 1.5 between the comparison groups (Fig. 1a and Supplementary Data 1). These transcripts were then tested for their coding potential capacity using CPC (coding potential calculator) to ensure that the obtained DE lncRNAs do not code for any peptide. CPC prediction results showed no peptide coding capability for lncRNAs (CPC score less than 0.37 represents noncoding transcript) and the DE PCGs showed high coding capability. There are certain PCGs seen towards noncoding portion due to their noncoding transcript isoforms that are previously known to function as both coding and noncoding RNAs (cncRNAs or bi-functional RNAs)23 (Fig. 1b). Detailed investigation of the top DE transcripts revealed lncRNAs involved in CNS related disorders (FOXG1-AS1 and LINC01551)24,25. In addition to the latter our analysis also revealed lncRNAs (DLX6-AS1) that regulate the expression of distant mRNAs (DLX6-AS1)26,27. Interestingly, previous brain related studies have also found some of our top DE lncRNAs, LBX2-AS128 and LUCAT129,30 as significantly co-expressed with other mRNAs expressed in brain (Fig. 1c). Most of the top DE PCGs where previously known to be involved in functions related to synaptic transmissions (SHANK1)31, brain abnormalities (FOXG1), immune response and cell-signaling (MSN and CARD6)32,33 (Fig. 1d). Thus, comparison using transcriptome sequencing identified most important and functionally relevant transcripts related to CCMs pathogenesis.
Expression status of known CCM genes in patients
Though CCMs patients are characterized based on genomic mutations that occur in KRIT1/CCM1, CCM2 and PDCD10/CCM3 genes6,34, there was no significant change in mRNA expression patterns of these PCGs compared to other top differentially expressed lncRNA and PCGs genes (Fig. 1e). These three CCM genes are known to be strongly expressed in neural cells of brain, cerebellum and spinal cord during embryonic development and postnatal brain development35. But in CCMs disease context, these genes are expressed at very low level, because the mutations (two nucleotide substitutions) in CCM1/KRIT1 gene causes CCM1/KRIT1 mRNA to decay due to abnormal splicing and premature termination codon (PTC)36. Hence, there is a possibility that mRNA expression of CCM genes may be reduced by these mutations from corresponding genes37 (Fig. 1e). There are also possibilities of other factors that can influence the expression of these CCMs genes. These observations show the importance of investigating transcription profiles of CCMs patients in addition to genomic alterations or aberrations. Therefore, studying expression patterns of lncRNAs in CCMs may provide us additional routes for non-invasive therapies.
Differentially expressed transcripts at CCM susceptible genomic locus
Previous studies have uncovered an association between tissue specific somatic mutations or chromosomal aberrations on transcriptional landscapes38,39,40,41. To characterize the downstream transcriptional effects of CCM-specific genomic aberrations, we scanned the genomic loci previously known for CCMs susceptibility and their effects on global transcriptional changes (Fig. 1f). Interestingly, we found 59 DE lncRNAs and 163 DE PCGs that map to three CCM mutated loci (CCM1: 7q11-q22, CCM2: 7p15-13 and CCM3: 3q25.2-q27): 21 DE lncRNAs and 71 DE PCGs on CCM1 locus; 21 DE lncRNAs and 41 DE PCGs on CCM2 locus; 17 DE lncRNAs and 51 DE PCGs on CCM3 locus (Fig. 1f and Supplementary Data 2). CCM locus DE lncRNAs such as DLX6-AS1 (CCM1 locus) and SOX2-OT (CCM3 locus) are known to be associated with brain function and disease related to central nervous system. In addition to that, lncRNA HOXA-AS2 (CCM2 locus) regulates malignant glioma and vasculogenic mimicry formation42.
CCM associated lncRNA-mRNA co-expression analysis
Since focus of our study was to investigate lncRNAs associated with CCMs pathogenesis, we observed that major DE lncRNAs belong to intergenic and natural antisense class of lncRNA (Fig. 2a). LncRNAs are also known to regulate the expression of other mRNAs via multiple mechanisms43,44. Therefore, comparison of the expression patterns of DE lncRNAs with PCGs was made to construct lncRNA-mRNA co-expression network. To achieve this, expression correlation analysis was performed to find significantly correlated lncRNA-mRNA pairs having Spearman coefficient above 0.9 and with correlation p-value < 0.05. This resulted in 122,112 lncRNA-mRNA significantly correlated pairs containing 1,838 DE lncRNAs and 4,379 DE PCGs. Among 1,838 DE lncRNAs, 326 lncRNAs were significantly correlated with more than 100 DE PCGs individually (Fig. 2b and Supplementary Data 1). Previous studies have shown that the expression of lncRNAs to be more tissue and cell type specific compared to PCGs. RNA-seq samples of 16 normal tissue types from human bodyMap 2.0 dataset revealed expression of these lncRNAs (n = 326) having more specificity towards brain tissue. As expected, the CCMs up-regulated DE lncRNAs showed lower expression and CCMs down-regulated DE lncRNAs showed higher expression in normal brain (Fig. 2c). Among these 326 lncRNAs, MEG3, MIAT and SENCR were already implicated in angiogenesis and vascular disease (angio-lncRNAs)45.

LncRNA-mRNA co-expression analysis and top lncRNA signatures. (a) Pie-chart with percentage of different classes of lncRNAs differentially expressed (DE) between CCMs and control group. (b) Scatterplot shows DE lncRNAs and its number of correlated or co-expressed DE PCGs. Left side of the pink dotted line denotes DE lncRNAs (n = 326) having more than 100 correlated DE PCGs. (c) Heatmap with expression status of top correlated DE lncRNAs (n = 326) in 16 normal tissues samples from human bodyMap dataset (each tissue contains 2 replicates). (d) IGV browser with RNA-seq read distribution of top two lncRNAs (LBX2-AS1 and SMIM25) in CCM patients and control samples. Nucleotide sequences for forward and reverse primers (pink) and location of primer sequences used for qRT-PCR validations are highlighted. (e) qRT-PCR validation of top two DE lncRNAs. qRT-PCR graphs are presented as mean ± standard error of the mean. (f) Boxplots showing expression status of LBX2-AS1 and SMIM25 in current cohort (CCMs n = 10, controls n = 4) and cohort from Koskimäki J. et al. (CCMs n = 5, controls n = 3). P-values for qRT-PCR experiments are calculated using two-tailed student t-test. *p-value < 0.05 and **p-value < 0.01.
Validation of LBX2-AS1 and SMIM25 lncRNAs
LncRNAs LBX2-AS1 and SMIM25/LINC01272 were top differentially expressed lncRNAs with more than 100 co-expressed PCGs and up-regulated in CCMs compared to control group (Fig. 2d). Further validation using RT-qPCR was performed on LBX2-AS1 and SMIM25 lncRNAs. Expression patterns of lncRNAs from RNA-seq analysis was successfully validated using qRT-PCR. In agreement with the findings from RNA-sequencing data, the expression of both LBX2-AS1 and SMIM25 transcripts were significantly up-regulated in qRT-PCR analysis. LBX2-AS1 and SMIM25 expressions were normalized to HPRT housekeeping gene and the normalized values were compared between control and CCMs groups (Fig. 2e). Expression status of these validated lncRNAs was also verified by analyzing RNA-seq data from a recent study from Koskimäki J et al. with cohorts of 5 CCM patients and 3 normal control samples2 (Fig. 2f). In addition to that, we have combined samples from our cohort (Controls = 4; CCMs = 10) with the samples from Koskimäki J et al. (Controls = 3; CCMs = 5) and performed differential expression analysis using 22 samples (7 controls vs 15 CCMs). We filtered the significant candidates using FDR/Padj <0.01 & absolute log-foldchange >1.5. Out of 190 lncRNAs from combined analysis we could find overlap of 150 lncRNAs (~79%) in our cohort’s DE analysis. Similarly, combined analysis gave 2,184 PCGs and out of which 2084 genes were overlapping with our cohort’s DE analysis. We found LBX2-AS1 and SMIM25 to be significantly deregulated with this combined analysis from two cohorts (Supplementary Data 1).
CCM locus PCGs co-expressed with DE lncRNA
There were 240 and 131 PCGs significantly correlated or co-expressed with LBX2-AS1 and SMIM25/LINC01272 respectively. Among those, 62 PCGs were commonly co-expressed with these two lncRNAs. Interestingly, several co-expressed PCGs of LBX2-AS1 and SMIM25 map to CCM hotspot loci CCM1: 7q11-q22, CCM2: 7p15-13 and CCM3: 3q25.2-q27 with crucial functions that have relevance to CCM pathogenesis (Fig. 3a and Supplementary Data 3). For example, ALDH3B1, ARPC3, LIMS1, LSP1, PTAFR and VAV3 have been previously reported to play a role in oxidative stress, adherens junction pathway, focal adhesion and VEGF-mediated angiogenesis (Fig. 3a). Collectively, these observations suggest that the lncRNAs and their co-expressed PCGs from CCM hotspot loci could serve as potential novel biomarkers and therapeutic targets in the treatment of CCM.

Top lncRNAs LBX2-AS1 and SMIM25 co-expression network and its potential co-regulatory functions. (a) Network shows top two lncRNAs and co-expressed DE PCGs determined by its expression correlation. Nodes and edges in the network are generated using cytoscape. Venn diagram shows common DE PCGs co-expressed between top validated lncRNAs LBX2-AS1 and SMIM25. (b) Bar graph with enriched biological process, pathways and phenotypes derived using GeneSCF. Different color codes indicate terms derived from different database repositories. Gene ontology terms are denoted as red (Biological Process, BP), dark blue (Cellular components, CC) and yellow (Molecular Function, MF); KEGG pathways are in in purple; Reactome pathways are shown in green; and Human Phenotype Ontology terms as light blue. Enrichment in the scale denotes −log10 (p-value) and number above each bar represents number of genes.
Co-expression analysis revealed functional lncRNA related to CCM pathogenesis
In order to identify functional lncRNAs in CCMs, the biological functions were predicted using GeneSCF for PCG pairs having similar expression patterns as DE lncRNAs46 (see methods for details). GeneSCF predicted enriched terms from various databases such as gene ontology, KEGG, The Mammalian Phenotype Ontology and Reactome pathway. Previously known important biological processes and vascular signaling pathways related to CCMs such as, VEGF receptor signaling pathway, cell junction assembly, angiogenesis, NF-kB transcription activity, focal adhesion, actin cytoskeleton, PI3K complex, Rho GTPase signaling, TLR-receptor binding and NAD(P)H oxidase activity were significantly enriched for the differentially co-expressed PCGs47,48,49 (p < 0.05) (Fig. 3b and Supplementary Data 4).
Discussion
The advancement of high-throughput technologies such as, next generation sequencing has facilitated the identification and characterization of lncRNAs in several diseases50. Despite extensive studies showing the potential role of lncRNAs in cerebrovascular pathologies17,18,51 their identification in CCMs disease remains unexplored.
Herein applying state-of-the-art RNA-seq approach we intent to profile lncRNAs signatures in CCMs disease. Our study applying comprehensive computational analysis identified several lncRNAs and protein coding genes (PCGs) differentially expressed (DE) in CCMs. Among them, top DE lncRNAs SMIM25 and LBX2-AS1 were having correlative expression patterns with significant number of differentially expressed (DE) PCGs. Co-expression analysis followed by gene set enrichment prediction revealed important signaling pathways that have been previously established to be pivotal in CCMs disease development and progression. We therefore, believe that our transcriptomics-wide approach might unfold uncovered functional roles of SMIM25 and LBX2-AS1 and their co-expressed PCGs in CCMs pathogenesis.
SMIM25, also known as LINC01272/GCRL1 belongs to the class of long intergenic non-coding RNA and is located in the chromosome 20. Although not well characterized in humans, recent emerging evidence suggests its association with various diseases, such as lung squamous cell carcinoma (LUSC), gastric cancer and in inflammatory bowel diseases52,53,54,55. Consistent with our data, the expression of SMIM25 was found to be strongly increased in inflammatory bowel diseases and gastric cancer. Additionally, Lin et al. showed that upregulation of SMIM25 is associated with gastric cell proliferation and metastasis while Haberman et al. demonstrated a myeloid pro-inflammatory function of SMIM25 in pediatric Crohn disease. Wang and co-authors indicated that SMIM25 may be used as potential diagnostic biomarkers in inflammatory bowel disease. We show here through RNA-Seq approach that SMIM25 transcript was strongly upregulated through RNA-Seq approach and its expression change was further confirmed by qRT-PCR analysis. Functional enrichment analysis revealed CCM-relevant signaling pathways such as Rho GTPase, ROS, toll-like receptor cascades, MAP3 activation and receptor tyrosine kinase targeted by SMIM25 as highly enriched entities. Among the total SMIM25-specific co-expressed PCGs, we identified FNDC3B, FYB1, AURKA, GMIP, COTL1, RETN, ARPC3, RPS6KA1, LSP1, VAV3, CD36, OS9, ETV6, MYO1F, GSTO1, ADAM9, PTAFR, LIMS1, CDK1, IRAK1, HSPA1A and CLDN23 to be involved in various molecular processes such as, oxidative stress, cell-cell adhesion, integrin signaling, angiogenesis, cell proliferation, MAP kinase pathway, endothelial cell dysfunction, RhoA-GTPase activity and PI3K signaling pathways that have been previously reported to be involved in CCMs disease. For example, CD36 known as cluster of differentiation 36, is an integral membrane protein which interacts with β1 integrins to mediate Thrombospondins TSP-1 mediated apoptosis by antagonizing pro-survival pathways56. Reduced TSP-1 expression has been reported to contribute to CCMs pathogenesis in acute Kriti inactivated brain microvascular endothelial cells (BMECs)57. We, therefore, believe that CD36 might have a role to play in CCM lesion and should be studied in further detail. Another crucial PCG co-expressed with SMIM25 was identified to be glutathione S-transferase omega-1 (GSTO1). GSTO1 is a pro-inflammatory molecule and is essential for the formation of reactive oxygen species31 through activation of the Toll-like receptor 4 (TLR4) cascade58. Recently, Tang and co-workers indicated that endothelial TLR4 and the gut microbiome as critical stimulators of CCMs formation while pharmacological blockage of TLR4 signaling reduced CCMs lesion burden in mice59. These data suggest that GSTO1 might also be involved in CCMs lesion formation and further studies should be conducted to uncover its potential functions. A third PCG, cyclin-dependent kinase 1 (CDK1) was identified as a potential candidate co-expressed with SMIM25. CDK1 function as a serine/threonine kinase and acts as a potent target for the bioactive ingredient, indirubin-3-monoxime (IR3mo)60. In a recent study, Otten and co-workers implemented a target prediction tools and identified IR3mo as a novel candidate in rescuing CCMs phenotype in zebrafish, mice and HUVEC models61. It would therefore be intriguing to conduct further studies and determine if CDK1 might have a functional role in CCMs pathogenesis.
LBX2-AS1 or ladybird homeobox 2 antisense RNA 1 belongs to antisense long non-coding RNA class and is located within the chromosome 2. Recent reports indicate its importance in gliomas, esophageal squamous cell carcinoma and lung adenocarcinoma28,62,63,64. Intriguingly, Liang and co-workers applied weighted gene co-expression network analysis (WGCNA) and showed that LBX2-AS1 was significantly associated with focal adhesion, extracellular matrix receptor (ECM) interaction and MAPK signaling pathways (Liang et al., 2018). Our results showing a strong up-regulation of LBX2-AS1 might suggest its significant role in CCMs disease. Furthermore, we also identified several PCGs such as ARHGAP25, BAX, MKI67, PFN1, TGFβ1, ANXA2, CELSR1, CCR1, CDCP1, CKLF, CLDN7, EMILIN2, FERMT3, GRN, HEXA, IGF2R, LGALS9, MAPKAPK3, P2RX4, RNF213, TMEM106A and TRADD known to be associated with various processes like, inflammation, angiogenesis, cell proliferation, Rho GTPase, β-catenin pathway, adherens junctions, apoptosis, cell adhesion, MAP kinase/Erk pathway and nitric oxide pathways. Among them, Bcl-2-associated protein, BAX, a pro-apoptotic regulator has been established to be involved in CCMs pathogenesis. Recent studies from Antognelli and co-workers demonstrated that the pro-apoptotic protein BAX was significantly increased in KRIT1−/− cells, which are characterized by an elevated apoptotic rate65. We also identified Antigen-KI-67 (MKI67), a cell proliferation marker co-expressed with LBX2-AS1. MKI67 has been reported to be strongly expressed in CCMs tissues, suggesting high proliferative index in these lesions66,67. Transforming growth factor beta 1 or TGF-β1, a polypeptide belonging to the transforming growth factor beta superfamily of cytokines was identified as a co-expressed PCG with LBX2-AS1. The role of TGF-β1 has been previously established in CCMs diseases. Maddaluno et al., demonstrated that the TGF-β1 signaling pathway activates endothelial to mesenchymal transition (EndMT) in CCM1-ablated endothelial cells and inhibition of the TGF-β1 signaling pathway prevented EndMT both in vitro and in vivo, subsequently reducing the size of vascular lesions in CCM1-deficient mice68. Additionally, Bravi and co-workers reported that TGF-β1 signaling is triggered downstream following β-catenin activation in CCM3-deficient endothelial cells both in vitro and in vivo and leads to endothelial-to-mesenchymal transition, implying TGF-β/BMP signaling as crucial regulators for CCMs disease progression69. Profilin 1 (PFN1) an actin monomer-binding protein belonging to the profilin family was identified to be a PCG co-expressed with LBX2-AS1. It has been previously established that profilin acts as a binding partner for the junctional multidomain protein, Afadin-6 (AF-6)70. Earlier Glading et al. have shown that KRIT-1 associates with Rap1 small GTPases and transfection of BAECs with activated Rap1 (RapV12) led to increased association of endogenous KRIT-1 with β-catenin and AF-671. Contrarily, their association was strongly inhibited following expression of the Rap activity inhibitor (Rap1GAP). Further studies will discern the role of profilin in CCMs pathogenesis.
Besides, we identified 62 PCGs that were commonly co-expressed with SMIM25 and LBX2-AS1. Among them, ALDH3B1, ARPC3, LIMS1, LSP1, PTAFR and VAV3 have been previously validated to play crucial roles in oxidative stress72, adherens junction pathway73, focal adhesion74, PECAM1-mediated expression in endothelial cells75, VEGF-mediated angiogenesis76 and vascular endothelial cell integrity77. These observations in particular warrants further studies to explore their functional relevance in CCMs disease.
The Gene Ontology for biological process (GOBP), cellular components (GOCC) and molecular functions (GOMF) analysis revealed VEGF receptor signaling pathway, cell junction assembly, angiogenesis, NF-kB transcription activity, focal adhesion, actin cytoskeleton, PI3K complex, TLR-receptor binding and NAD(P)H oxidase activity as highly enriched processes in CCMs pathogenesis. The Reactome pathway analysis identified immune system, TLR-receptor cascades, signaling by interleukins, Rho GTPases signaling, cytokine signaling and reactive oxygen species31 as significantly enriched pathways. Among this, VEGF is one of the key factors for CCMs pathogenesis which induces endothelial proliferation (Fig. 3b)78. There is also evidence showing that CCM1 expression resulted in significant reduction of VEGF-induced sprout formation during endothelial cell differentiation and capillary formation in a 3D spheroidal system79. The authors also detected that cell migration in CCM-expressing HUVECs was significantly delayed in a Boyden chamber using VEGF as a chemoattractant. A previous study has also shown that loss of CCM2 increased Rho GTPase activity in CCMs80. Consistent with this previous study our analysis also predicted Rho GTPase related pathways were enriched with CCMs up-regulated genes while there is no significant expression of CCM2 gene (Figs. 1e and 3b). Apart from these known pathways in CCMs, it is also important to thoroughly investigate other biological pathways reported in this study to understand the importance of these DE lncRNAs. Additionally, phenotype enrichment analysis reflected terms related to CCMs symptoms such as, seizures, neurological speech impairment (focal neurologic deficit) and venous thrombosis (Fig. 3b). All this evidence suggests that CCMs differentially expressed lncRNAs with similar expression patterns as DE PCGs might co-regulate important pathways relevant to CCMs pathogenesis.
We provide here a comprehensive transcriptomic profile identifying lncRNAs and their co-expressed PCGs in human resected CCM lesions. To the best of our knowledge, this is the first high-throughput study to show their differential expression patterns in brainstem CCMs. Our results identified lncRNAs, SMIM25 and LBX2-AS1 and their commonly co-expressed PCGs; ALDH3B1, ARPC3, LIMS1, LSP1, PTAFR and VAV3 as strong candidates in human CCMs disease. Future studies understanding their functional role in CCMs biology will open new perspectives for better therapeutic targets for disease prevention and treatment.
Materials and Methods
Ethical statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the responsible committee (institutional and national) and with the 1964 Helsinki declaration and its later amendments. The study protocol was approved by the local ethical committee at the Hannover Medical School, Germany (Approval Number 6960) and the Ethics Committee of University Bonn Medical Center (Approval Number 076/08 and 042/08). Written informed consent was collected from all the patients involved in this study.
Patients and clinical data
Fresh tissue biopsies were obtained intraoperatively from 10 adult patients (CCM1-10) affected due CCMs within the brainstem and stored at −80 °C until further experiments. Normal brain tissues from 4 subjects who underwent temporal lobe epilepsy (TLE) surgery at the University Hospital Bonn, Germany served as corresponding controls. The control tissue samples were treated in an identical manner as CCMs tissues. The clinical diagnosis of the CCMs was based on MRI and histopathological characteristics as previously described81. Each CCMs patient manifested at least one or more clinical episodes of seizures, hemorrhage and focal neurological deficits. The patient clinical characteristics are described in Tables 1 and 2. For research involving participant or patient under the age of 18 years (including donors of tissue samples), informed consent was taken from the parent.
RNA isolation
Tissue sections were stored in at least 10 volumes of RNAlater-ICE (AM7030; Ambion) at −80 °C before total RNA extraction. For RNA extraction, brain tissue specimens were cut off and further slit to smaller pieces. These pieces were transferred into vials containing ceramic beads (Precellys system, CK14L; Peqlab) and 700 µl of RLT lysis buffer from RNeasy Micro Kit (74004; Qiagen) including 1% of beta-mercaptoethanol. Homogenization was performed by use of the Precellys 24 Homogenizer (Peqlab) with 2–4 pulses of 5 sec at 6000 rpm. Total RNA was extracted with RNeasy Micro Kit according to the company’s recommendations including an on-column DNase-I digestion step (5 minutes at RT). Total RNA was finally eluted with 14 µl of RNase free water. RNA samples were quantified and purity was determined with the Nanodrop ND-1000 spectrophotometer (Peqlab). RNA integrity was determined with an Agilent Bioanalyzer 2100 using the RNA 6000-Nano assay (5067-1511; Agilent Technologies). RNA Integrity Numbers (RINs) of samples used for RNA-Sequencing experiments ranged from 7.0 to 8.4 with an average RIN of 7.6 for the CCMs samples and 7.5 for the control samples.
RNA library preparation, quality control, and quantification
200 ng of total RNA per sample were utilized as input for rRNA depletion procedure with ‘NEBNext® rRNA Depletion Kit (Human/Mouse/Rat), 96 rxns’ (E6310X; New England Biolabs) followed by stranded cDNA library generation using ‘NEBNext® Ultra Directional RNA Library Prep Kit for Illumina’ (E7420L; New England Biolabs). All steps were performed as recommended in user manualE7420 (Version 6.0_08-2015; NEB) except that all reactions were downscaled to 2/3 of initial volumes. Furthermore, one additional purification step was introduced at the end of the standard procedure, using 1x ‘Agencourt® AMPure® XP Beads’ (#A63881; Beckman Coulter, Inc.).
cDNA libraries were barcoded by single indexing approach, using ‘NEBNext Multiplex Oligos for Illumina – Set 1’ (Index Primer 2, 3, 4, 5, 6, 7, and 12). All generated cDNA libraries were amplified by 11 cycles of final PCR. Fragment length distribution of individual libraries was monitored using ‘Bioanalyzer High Sensitivity DNA Assay’ (5067-4626; Agilent Technologies). Quantification of libraries was performed by use of the ‘Qubit® dsDNA HS Assay Kit’ (Q32854; ThermoFisher Scientific).
Library denaturation and sequencing run
Equal molar amounts of seven individually barcoded libraries were pooled. This library pool was denatured with NaOH and was finally diluted to 1.5pM according to the Denature and Dilute Libraries Guide (Document #15048776 v02; Illumina). 1.3 ml of denatured pool was loaded on an Illumina NextSeq. 550 sequencer using a High Output Flowcell for 75 bp single read sequencing (#FC-404-2005; Illumina).
Raw data processing and quality control
BCL files were converted to FASTQ files using bcl2fastq Conversion Software version 2.16.0.10 (Illumina). The FASTQ files were adapter and quality trimmed using Trim Galore (version 0.4.1) with default settings as described in the User Guide except for the setting of the quality cutoff (−q/−quality) which was set to a Phred score of 15. Trim Galore used Cutadapt (version 1.9.1) as subroutine. Quality control of FASTQ files was performed by FastQC (version 0.11.4) before and after trimming.
Transcriptome mapping and assembly
After trimming, FASTQ files were mapped against a reference human genome hg38 with the splice-aware aligner STAR (version 2.5.0c)82 to generate BAM files. The BAM files were built in a 2-pass Mapping (–twopassMode Basic) and were finally sorted (–outSAMtype BAM SortedByCoordinate). All other setting have been left as default as described in the manual. Homo sapiens sequence and annotation data (UCSC, build hg38) used from illumina’s iGenome site (http://support.illumina.com/sequencing/sequencing_software/igenome.html).
Differential expression and lncRNA-mRNA co-expression analysis
Obtained aligned files (BAM format) were subjected to quantification using featureCounts from Subread package83. The reads were quantified for Ensembl transcript annotation release GRCh38.93 (corresponds to genome hg38)84 with featureCounts parameters ‘–minOverlap 10-Q30-s2-ignoreDup -J’. Generated matrix file from read quantification was then used for differential expression (DE) analysis. DE analysis was performed using Bioconductor package DESeq285 with contrast of normal and CCMs groups. Transcripts (lncRNAs and protein coding genes) having corrected p-value or Padj <0.01 (FDR) and absolute log-fold change greater than 1.5 were considered to be significantly differentially expressed (DE). For calculating the coding potential of DE lncRNAs and PCGs we started by extracting nucleotide sequence (FASTA) of the DE transcripts using BioMart from Ensembl. Obtained sequences (FASTA) are used as an input for CPC2 (Coding potential calculator v2) tool to predict coding probability of the transcripts86.
Expression patterns of DE lncRNAs were compared against DE protein coding genes (PCGs) to look for expression correlation. Significant lncRNA-mRNA correlated pairs were calculated using Spearman correlation coefficient by considering R value above 0.9 and with p-value < 0.05. The co-expression network linking lncRNA-mRNA pairs were constructed using Cytoscape87.
Gene set enrichment analysis
PCGs co-expressed (significantly correlated) with top validated lncRNAs was used for gene set enrichment analysis. These PCGs were used to predict phenotype (Human Phenotype Ontology), gene ontology (GOBP, GOCC, GOMF) and molecular pathways (Reactome) using GeneSCF tool46. Significant terms were considered, if having p-value < 0.05.
Processing RNA-seq public dataset from multi-tissues
Human BodyMap transcriptome sequencing data (E-MTAB-513) was processed by aligning with STAR82, quantified with featureCounts83 and normalized as Transcript Per Million (TPM). Expression of CCMs differentially expressed and top correlated lncRNAs were tested in 2 × 16 (two replicates each) normal tissue samples including brain. The heatmap from Fig. 2c is plotted by calculating z-score or standard score from the normalized TPM values to check specificity of DE lncRNAs in different tissues.
Processing CCM patient RNA-seq public dataset
RNA-seq samples of 5 CCM patients and 3 healthy samples from neurovascular units (NVU) were downloaded from ‘GSE123968’. Samples were processed and treated in similar way as cohort from current study and also similar to Human BodyMap RNA-seq samples. We combined RNA-seq samples from our cohort (Controls = 4; CCMs = 10) with this Koskimäki J et al. 2019 cohort (Controls = 3; CCMs = 5). Differential expression analysis was performed using Bioconductor package DESeq2 with contrast of normal (7 controls) and CCMs groups (15 CCMs). Transcripts (lncRNAs and protein coding genes) having corrected p-value or Padj <0.01 (FDR) and absolute log-fold change greater than 1.5 were considered to be significantly differentially expressed (DE).
Technical validation of lncRNA expression by qRT-PCR
In order to validate the reliability of RNA-sequencing data, we randomly selected SMIM25 and LBX2-AS1, the two most strongly upregulated lncRNAs for further analysis. SYBR green-based quantitative polymerase chain reaction (qRT-PCR) assay was used for this purpose. Briefly, DNase1-treated total RNA of 2 µg was reverse transcribed to cDNA using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). The expression analysis of the SMIM25 and LBX2-AS1 primer was carried out using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems) on a StepOnePlus Real-time PCR (Applied Biosystems). The specific forward and reverse primers for the lncRNAs were designed using Primer 5.0. Thermal cycling conditions consisted of an initial denaturing step at 95 °C for 10 min and then 40 cycles. The relative expression and fold change for the SMIM25 and LBX2-AS1 lncRNA was normalized by HPRT using the 2−ΔΔCt method and each qRT-PCR assay was repeated in triplicates.
Statistical analysis
All experimental graphs from qRT-PCR are presented as mean ± standard error of the mean and a p value less than 0.05 value was considered statistically significant. The p-values of qRT-PCR was derived using two-tailed student t-test. From RNA-seq, the significantly differentially expressed lncRNAs and protein coding genes were determined by DESeq285 and filtered using adjusted p value and log-fold change values.
Ethics declarations
Written informed consent was collected from all the patients involved in this study.
Data availability
The RNA-seq data used in this publication can be accessed from GEO repository with accession GSE137596 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137596).
References
Kar, S. et al. Genome-Wide Sequencing Reveals MicroRNAs Downregulated in Cerebral Cavernous Malformations. J Mol Neurosci 61, 178–188, https://doi.org/10.1007/s12031-017-0880-6 (2017).
Koskimaki, J. et al. Comprehensive transcriptome analysis of cerebral cavernous malformation across multiple species and genotypes. JCI Insight 4, https://doi.org/10.1172/jci.insight.126167 (2019).
Kar, S. et al. Genome-Wide Sequencing Reveals Small Nucleolar RNAs Downregulated in Cerebral Cavernous Malformations. Cell Mol Neurobiol 38, 1369–1382, https://doi.org/10.1007/s10571-018-0602-9 (2018).
Velz, J., Bozinov, O., Sarnthein, J., Regli, L. & Bellut, D. The current management of spinal cord cavernoma. J Neurosurg Sci 62, 383–396, https://doi.org/10.23736/S0390-5616.18.04305-9 (2018).
Draheim, K. M., Fisher, O. S., Boggon, T. J. & Calderwood, D. A. Cerebral cavernous malformation proteins at a glance. J Cell Sci 127, 701–707, https://doi.org/10.1242/jcs.138388 (2014).
Baranoski, J. F., Kalani, M. Y., Przybylowski, C. J. & Zabramski, J. M. Cerebral Cavernous Malformations: Review of the Genetic and Protein-Protein Interactions Resulting in Disease Pathogenesis. Front Surg 3, 60, https://doi.org/10.3389/fsurg.2016.00060 (2016).
Cianfruglia, L. et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins. Antioxidants (Basel) 8, https://doi.org/10.3390/antiox8010027 (2019).
Subhash, S. et al. H3K4me2 and WDR5 enriched chromatin interacting long non-coding RNAs maintain transcriptionally competent chromatin at divergent transcriptional units. Nucleic Acids Res 46, 9384–9400, https://doi.org/10.1093/nar/gky635 (2018).
Mondal, T. et al. Sense-Antisense lncRNA Pair Encoded by Locus 6p22.3 Determines Neuroblastoma Susceptibility via the USP36-CHD7-SOX9 Regulatory Axis. Cancer Cell 33, 417–434 e417, https://doi.org/10.1016/j.ccell.2018.01.020 (2018).
Kataruka, S., Akhade, V. S., Kayyar, B. & Rao, M. R. S. Mrhl Long Noncoding RNA Mediates Meiotic Commitment of Mouse Spermatogonial Cells by Regulating Sox8 Expression. Mol Cell Biol 37, https://doi.org/10.1128/MCB.00632-16 (2017).
Kapranov, P. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488, https://doi.org/10.1126/science.1138341 (2007).
Ransohoff, J. D., Wei, Y. & Khavari, P. A. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 19, 143–157, https://doi.org/10.1038/nrm.2017.104 (2018).
Ren, W. & Yang, X. Pathophysiology of Long Non-coding RNAs in Ischemic Stroke. Front Mol Neurosci 11, 96, https://doi.org/10.3389/fnmol.2018.00096 (2018).
Zhang, L. et al. LncRNA SNHG1 regulates cerebrovascular pathologies as a competing endogenous RNA through HIF-1alpha/VEGF signaling in ischemic stroke. J Cell Biochem 119, 5460–5472, https://doi.org/10.1002/jcb.26705 (2018).
Dykstra-Aiello, C. et al. Altered Expression of Long Noncoding RNAs in Blood After Ischemic Stroke and Proximity to Putative Stroke Risk Loci. Stroke 47, 2896–2903, https://doi.org/10.1161/STROKEAHA.116.013869 (2016).
Dharap, A., Nakka, V. P. & Vemuganti, R. Effect of focal ischemia on long noncoding RNAs. Stroke 43, 2800–2802, https://doi.org/10.1161/STROKEAHA.112.669465 (2012).
Torring, P. M. et al. Long non-coding RNA expression profiles in hereditary haemorrhagic telangiectasia. PLoS One 9, e90272, https://doi.org/10.1371/journal.pone.0090272 (2014).
Li, H. et al. Identification of a Long Noncoding RNA-Associated Competing Endogenous RNA Network in Intracranial Aneurysm. World Neurosurg 97, 684–692 e684, https://doi.org/10.1016/j.wneu.2016.10.016 (2017).
Guo, Q. et al. Comprehensive analysis of lncRNA-mRNA co-expression patterns identifies immune-associated lncRNA biomarkers in ovarian cancer malignant progression. Sci Rep 5, 17683, https://doi.org/10.1038/srep17683 (2015).
Wu, W. et al. Tissue-specific Co-expression of Long Non-coding and Coding RNAs Associated with Breast Cancer. Sci Rep 6, 32731, https://doi.org/10.1038/srep32731 (2016).
Luo, S. et al. Divergent lncRNAs Regulate Gene Expression and Lineage Differentiation in Pluripotent Cells. Cell Stem Cell 18, 637–652, https://doi.org/10.1016/j.stem.2016.01.024 (2016).
Starke, R. M. et al. Developments in Neurovascular Diseases and Treatments. ScientificWorldJournal 2015, 608607, https://doi.org/10.1155/2015/608607 (2015).
Sampath, K. & Ephrussi, A. CncRNAs: RNAs with both coding and non-coding roles in development. Development 143, 1234–1241, https://doi.org/10.1242/dev.133298 (2016).
Velmeshev, D., Magistri, M. & Faghihi, M. A. Expression of non-protein-coding antisense RNAs in genomic regions related to autism spectrum disorders. Mol Autism 4, 32, https://doi.org/10.1186/2040-2392-4-32 (2013).
Baranzini, S. E. et al. Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 133, 2603–2611, https://doi.org/10.1093/brain/awq192 (2010).
Feng, J. et al. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev 20, 1470–1484, https://doi.org/10.1101/gad.1416106 (2006).
Cajigas, I. et al. The Evf2 Ultraconserved Enhancer lncRNA Functionally and Spatially Organizes Megabase Distant Genes in the Developing Forebrain. Mol Cell 71, 956–972 e959, https://doi.org/10.1016/j.molcel.2018.07.024 (2018).
Liang, R. et al. Analysis of long non-coding RNAs in glioblastoma for prognosis prediction using weighted gene co-expression network analysis, Cox regression, and L1-LASSO penalization. Onco Targets Ther 12, 157–168, https://doi.org/10.2147/OTT.S171957 (2019).
Gao, Y. S., Liu, X. Z., Zhang, Y. G., Liu, X. J. & Li, L. Z. Knockdown of Long Noncoding RNA LUCAT1 Inhibits Cell Viability and Invasion by Regulating miR-375 in Glioma. Oncol Res 26, 307–313, https://doi.org/10.3727/096504017X15088061795756 (2018).
Cao, Y. P. et al. Long Non-Coding RNA Expression Profiles for the Characterization of Different Bladder Cancer Grade. Cell Physiol Biochem 50, 1154–1163, https://doi.org/10.1159/000494542 (2018).
Mao, W. et al. Shank1 regulates excitatory synaptic transmission in mouse hippocampal parvalbumin-expressing inhibitory interneurons. Eur J Neurosci 41, 1025–1035, https://doi.org/10.1111/ejn.12877 (2015).
Serrador, J. M. et al. Moesin interacts with the cytoplasmic region of intercellular adhesion molecule-3 and is redistributed to the uropod of T lymphocytes during cell polarization. J Cell Biol 138, 1409–1423, https://doi.org/10.1083/jcb.138.6.1409 (1997).
Dufner, A. et al. CARD6 is interferon inducible but not involved in nucleotide-binding oligomerization domain protein signaling leading to NF-kappaB activation. Mol Cell Biol 28, 1541–1552, https://doi.org/10.1128/MCB.01359-07 (2008).
Choquet, H., Pawlikowska, L., Lawton, M. T. & Kim, H. Genetics of cerebral cavernous malformations: current status and future prospects. J Neurosurg Sci 59, 211–220 (2015).
Petit, N., Blecon, A., Denier, C. & Tournier-Lasserve, E. Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr Patterns 6, 495–503, https://doi.org/10.1016/j.modgep.2005.11.001 (2006).
Cave-Riant, F. et al. Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with Cerebral Cavernous Malformations. Eur J Hum Genet 10, 733–740, https://doi.org/10.1038/sj.ejhg.5200870 (2002).
Mort, M., Ivanov, D., Cooper, D. N. & Chuzhanova, N. A. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat 29, 1037–1047, https://doi.org/10.1002/humu.20763 (2008).
Benton, M. C. et al. Mapping eQTLs in the Norfolk Island genetic isolate identifies candidate genes for CVD risk traits. Am J Hum Genet 93, 1087–1099, https://doi.org/10.1016/j.ajhg.2013.11.004 (2013).
Gamazon, E. R. et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 50, 956–967, https://doi.org/10.1038/s41588-018-0154-4 (2018).
Huang, J. et al. eQTL mapping identifies insertion- and deletion-specific eQTLs in multiple tissues. Nat Commun 6, 6821, https://doi.org/10.1038/ncomms7821 (2015).
Zhang, W. et al. A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat Genet 50, 613–620, https://doi.org/10.1038/s41588-018-0091-2 (2018).
Gao, Y. et al. Long Non-Coding RNA HOXA-AS2 Regulates Malignant Glioma Behaviors and Vasculogenic Mimicry Formation via the MiR-373/EGFR Axis. Cell Physiol Biochem 45, 131–147, https://doi.org/10.1159/000486253 (2018).
Bhat, S. A. et al. Long non-coding RNAs: Mechanism of action and functional utility. Noncoding RNA Res 1, 43–50, https://doi.org/10.1016/j.ncrna.2016.11.002 (2016).
Marchese, F. P., Raimondi, I. & Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol 18, 206, https://doi.org/10.1186/s13059-017-1348-2 (2017).
Yu, B. & Wang, S. Angio-LncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics 8, 3654–3675, https://doi.org/10.7150/thno.26024 (2018).
Subhash, S. & Kanduri, C. GeneSCF: a real-time based functional enrichment tool with support for multiple organisms. BMC Bioinformatics 17, 365, https://doi.org/10.1186/s12859-016-1250-z (2016).
Padarti, A. & Zhang, J. Recent advances in cerebral cavernous malformation research. Vessel Plus 2, https://doi.org/10.20517/2574-1209.2018.34 (2018).
Baxter, S. S. et al. Role of cytoskeletal proteins in cerebral cavernous malformation signaling pathways: a proteomic analysis. Mol Biosyst 10, 1881–1889, https://doi.org/10.1039/c3mb70199a (2014).
Fisher, O. S. & Boggon, T. J. Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell Mol Life Sci 71, 1881–1892, https://doi.org/10.1007/s00018-013-1532-9 (2014).
Tsoi, L. C. et al. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol 16, 24, https://doi.org/10.1186/s13059-014-0570-4 (2015).
Huang, F. et al. Toward Understanding Non-coding RNA Roles in Intracranial Aneurysms and Subarachnoid Hemorrhage. Transl Neurosci 8, 54–64, https://doi.org/10.1515/tnsci-2017-0010 (2017).
Chen, W. J. et al. Clinical roles of the aberrantly expressed lncRNAs in lung squamous cell carcinoma: a study based on RNA-sequencing and microarray data mining. Oncotarget 8, 61282–61304, https://doi.org/10.18632/oncotarget.18058 (2017).
Lin, Z. et al. Long noncoding RNA gastric cancer-related lncRNA1 mediates gastric malignancy through miRNA-885-3p and cyclin-dependent kinase 4. Cell Death Dis 9, 607, https://doi.org/10.1038/s41419-018-0643-5 (2018).
Wang, S. et al. KIF9AS1, LINC01272 and DIO3OS lncRNAs as novel biomarkers for inflammatory bowel disease. Mol Med Rep 17, 2195–2202, https://doi.org/10.3892/mmr.2017.8118 (2018).
Haberman, Y. et al. Long ncRNA Landscape in the Ileum of Treatment-Naive Early-Onset Crohn Disease. Inflamm Bowel Dis 24, 346–360, https://doi.org/10.1093/ibd/izx013 (2018).
Lawler, P. R. & Lawler, J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med 2, a006627, https://doi.org/10.1101/cshperspect.a006627 (2012).
Lopez-Ramirez, M. A. et al. Thrombospondin1 (TSP1) replacement prevents cerebral cavernous malformations. J Exp Med 214, 3331–3346, https://doi.org/10.1084/jem.20171178 (2017).
Menon, D., Coll, R., O’Neill, L. A. & Board, P. G. GSTO1-1 modulates metabolism in macrophages activated through the LPS and TLR4 pathway. J Cell Sci 128, 1982–1990, https://doi.org/10.1242/jcs.167858 (2015).
Tang, A. T. et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 545, 305–310, https://doi.org/10.1038/nature22075 (2017).
Marko, D. et al. Inhibition of cyclin-dependent kinase 1 (CDK1) by indirubin derivatives in human tumour cells. Br J Cancer 84, 283–289, https://doi.org/10.1054/bjoc.2000.1546 (2001).
Otten, C. et al. Systematic pharmacological screens uncover novel pathways involved in cerebral cavernous malformations. EMBO Mol Med 10, https://doi.org/10.15252/emmm.201809155 (2018).
Wu, F. et al. Expression profile analysis of antisense long non-coding RNA identifies WDFY3-AS2 as a prognostic biomarker in diffuse glioma. Cancer Cell Int 18, 107, https://doi.org/10.1186/s12935-018-0603-2 (2018).
Zhang, Y. et al. LBX2-AS1 is activated by ZEB1 and promotes the development of esophageal squamous cell carcinoma by interacting with HNRNPC to enhance the stability of ZEB1 and ZEB2 mRNAs. Biochem Biophys Res Commun 511, 566–572, https://doi.org/10.1016/j.bbrc.2019.02.079 (2019).
Tian, Z. et al. Identification of dysregulated long non-coding RNAs/microRNAs/mRNAs in TNM I stage lung adenocarcinoma. Oncotarget 8, 51703–51718, https://doi.org/10.18632/oncotarget.18512 (2017).
Antognelli, C. et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic Biol Med 115, 202–218, https://doi.org/10.1016/j.freeradbiomed.2017.11.014 (2018).
Maiuri, F. et al. Clinical progression and familial occurrence of cerebral cavernous angiomas: the role of angiogenic and growth factors. Neurosurg Focus 21, e3 (2006).
Li, Q. F., Decker-Rockefeller, B., Bajaj, A. & Pumiglia, K. Activation of Ras in the Vascular Endothelium Induces Brain Vascular Malformations and Hemorrhagic Stroke. Cell Rep 24, 2869–2882, https://doi.org/10.1016/j.celrep.2018.08.025 (2018).
Maddaluno, L. et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498, 492–496, https://doi.org/10.1038/nature12207 (2013).
Bravi, L. et al. Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc Natl Acad Sci USA 112, 8421–8426, https://doi.org/10.1073/pnas.1501352112 (2015).
Boettner, B., Govek, E. E., Cross, J. & Van Aelst, L. The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc Natl Acad Sci USA 97, 9064–9069, https://doi.org/10.1073/pnas.97.16.9064 (2000).
Glading, A., Han, J., Stockton, R. A. & Ginsberg, M. H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 179, 247–254, https://doi.org/10.1083/jcb.200705175 (2007).
Marchitti, S. A., Brocker, C., Orlicky, D. J. & Vasiliou, V. Molecular characterization, expression analysis, and role of ALDH3B1 in the cellular protection against oxidative stress. Free Radic Biol Med 49, 1432–1443, https://doi.org/10.1016/j.freeradbiomed.2010.08.004 (2010).
Yi, S., Wang, X. H. & Xing, L. Y. Transcriptome analysis of adherens junction pathway-related genes after peripheral nerve injury. Neural Regen Res 13, 1804–1810, https://doi.org/10.4103/1673-5374.237127 (2018).
Sandfort, V., Eke, I. & Cordes, N. The role of the focal adhesion protein PINCH1 for the radiosensitivity of adhesion and suspension cell cultures. PLoS One 5, https://doi.org/10.1371/journal.pone.0013056 (2010).
Hossain, M. et al. Endothelial LSP1 Modulates Extravascular Neutrophil Chemotaxis by Regulating Nonhematopoietic Vascular PECAM-1 Expression. J Immunol 195, 2408–2416, https://doi.org/10.4049/jimmunol.1402225 (2015).
Bhosle, V. K. et al. Nuclear localization of platelet-activating factor receptor controls retinal neovascularization. Cell Discov 2, 16017, https://doi.org/10.1038/celldisc.2016.17 (2016).
Hilfenhaus, G. et al. Vav3-induced cytoskeletal dynamics contribute to heterotypic properties of endothelial barriers. J Cell Biol 217, 2813–2830, https://doi.org/10.1083/jcb.201706041 (2018).
Jung, K. H. et al. Cerebral cavernous malformations with dynamic and progressive course: correlation study with vascular endothelial growth factor. Arch Neurol 60, 1613–1618, https://doi.org/10.1001/archneur.60.11.1613 (2003).
Wustehube, J. et al. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci USA 107, 12640–12645, https://doi.org/10.1073/pnas.1000132107 (2010).
Whitehead, K. J. et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med 15, 177–184, https://doi.org/10.1038/nm.1911 (2009).
Bertalanffy, H. et al. Cerebral cavernomas in the adult. Review of the literature and analysis of 72 surgically treated patients. Neurosurg Rev 25, 1–53; discussion 54–55 (2002).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, https://doi.org/10.1093/bioinformatics/btt656 (2014).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res 46, D754–D761, https://doi.org/10.1093/nar/gkx1098 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Kang, Y. J. et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res 45, W12–W16, https://doi.org/10.1093/nar/gkx428 (2017).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504, https://doi.org/10.1101/gr.1239303 (2003).
Acknowledgements
This work was supported by the grants from Knut and Alice Wallenberg Foundation [KAW2014.0057]; Swedish Foundation for Strategic Research [RB13-0204]; Swedish Cancer Research foundation [Cancerfonden: Kontraktno. CAN2018/591]; Swedish Research Council [2017-02834]; Barncancerfonden [PR2018-0090]; Ingabritt Och Arne Lundbergsforskningsstiftelse and LUA/ALF (to C.K.). Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
S.S., S.K. and C.K. conceptualized and designed the study; S.S. performed the computational analysis; S.S. and S.K. contributed to the data interpretation; S.S. and S.K. wrote the manuscript; N.K. and K.K.B. performed the qRT-PCR experiments and helped in the analysis; C.H. helped in the patient clinical data analysis; T.G. and O.D.-B. carried out the RNA-seq; H.B. provided the CCMs tissue samples; W.S.K. provided the control TLE tissues; H.B., A.S., F.W., S.P. and C.K. critically edited the manuscript. All authors approved the manuscript in its current form.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Subhash, S., Kalmbach, N., Wegner, F. et al. Transcriptome-wide Profiling of Cerebral Cavernous Malformations Patients Reveal Important Long noncoding RNA molecular signatures. Sci Rep 9, 18203 (2019). https://doi.org/10.1038/s41598-019-54845-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54845-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
