
Abstract
The giant river prawn (Macrobrachium rosenbergii) is one of the most widely cultured crustacean species. In recent years, its aquaculture has faced challenges, including the degradation of germplasm resources and the emergence of viral diseases. Genomic information can be a valuable resource for developing molecular breeding programs for this important aquaculture species. Here we constructed a high-quality reference genome for M. rosenbergii by integrating Nanopore, Illumina, and high-throughput chromosome conformation capture (Hi-C) technologies. The final genome assembly is 3.18 Gb in size, with scaffold N50 and contig N50 of 62.73 Mb and 8.92 Mb, respectively. Approximately 98.6% of the assembled sequences were anchored into 59 pseudo-chromosomes. Benchmarking Universal Single-Copy Orthologs (BUSCO) benchmark of the genome assembly reached 94.5%. Repetitive sequences comprise 43.77% of the assembled genome, and 17,436 protein-coding genes were annotated. The high-quality genome of M. rosenbergii will empower molecular breeding efforts and provide invaluable resources for comparative genomic analysis of decapod species.
Similar content being viewed by others
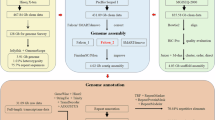

Background & Summary
The order Decapoda represents one of the largest taxa within the subphylum Crustacea, encompassing at least 180 extant families and 14,756 species1. Giant river prawn (Macrobrachium rosenbergii), native to Indo-West Pacific from northwest India to Vietnam, Philippines, New Guinea and northern Australia, is the largest known palaemonid in the world2,3. M. rosenbergii inhabits tropical freshwater environments influenced by adjacent brackish water areas, exhibiting predominantly omnivorous feeding habits throughout its life cycle4. As an active predator of freshwater ecosystems, M. rosenbergii has been utilized as an indicator for water quality and metal accumulation assessment.
Certain decapod crustaceans, such as shrimp, crabs, and lobsters, are economically important aquatic species that contribute to global food production5. M. rosenbergii is one of the most important cultured decapod species, with the global production reaching 294 thousand tons in 20225,6. However, the decline in M. rosenbergii production from 2006 to 2012 raised concerns about inbreeding depression and a reduction in effective population size7,8. Moreover, the emergence of viral diseases poses a significant challenge to the sustainable development of M. rosenbergii aquaculture9,10. Decapod iridescent virus 1 (DIV1), a recently reported member of the Iridoviridae family, has been highlighted by the World Organisation for Animal Health (WOAH) due to its pathogenic risk to a range of economically important crustaceans11,12,13. It has been reported that DIV1 infect M. rosenbergii, leading to cumulative mortalities of over 80%14. Thus, breeding new strains is urgently needed for this important aquaculture species. Genome-wide association study (GWAS), based on high-quality genome assembly, aim to identify associations between phenotypic traits and genetic variants15. The trait-linked genetic variants identified by GWAS can be directly used as molecular markers in marker-assisted selection (MAS) and genomic selection (GS) of economically important species16. Whole-genome assembly provides essential genetic resources for developing molecular breeding programs and investigating of virus-host interaction in M. rosenbergii.
Here we assembled a high-quality chromosome-level genome of M. rosenbergii by integrating Nanopore, Illumina, and high-throughput chromosome conformation capture (Hi-C) sequencing technologies. The assembled genome size is 3.18 Gb with a scaffold N50 of 62.73 Mb. Approximately 3.13 Gb (98.6%) of assembled sequences were anchored to 59 pseudo-chromosomes. A total of 17,436 protein-coding genes were annotated. Benchmarking Universal Single-Copy Orthologs (BUSCO)17 evaluation showed that the final assembly achieved a completeness of 94.5%, with annotation completeness reaching 91%. This genome assembly will provide valuable resources for breeding programs and evolutionary studies of this species.
Methods
Sample collection and sequencing
One adult male M. rosenbergii, which was collected from the experimental ponds of Zhejiang Institute of Freshwater Fisheries in Huzhou, Zhejiang Province, China, was used for genome sequencing. High-quality DNA was extracted using a DNeasy Blood & Tissue Kit (Qiagen, Germany) in accordance with the manufacturer’s protocols. DNA quality and quantity were measured through standard agarose-gel electrophoresis and a Qubit 3.0 fluorometer (Invitrogen, USA), respectively. Nanopore sequencing libraries of M. rosenbergii were constructed and sequenced using the Nanopore PromethION platform (Oxford Nanopore Technologies, UK). A total of 296.45 Gb Nanopore reads were generated for genome assembly. For Illumina sequencing, short-insert paired-end (PE) (150 bp) DNA libraries of M. rosenbergii were constructed in accordance with the manufacturer’s instructions. Sequencing of PE libraries was performed (2 × 150 bp) on the Illumina NovaSeq. 6000 platform (Illumina, USA), resulting in a total output of 161.13 Gb paired-end sequencing data. Muscle sample of M. rosenbergii were collected to construct Hi-C libraries. Hi-C library was constructed using the previously published approach18, and sequenced with 2 \(\times \) 150 bp chemistry on the Illumina Novaseq 6000 platform. In total, 277.46 Gb of Hi-C sequencing data was generated to scaffold chromosomes.
Hepatopancreas, heart, gills, eyes, and muscle samples were collected from the M. rosenbergii specimen to construct sequencing libraries for strand-specific RNA-sequencing (RNA-seq). Total RNA was extracted with TRIzol reagent (Invitrogen, USA). The mRNA was enriched from total RNA using poly-T oligo-attached magnetic beads. rRNA was removed using a TruSeq Stranded Total RNA Library Prep Kit (Illumina, USA). A PE library was constructed using a VAHTSTM mRNA-seq V2 Library Prep Kit for Illumina (Vazyme, China) and sequenced (2 × 150 bp) using the Illumina HiSeq NovaSeq.6000 platform (Illumina, USA).
Genome size estimation
Low-quality reads (≥10% unidentified nucleotides and/or ≥50% nucleotides with a Phred score <5) and sequencing adapter-contaminated Illumina reads were filtered and trimmed with Fastp (v0.21.0)19. The genome size and heterozygosity were estimated using high-quality Illumina reads based on k-mer frequency distribution. The number of k-mers and peak depth of k-mer size at k = 17 were obtained using Jellyfish (v2.3.0)20 with the -C setting. The Jellyfish results were then input into GenomeScope2 (v1.0.0)21 to estimate genome size and heterozygosity rate. The genome size of M. rosenbergii was estimated to be 3,042,399,425 bp, with a heterozygosity ratio of 1.1% (Fig. 1a).

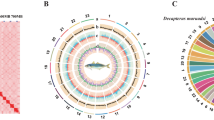
Genome assembly of M. rosenbergii. (a) Distribution of 17-mer frequency in M. rosenbergii genome. The genome size of M. rosenbergii was estimated to be 3.04 Gb. The heterozygous rate of M. rosenbergii was estimated to be 1.1%. (b) Concentric circle illustrates structural, functional, and evolutionary aspects of M. rosenbergii. a. GC content, b. Gene density, c. simple sequence repeat (SSR) density, d. Collinear regions detected within the genome.
Genome assembly
Low-quality Nanopore reads were filtered using a previously published Python script22. Three draft-genome assemblies were then generated using the filtered Nanopore reads with Wtdbg2 (v2.5)23, Flye (v2.7)24, and Shasta (v0.4.0)25, respectively. The contigs of the three draft assemblies were subjected to error correction using filtered Nanopore reads with Racon (v1.4.16)26. The corrected contigs were subsequently polished using high-quality Illumina reads with Pilon (v1.22)27. The N50 of the error-corrected contigs for the Wtdbg2, Flye, and Shasta assemblies were 0.66 Mb, 1.21 Mb and 1.22 Mb respectively. The error-corrected contigs of the three assemblies were merged into longer sequences using quickmerge (v0.3)28.
We used Hi-C to correct misjoins, to order and orient contigs, and to merge overlaps. Low-quality Hi-C reads were filtered using Fastp (v0.21.0)19. Filtered Hi-C reads were aligned to the assembled contigs using Juicer (v1.5.7)29. Scaffolding was accomplished using 3D-DNA pipeline (v180419)30. Juicebox (v2.16.0)29 was used to modify the order and direction of certain scaffolds in a Hi-C contact map and to help determine chromosome boundaries. Approximately 98.6% of the contig sequences were anchored to 59 chromosomes (Fig. 1b; Table 1). The longest and shortest chromosomes were 118 Mb and 0.73 Mb in length, respectively. The scaffold N50 reached 62.73 Mb for the final genome assembly (Table 1). BUSCO analysis with arthropod (obd10) gene set showed that the assembled M. rosenbergii genome contained 94.5% complete single-copy orthologs17 (Table 2).
Genome annotation
Repetitive elements in the M. rosenbergii genome assembly were identified by de novo predictions using RepeatMasker (v4.1.0)31. RepeatModeler (v2.0.1)32 was used to build the de novo repeat libraries of M. rosenbergii. To identify repetitive elements, sequences from the assembly were aligned to the de novo repeat library using RepeatMasker (v4.1.0). Additionally, repetitive elements in the M. rosenbergii genome assembly were identified by homology searches against known repeat databases using RepeatMasker (v4.1.0). Finally, a total of 1.39 Gb (43.77%) repetitive sequences were identified in the M. rosenbergii genome (Table 3). Long interspersed nuclear elements (LINEs) and long terminal repeats (LTR) retrotransposons were the largest class of annotated transposable elements (TEs), accounting for 7.11% and 5.93% of the M. rosenbergii genome.
Protein-coding genes in the M. rosenbergii genome were predicted using RNA-seq-based prediction. RNA-seq reads of M. rosenbergii were aligned to the reference sequence using HISAT2 (v2.1.0)33. Gene models were predicted based on the alignment results of HISAT2 using StringTie (v2.1.4)34, and coding regions were identified using TransDecoder (v5.5.0) (https://github.com/TransDecoder/TransDecoder). In total, 17,436 protein-coding genes were identified in the M. rosenbergii genome. Completeness of the predicted gene models was evaluated using BUSCO (v5.0.1)17 against the conserved Arthropoda dataset (odb10). In the predicted gene models, BUSCO analysis identified 91.0% complete conserved single-copy arthropod genes (Table 4). To assign functions to the predicted proteins, we aligned the M. rosenbergii protein models against the NCBI nonredundant (NR) amino acid sequences, Swiss-Prot, and Translated EMBL-Bank (trEMBL) using Diamond (v0.9.24)35 with an E-value cutoff of 10−5. Protein models were also aligned against the InterPro database using InterProScan (v5.63)36. In total, 98.3% (17,140) of predicted protein models were functional annotated. Specifically, 15,735 protein models were annotated in the NCBI NR database, 11,766 protein models were annotated in the Swiss-Prot database, 16,471 protein models were annotated in the InterPro database, and 15,490 protein models were annotated in the TrEMBL database.
Data Records
Genomic Illumina sequencing data and Transcriptomic sequences can be accessed in the NCBI Sequence Read Archive with accession numbers SRR3012040137 and SRR2931455438. The annotation file of the M. rosenbergii genome has been deposited at figshare (https://figshare.com/articles/dataset/Macrobrachium_rosenbergii_genome_assembly_and_gene_annotation/26068237)39. The final assembled M. rosenbergii genome has been deposited in the NCBI GenBank with accession number GCA_040412425.140.
Technical Validation
The completeness of the M. rosenbergii genome assembly was first evaluated using BUSCO (v5.1.0)34 against the conserved Arthropoda dataset (obd10). The BUSCO analysis indicated that 94.5% of conserved single-copy arthropod genes were captured in the M. rosenbergii genome (Table 2). Merqury (v1.3)41 was subsequently used to assess the quality of the assembly. The consensus quality value (QV) and k-mer completeness of the assembly was 30.29, thus suggesting good quality (Table 5). Lastly, the quality of the genome annotation was evaluated using the BUSCO software, based on the arthropoda_odb10 datasets (Table 4). This assessment revealed that the final genome annotation encompassed 91% of the arthropoda_odb10 genes, demonstrating a high completeness rate in gene predictions.
To evaluate the reliability of genome assembly and annotation of M. rosenbergii, phylogenetic tree was constructed for M. rosenbergii and 11 arthropod species. Protein sequences of the 11 species (Drosophila melanogaster, Daphnia magna, Bathynomus jamesi, Macrobrachium nipponense, Litopenaeus vannamei, Penaeus japonicus, Procambarus clarkii, Cherax quadricarinatus, Scylla paramamosain, Portunus trituberculatus and Callinectes sapidus) were downloaded for phylogenetic analysis. Orthofinder (v2.5.4)42 was applied to identify and cluster gene families among 11 reference species and M. rosenbergii. Single-copy orthologs in each gene cluster were aligned using MAFFT (v7.310)43. The alignments were trimmed using trimAL (v1.4.rev15)44. The trimmed alignments of single-copy orthologs were concatenated using PhyloSuite (v1.2.3)45. The maximum likelihood (ML) tree was generated based on the concatenated alignments using IQ-TREE (v1.6.12)46, with D. melanogaster as the outgroup (Fig. 2). Branch support was estimated using both the SH-like approximate likelihood ratio test (SH-aLRT) and ultrafast bootstrap approximation47,48. Divergence time was estimated using the MCMCTree module in the PAML (v4.9)49 package. MCMCTree analysis was performed using the maximum-likelihood tree constructed by IQ-TREE as a guide tree, and calibrated with divergence times obtained from the and TimeTree database50 (minimum = 95 million years and soft maximum = 132 million years between P. japonicus and L. vannamei; minimum = 31 million years and soft maximum = 100 million years between P. trituberculatus and C. sapidus). M. rosenbergii and M. nipponense form a clade, with an estimated divergence time of approximately 120.63 million years ago (MYA) (CI: 57.58–201.66 MYA).

Species tree of 12 arthropod species with Drosophila melanogaster as the outgroup. Bootstrap values are listed in red next to each node. Divergence time between species pairs is listed beside each node, and 95% confidence interval of estimated divergence time is listed in the parentheses. MYA, million years ago.
Code availability
No specific code or script was used in this work. Commands used for data processing were all executed according to the manuals and protocols of the corresponding software.
References
De Grave, S. et al. A Classification of Living and Fossil Genera of Decapod Crustaceans. Raffles B Zool supplement 21, 1-109, https://api.semanticscholar.org/CorpusID:80915884 (2009).
Wowor, D. & Ng, P. The giant freshwater prawns of the Macrobrachium rosenbergii species group (Crustacea, Decapoda, Caridea, Palaemonidae). Raffles B Zool 55, 321–336, https://scholarbank.nus.edu.sg/handle/10635/101924 (2007).
New, M. B. in Freshwater Prawn Culture: The Farming of Macrobrachium Rosenbergii Ch. 1, 1-11 (Blackwell Science, 2000).
FAO. Macrobrachium rosenbergii. In Cultured aquatic species fact sheets. Rome, FAO https://www.fao.org/fishery/docs/DOCUMENT/aquaculture/CulturedSpecies/file/en/en_giantriverprawn.htm (2009).
FAO. The State of World Fisheries and Aquaculture 2022. Towards Blue Transformation. Rome, FAO. https://doi.org/10.4060/cc0461en (2022).
New, M. B. & Nair, C. M. Global scale of freshwater prawn farming. Aquac. Res. 43, 960–969, https://doi.org/10.1111/j.1365-2109.2011.03008.x (2012).
Josileen, J. Freshwater prawns: biology and farming. Crustaceana 84, 509–511, https://doi.org/10.1163/001121611x564020 (2011).
Pillai, B. R. et al. Selective breeding of giant freshwater prawn (Macrobrachium rosenbergii) in India: Response to selection for harvest body weight and on-farm performance evaluation. Aquac. Res. 51, 4874–4880, https://doi.org/10.1111/are.14801 (2020).
Kibenge, F. S. Emerging viruses in aquaculture. Curr Opin Virol 34, 97–103, https://doi.org/10.1016/j.coviro.2018.12.008 (2019).
Hooper, C. et al. Diseases of the giant river prawn Macrobrachium rosenbergii: A review for a growing industry. Rev Aquacult 15, 738–758, https://doi.org/10.1111/raq.12754 (2022).
Qiu, L. et al. Characterization of a new member of Iridoviridae, Shrimp hemocyte iridescent virus (SHIV), found in white leg shrimp (Litopenaeus vannamei). Sci Rep 7, 11834, https://doi.org/10.1038/s41598-017-10738-8 (2017).
Liao, X. Z., He, J. G. & Li, C. Z. Decapod iridescent virus 1: An emerging viral pathogen in aquaculture. Rev Aquacult 14, 1779–1789, https://doi.org/10.1111/raq.12672 (2022).
Huang, Q. J. et al. Establishment of a real-time Recombinase Polymerase Amplification (RPA) for the detection of decapod iridescent virus 1 (DIV1). J. Virol. Methods 300, 114377, https://doi.org/10.1016/j.jviromet.2021.114377 (2022).
Qiu, L. et al. Description of a Natural Infection with Decapod Iridescent Virus 1 in Farmed Giant Freshwater Prawn, Macrobrachium rosenbergii. Viruses 11, 354, https://doi.org/10.3390/v11040354 (2019).
Uffelmann, E. et al. Genome-wide association studies. Nat Rev Method Prime 1, 59, https://doi.org/10.1038/s43586-021-00056-9 (2021).
Hayes, B. & Goddard, M. Genome-wide association and genomic selection in animal breeding. Can Sci Publ 53, 876–883, https://doi.org/10.1139/g10-076 (2010).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Zhang, L. et al. The genome of an apodid holothuroid (Chiridota heheva) provides insights into its adaptation to a deep-sea reducing environment. Commun Biol 5, 224, https://doi.org/10.1038/s42003-022-03176-4 (2022).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158, https://doi.org/10.1038/s41592-019-0669-3 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546, https://doi.org/10.1038/s41587-019-0072-8 (2019).
Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 38, 1044–1053, https://doi.org/10.1038/s41587-020-0503-6 (2020).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res 44, e147, https://doi.org/10.1093/nar/gkw654 (2016).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4 10 11–14 10 14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Mulder, N. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol 396, 59–70, https://doi.org/10.1007/978-1-59745-515-2_5 (2007).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30120401 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29314554 (2024).
Yang, Z. Macrobrachium rosenbergii genome assembly and gene annotation. Figshare https://doi.org/10.6084/m9.figshare.26068237.v1 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_040412425.1 (2024).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066, https://doi.org/10.1093/nar/gkf436 (2002).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973, https://doi.org/10.1093/bioinformatics/btp348 (2009).
Zhang, D. et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Bioinformatics 20, 348–355, https://doi.org/10.1111/1755-0998.13096 (2020).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274, https://doi.org/10.1093/molbev/msu300 (2015).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522, https://doi.org/10.1093/molbev/msx281 (2017).
Guindon, S. et al. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 59, 307–321, https://doi.org/10.1093/sysbio/syq010 (2010).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591, https://doi.org/10.1093/molbev/msm088 (2007).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972, https://doi.org/10.1093/bioinformatics/btl505 (2006).
Acknowledgements
This study was supported by Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (SML2023SP234), Guangdong S&T programme (2024B1212060001), and the earmarked fund of China Agriculture Research System for CARS-48.
Author information
Authors and Affiliations
Contributions
J.G.H. and M.W. conceived of the project and designed research; Q.G., D.Z., S.P.W., C.L. collected the sample; Y.Z., Y.L., G.T. assembled the genome; G.G., L.Z. conducted the evolutionary analyses; J.G.H. and M.W. wrote the paper with contribution from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, Y., Guo, G., Lv, Y. et al. A Chromosome-level genome assembly of giant river prawn (Macrobrachium rosenbergii). Sci Data 11, 935 (2024). https://doi.org/10.1038/s41597-024-03804-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03804-0
