
Abstract
Activation of the chemokine receptor CXCR4 by its chemokine ligand CXCL12 regulates diverse cellular processes. Previously reported crystal structures of CXCR4 revealed the architecture of an inactive, homodimeric receptor. However, many structural aspects of CXCR4 remain poorly understood. Here, we use cryo-electron microscopy to investigate various modes of human CXCR4 regulation. CXCL12 activates CXCR4 by inserting its N terminus deep into the CXCR4 orthosteric pocket. The binding of US Food and Drug Administration-approved antagonist AMD3100 is stabilized by electrostatic interactions with acidic residues in the seven-transmembrane-helix bundle. A potent antibody blocker, REGN7663, binds across the extracellular face of CXCR4 and inserts its complementarity-determining region H3 loop into the orthosteric pocket. Trimeric and tetrameric structures of CXCR4 reveal modes of G-protein-coupled receptor oligomerization. We show that CXCR4 adopts distinct subunit conformations in trimeric and tetrameric assemblies, highlighting how oligomerization could allosterically regulate chemokine receptor function.
Similar content being viewed by others


Main
Chemokine receptors are a family of class A G-protein-coupled receptors (GPCRs) that mediate cell migration in response to the binding of chemokine ligands1. CXCR4 is a well-studied chemokine receptor that is activated by the chemokine ligand CXCL12 (also called stromal cell-derived factor 1 (SDF1)) and signals primarily through coupling with Gi protein2, regulating cell migration in hematopoiesis, neovascularization, angiogenesis and various other physiological processes3. CXCR4 is involved in numerous diseases, including roles as a cancer marker implicated in tumor proliferation4 and as a coreceptor for X4-tropic human immunodeficiency virus (HIV) strains5. Mutations in CXCR4 that result in enhanced and prolonged signaling result in a rare immune disorder called WHIM (warts, hypogammaglobulinemia, infections and myelokathexis) syndrome6. The roles of CXCR4 in health and disease have made the receptor an intensely investigated drug target7. The small-molecule CXCR4 antagonist AMD3100 (plerixafor), initially developed as an HIV entry inhibitor8, was approved by the US Food and Drug Administration (FDA) as a hematopoietic stem cell mobilizer for autologous transplantation in patients with non-Hodgkin’s lymphoma or multiple myeloma9,10. Numerous additional CXCR4-targeting therapeutics have been developed7, notably including monoclonal antibodies with improved pharmacokinetic properties and, thus, potentially greater efficacy compared to small molecules and peptides11,12,13.
Structural studies of class A GPCRs have focused on isolated monomeric forms of the receptors bound to various ligands, pharmacological modulators and transducer proteins14. However, increasing evidence suggests that GPCRs can form dimers and higher-order oligomers in the plasma membrane, with implications for signaling and therapeutic action15. Chemokine receptors are no exception; a multitude of studies have indicated the existence of homo-oligomers and hetero-oligomers16,17,18, including crystal structures of antagonist-bound CXCR4 consistently revealing homodimeric forms19,20. Interestingly, CXCR4 has also shown a propensity to form higher-order oligomers using a mechanism that can be separated from dimerization21.
Despite its critical roles in health and disease, many mechanistic aspects of CXCR4 remain poorly understood because of a lack of structural information. These include its activation by CXCL12, the binding mode of AMD3100, its coupling to Gi protein, the inhibitory action of antibodies and mechanisms of higher-order oligomerization. Here, we address these open questions by reporting a series of cryo-electron microscopy (cryo-EM) structures of CXCR4 complexes.
Results
Structural basis of CXCL12 and AMD3100 action on CXCR4
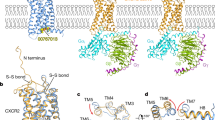
To stabilize active-state signaling complexes and improve protein yields, we made the following modifications to wild-type CXCR4: we replaced the N-terminal methionine with a hemagglutinin signal peptide22, included a previously characterized constitutively active substitution (N119S)23 and fused monomeric enhanced green fluorescent protein (GFP)24 and a FLAG tag to the receptor C terminus (Extended Data Fig. 1a). We refer to this construct as CXCR4EM. We also used a Gαi construct harboring dominant negative substitutions25 to facilitate the isolation of receptor–Gi complexes in the absence of stabilizing antibody fragments26. Fluorescence-detection size-exclusion chromatography (FSEC)27 experiments indeed indicated complex formation between CXCR4EM and Gi in the absence of agonist (Extended Data Fig. 1b). We prepared detergent-solubilized CXCR4EM–Gi complexes and first determined cryo-EM structures in apo, CXCL12-bound and AMD3100-bound states at overall resolutions of 2.7, 3.3, and 3.2 Å, respectively (Fig. 1, Extended Data Figs. 2 and 3, Table 1 and Supplementary Fig. 1). Each of the structures shows a prototypical arrangement of an active receptor coupled to a heterotrimeric G protein, including a hallmark kink of transmembrane helix 6 (TM6) relative to previously reported crystal structures of antagonist-bound CXCR4 (refs. 19,20) (Extended Data Fig. 4a). We, therefore, refer to the CXCR4 conformation in these structures as active.

a, Apo CXCR4–Gi complex. b, CXCR4–Gi–CXCL12 complex. Inset, the fit of the CXCL12 N-terminal tail (residues 1–10) in cryo-EM map, represented as a semitransparent surface. The locations of chemokine recognition sites 1 and 2 are labeled. The curved dotted line represents missing density for the distal N terminus of CXCR4, which has been reported to interact with CXCL12. The gray density on the extracellular side may correspond to a partially occupied second protomer of dimeric CXCL12. c, CXCR4–Gi–AMD3100 complex. Inset, the fit of the AMD3100 compound in cryo-EM map.
Our cryo-EM reconstruction of CXCR4EM–Gi–CXCL12 revealed a clear signal for the chemokine bound at the extracellular side of the receptor (Fig. 1a). Density for the chemokine N terminus (residues 1–12) was sufficiently resolved to build side chains (Fig. 1b), whereas the remainder of the ligand was less resolved because of flexibility and only permitted main-chain tracing (Extended Data Fig. 3i). Consequently, interactions between the chemokine N-terminal region and receptor orthosteric pocket (chemokine recognition site 2) were readily discernible, while interactions between the globular portion of the ligand and the N terminus of CXCR4 (ref. 28) (chemokine recognition site 1) were unclear. CXCL12 is known to exist in monomeric and dimeric forms that have been shown to yield distinct signaling outcomes upon CXCR4 binding29,30. A weak signal corresponding to a second protomer of the CXCL12 dimer could be observed in our cryo-EM reconstruction, consistent with the notion that dimeric forms of CXC ligands act on single receptor subunits20,31 (Extended Data Fig. 3i).
The binding mode of CXCL12 onto CXCR4 is overall similar to that found in published structures of CC and CXC chemokine–chemokine receptor complexes31,32,33,34,35,36 (Fig. 2 and Extended Data Fig. 4b). However, the CXCL12 binding pose observed in our structure notably differs from that of CXCL12 bound to atypical chemokine receptor 3 (ACKR3, formerly known as CXCR7)37, a promiscuous receptor that has been suggested to function as a chemokine ‘scavenger’ and has approximately tenfold higher affinity for CXCL12 than CXCR4 (refs. 38,39). The CXCL12 C-terminal α-helix is rotated approximately 70° when bound to ACKR3 relative to CXCR4 (Extended Data Fig. 4c). Correspondingly, the 40s loop of CXCL12 is situated proximal to the N-terminal region in CXCR4, while it is nearby extracellular loop 3 (ECL3) in ACKR3. In addition to the distinct overall chemokine–receptor docking orientations, the binding geometries of the CXCL12 N terminus within the orthosteric pockets of each receptor are unique (Extended Data Fig. 4d).

a, Expanded view of interaction between the CXCL12 N-terminal tail and CXCR4 orthosteric pocket. Hydrogen-bonding and electrostatic interactions are depicted as dashed lines. b, Expanded view of AMD3100 binding at CXCR4 orthosteric pocket. Asterisks indicate the positions of the two lactam rings, each of which interact with acidic residues. c, Cutaway surface view of CXCR4 orthosteric pocket. The CXCL12 N terminus is shown in stick representation and AMD3100 is shown in sphere representation to illustrate their relative binding positions in the orthosteric pocket.
Amino acid substitutions at the distal N terminus of CXCL12 can convert the chemokine into an antagonist40, highlighting the importance of the N terminus for receptor activation. Our structure shows how the CXCL12 N terminus protrudes into the orthosteric pocket of CXCR4 and makes extensive contacts with the TM core (Fig. 2a). The distal CXCL12 N terminus is positioned overall deeper into the pocket compared to the antagonistic viral chemokine vMIP-II (ref. 20) (Extended Data Fig. 4e), consistent with their respective ligand functions. P2CXCL12 penetrates deepest into the orthosteric pocket, contacting the side chain of Y1163.32 (Ballesteros–Weinstein numbering41 in superscript). The side chain of K1CXCL12 projects upward from the TM core to the extracellular side of the receptor and is positioned to interact electrostatically with D972.63 and possibly D187ECL2. S4CXCL12 makes an apparent hydrogen bond interaction with E2887.39. L5CXCL12 packs onto a mainly hydrophobic surface composed of L411.35, Y451.39, W942.60 and A982.64. R8CXCL12 appears poised to make a charge–charge interaction with D2626.58, as predicted previously on the basis of charge-swap experiments28. Several of the CXCR4 residues mentioned above (W942.60, D972.63, Y1163.32, D187ECL2 and E2887.39) have been shown to be important for CXCL12–CXCR4 signaling28,42,43, underscoring the functional relevance of the interactions observed in our cryo-EM structure. We expand on the structural basis of CXCL12 activation of CXCR4 in a later section.
We observed unambiguous density for the bilobed AMD3100 molecule in our cryo-EM reconstruction of CXCR4EM–Gi–AMD3100 (Fig. 1c). Although it has primarily been described as an antagonist44, our observation that AMD3100 binds to the active CXCR4EM–Gi complex without disrupting G-protein coupling is consistent with the compound acting as a weak partial agonist on constitutively active mutants of CXCR4 (ref. 23). AMD3100 binds the orthosteric pocket using a diagonal orientation and directly blocks CXCL12 docking, although its overall binding mode is shifted toward TM5 and TM6 relative to the CXCL12 N terminus (Fig. 2b,c). Each of the two positively charged cyclam rings of AMD3100 (ref. 45) is stabilized electrostatically by an acidic side chain pointed toward the center of the ring; the cyclam moiety closer to the extracellular side interacts with D2626.58 while the cyclam proximal to the TM core interacts with E2887.39. The closely matched spacings between the side chains of D2626.58 and E2887.39 residues and the cyclam rings, therefore, appear be the main binding determinant of AMD3100 and other bicyclam analogs. Consistent with our structure, a previous study showed that that D262N and E288A mutants each reduced the affinity of AMD3100 to CXCR4 more than 50-fold45. The central aromatic ring of the phenylenebis(methylene) linker connecting the two cyclam moieties makes hydrophobic contacts with I2847.35, which is positioned directly in between D2626.58 and E2887.39 in the orthosteric pocket. This interaction may contribute to the increased potency of bicyclams with an aromatic linker relative to those with an aliphatic linker46. The rigidity imposed by the aromatic linker on the relative positions of the two cyclam moieties may also have a role in stabilizing the binding pose of AMD3100.
Antagonism of CXCR4 by REGN7663 monoclonal antibody
Antibody-based therapeutics against CXCR4 and other GPCRs are a promising alternative to small molecules because of their high specificity to the target, opportunity for Fc effector functions and favorable pharmacokinetic properties11,13,47,48. REGN7663 is a fully human anti-CXCR4 monoclonal antibody generated using VelocImmune mice49,50. We showed using a cyclic adenosine monophosphate response element (CRE) luciferase reporter assay that REGN7663 is a potent blocker (half-maximal inhibitory concentration (IC50) = 2.7 ± 0.1 nM, calculated from n = 3 independent experiments) of CXCL12-induced signaling in HEK293 cells engineered to overexpress CXCR4 (Fig. 3a). Furthermore, in the absence of CXCL12, REGN7663 decreased the apparent basal activity (half-maximal effective concentration (EC50) = 1.3 ± 0.4 nM, calculated from n = 3 independent experiments), indicating inverse agonism in the setting of CXCR4 overexpression (Fig. 3b). Notably, we observed that the dose–response curve for REGN7663 showed a Hill slope of approximately 1 in agonist mode (in the absence of CXCL12) and a Hill slope of approximately 2 in antagonist mode (in the presence of 0.5 nM CXCL12). Understanding the molecular basis for this apparent difference in cooperativity will require additional study.

a, CRE luciferase reporter assay showing CXCL12-dependent decrease in signal and block of CXCL12 activity (at 0.5 nM CXCL12) by REGN7663 (light blue). The negative control monoclonal antibody (violet) showed no effect. The IC50 for REGN7663 was calculated to be 2.7 ± 0.1 nM (mean ± s.d.) in antagonist mode from n = 3 independent experiments. RLU, relative luminescence units. b, REGN7663 shows a concentration-dependent increase in signal relative to baseline in the absence of CXCL12, demonstrating inverse agonism. The EC50 for REGN7663 was calculated to be 1.3 ± 0.4 nM in agonist mode (absence of CXCL12) from n = 3 independent experiments. In a,b, representative data from one experiment are shown (the same data for CXCL12 are shown as solid black circles in a,b to allow a comparison to monoclonal antibody data). c, Cryo-EM reconstruction of CXCR4EM–Gi–REGN7663 Fab complex, with each polypeptide chain colored differently. d, Top-down view of CXCR4 (yellow) with CDR loops of bound REGN7663 shown (blue, heavy chain (HC); cyan, light chain (LC)). e, Electrostatic interaction between CDR-H3 of REGN7663 and CXCR4 orthosteric pocket-facing residue E288.
To understand how REGN7663 binds and inhibits CXCR4, we determined a 3.4-Å-resolution cryo-EM structure of REGN7663 Fab in complex with CXCR4EM–Gi (Fig. 3c, Extended Data Fig. 5a,d, Table 1 and Supplementary Fig. 1). The structure revealed that REGN7663 binds directly onto the extracellular face of CXCR4, antagonizing the receptor by steric blockade of CXCL12 binding. Most of the REGN7663 epitope resides at the extracellular N-terminal region and ECL2 (Extended Data Fig. 5e,f). The REGN7663 heavy chain dominates the binding interactions, burying more surface area (~1,100 Å2) than the light chain (~300 Å2). Although the overall architecture of the complex is similar to the apo, CXCL12-bound and AMD3100-bound CXCR4EM–Gi structures, REGN7663 binding induces distinct conformations of the N terminus and ECL2, suggesting that their flexibility is important for specific monoclonal antibody binding (Extended Data Fig. 5g). Heavy-chain complementarity-determining regions (CDRs) 1 and 2 of REGN7663 are oriented toward the extracellular ends of TM4 and TM5, while light-chain CDRs are oriented extracellular to TM1 and TM2 (Fig. 3d). Remarkably, the CDR-H3 loop of REGN7663 wedges between the CXCR4 N terminus and ECL2, exhibiting a partial insertion into the CXCR4 orthosteric pocket. The side chain of REGN7663 residue R105 protrudes deepest into the orthosteric pocket, making an apparent charge–charge interaction with E2887.39(Fig. 3e). The insertion of the CDR-H3 loop, albeit not activating in the case of REGN7663, is reminiscent of how the CDR3 loop of the single-domain antagonist antibody JN241 occupies the orthosteric pocket of apelin receptor51. Taken together with the finding that JN241 was converted into a full agonist through subtle engineering of CDR3 (ref. 51), our structure of the REGN7663–CXCR4 complex illustrates the potential for full antibodies (containing light and heavy chains) functionally modulating GPCRs by inserting CDR loop(s) into the orthosteric pocket.
CXCR4 activation and Gαi protein docking
We next sought to assess the conformational changes associated with CXCR4 activation. Available crystal structures of inactive, antagonist-bound CXCR4 contain construct modifications (namely T4 lysozyme (T4L) inserted at intracellular loop 3 (ICL3) and a thermostabilizing amino acid substitution in TM3) that could confound comparison to our current structures. We, therefore, determined a 3.1-Å-resolution cryo-EM structure of CXCR4EM in the absence of Gi protein, using REGN7663 Fab as a fiducial mark (Fig. 4a, Extended Data Fig. 5h–k, Table 1 and Supplementary Fig. 2). Structural alignment of the REGN7663 Fab–CXCR4EM–Gi structure with the Gi-free REGN7663 Fab–CXCR4EM structure showed nearly identical conformations at the REGN7663 epitope and paratope regions but distinct conformations at the intracellular half of the receptor, including the characteristic movement of TM6 underlying receptor activation (Fig. 4b and Extended Data Fig. 5l). Additional conformational changes upon activation and Gi binding include the movement of TM5 toward TM6, subtle displacement of TM2 outward, an inward kink of TM7 and loss of ordered structure in H8. We note that H8 was also unresolved in previously determined antagonist-bound CXCR4 crystal structures19,20, suggesting that this is a consistent feature of the inactive receptor.

a, Cryo-EM reconstruction of inactive CXCR4EM–REGN7663 Fab complex (CXCR4, pink; REGN7663 heavy chain, gray; REGN7663 light chain, white). b, Structural alignment of inactive CXCR4 (pink) and active CXCR4 (yellow); the CXCR4EM–Gi–REGN7663 Fab complex was used for alignment. Left, side view; right, bottom-up view. The green block arrows depict conformational transitions from inactive to active CXCR4. c, Expanded view showing CXCL12 N terminus (cyan) binding to active CXCR4 (yellow). Inactive CXCR4 (pink) is shown for comparison and residues important for transmitting chemokine binding into activation are shown in stick representation. d, Expanded view of Gαi (light green) binding to active CXCR4 (yellow). Residues participating in the interaction are shown in stick representation and labeled (Gαi residue labels are underlined). Electrostatic interactions are highlighted with dashed lines.
We further compared the conformations of the inactive and CXCL12-bound structures to analyze how CXCL12 binding results in activation (Fig. 4c). Binding of the CXCL12 N-terminal coil to the orthosteric pocket requires structural changes to the inactive state pocket. Residues P2 and S4 at the CXCL12 N terminus push E2887.39 outward and toward the cytoplasmic side, while V3CXCL12 forces a downward displacement of Y2556.51. The movements of E2887.39 and Y2556.51 are in turn transmitted to F2927.43, which was previously implicated in CXCR4 signal transmission43, and conserved toggle switch residue52 W2526.48, respectively. Together, these conformational changes trigger further structural rearrangements that ultimately stabilize the active, Gi-bound conformation of CXCR4. Furthermore, a close comparison revealed that, because of binding of the CXCL12 N terminus in the orthosteric pocket, the E2887.39 side chain reorients, along with a small, ~0.7–1 Å outward movement of the extracellular half of the TM7 helix relative to our AMD3100–CXCR4EM–Gi, REGN7663 Fab–CXCR4EM–Gi and apo CXCR4EM–Gi structures (Extended Data Fig. 6a). This slight conformational difference at TM7 induced by CXCL12 may explain why it is a full agonist while the other ligands are not. Similar structural mechanisms of chemokine activation to that described above for CXCL12 were observed for the CCR2–CCL2 complex32 and CCR5–MIP-1α complex34.
Like other class A GPCRs, coupling of Gαi to CXCR4 is mediated by insertion of the Gαi α5 helix and C-terminal ‘wavy hook’ into the cytoplasmic-facing core of the receptor TM domain (Fig. 4d). Wavy hook residues L353 and F354 bury deepest into CXCR4 and contact R1343.50, Q233ICL3, K2366.32, A2376.33, T2406.36 and A307 mainly through van der Waals and hydrophobic interactions. The Gαi α5 helix makes numerous additional contacts with TM2, TM3, ICL2, TM5, ICL3 and TM6. Salt-bridge interactions involving D341(Gαi)–K2346.30 and E28(Gαi)–K1494.38 probably have an important role in stabilizing the docking of Gi protein onto CXCR4. Although the overall Gi binding mode of CXCR4 and other chemokine receptors is shared, the angle at which the Gαi α5 helix docks into the TM bundle differs slightly (Extended Data Fig. 6b). While CXCR4, CXCR1 (ref. 36) and CXCR2 (ref. 31) show highly similar α5 docking angles, the docking angles in CCR1 (ref. 33), CCR2 (ref. 32) and CCR5 (ref. 34) are similar to each other and shifted relative to CXCR4 because of distinct intracellular loop conformations and receptor interactions with Gαi (Extended Data Fig. 6c). More specifically, in the CC chemokine receptors, the Gαi α5 helix is shifted toward ICL2 and further from ICL3. Available data, therefore, indicate that CXC and CC chemokine receptors have slightly different Gi docking geometries.
Oligomeric structures of CXCR4
Although GPCRs are generally understood to function as monomeric units, numerous studies have reported that chemokine receptors form dimers and higher-order oligomers at the cell surface as expression levels increase53,54,55,56. Homo-oligomerization and hetero-oligomerization have been proposed to add complexity to chemokine receptor function, perhaps through allosteric communication between interacting subunits57,58. Multiple structures of CXCR4 from different crystal forms showed the same homodimeric architecture19,20, demonstrating that the detergent-solubilized receptor has the propensity to dimerize using specific intersubunit interactions mainly involving TM5 and TM6. Our SEC data of CXCR4EM consistently showed multiple peaks with different elution volumes, including peaks corresponding to oligomeric species larger than monomeric CXCR4EM or CXCR4EM–Gi (Extended Data Figs. 1b and 2a). Wild-type CXCR4 fused to GFP showed a similar FSEC profile to CXCR4EM, indicating that the apparent oligomerization was not specific to the constitutively active N119S substitution present in CXCR4EM.
We isolated and characterized a presumed oligomeric SEC peak (Extended Data Fig. 2a) of CXCR4EM using cryo-EM. Initial cryo-EM data yielded clear top and bottom views of trimeric and tetrameric species but the preferred orientation precluded structure determination. After screening various sample preparation conditions, we ultimately used stage-tilted data collection59 to obtain 3.4-Å-resolution reconstructions of CXCR4EM homotrimers and homotetramers in complex with REGN7663 Fab (Fig. 5, Extended Data Fig. 7a–j, Table 1 and Supplementary Fig. 2). According to three-dimensional (3D) classification, our data contained a roughly 1:3 ratio of trimers to tetramer particles (Extended Data Fig. 7k). We did not observe two-dimensional (2D) or 3D class averages consistent with dimeric CXCR4, except for nonphysiological antiparallel dimers in our samples prepared in the presence of Gi (Extended Data Fig. 7i). The trimer and tetramer both show CXCR4 subunits arranged symmetrically around a cavity at the central axis, at first glance evoking structural similarity to homomeric ion channels, although CXCR4 has no known channel function. In the case of the CXCR4 oligomers, we found evidence for numerous bound lipids at the central axis in the cryo-EM maps (Fig. 5c,f and Extended Data Fig. 8). Because of matching shape features, we tentatively built three phosphatidic acids and three cholesterol molecules in the trimeric map central cavity and four phosphatidic acids and eight cholesterols in the tetrameric cavity (Extended Data Fig. 8d,h). Although the presumed cholesterol molecules could, in principle, correspond to exogenously added cholesteryl hemisuccinate present in the purification buffers, the EM density we modeled as phosphatidic acid strongly resembles a phospholipid and not the LMNG detergent used for purification. This implies that the central cavity lipids were carried over from the cell membrane and remained stably bound through purification, indicating that the oligomeric structures reported here are representative of species present in the CXCR4-expressing cells used in this study and not an artifact of the purification process. The presence of ordered lipids plugging the central axis of CXCR4 oligomers is reminiscent of microbial channelrhodopsin trimers, although the quaternary arrangement of the seven-TM-helix protomers differs60,61.

a, Cryo-EM reconstruction of CXCR4 trimer in complex with REGN7663 Fab. b,c, Side (b) and top-down (c) views of CXCR4 trimer structure. TM helices are shown in cylinder representation and bound lipids are shown in stick representation. Fab molecules are omitted for clarity. d, Cryo-EM reconstruction of CXCR4 tetramer in complex with REGN7663 Fab. e,f, Side (e) and top-down (f) views of CXCR4 tetramer structure. g, Side (left) and top (right) views of previously reported dimeric crystal structure of CXCR4. h, Top-down view of a CXCR4 protomer (gray) showing the positions of neighboring subunits from a dimer (orange), trimer (cyan) and tetramer (magenta).
The comparable interprotomer interfaces of trimeric and tetrameric CXCR4 are composed of TM5, TM6 and TM7 of one protomer interacting with TM1 and TM7 of the neighboring protomer (Fig. 5c,f). A ~20° rotation of the angle between neighboring subunits underlies the distinct oligomeric states (Fig. 5h). This oligomeric interface does not overlap with the dimeric interface observed in crystal structures of CXCR4 (refs. 19,20) (Fig. 5h), speculatively allowing for ‘superclustering’ of CXCR4 protomers mediated by a combination of trimeric or tetrameric and dimeric interfaces (Extended Data Fig. 9a,b). The structural superposition indicates that the steric clash caused by the T4L fusion in the crystallization construct may have precluded the assembly of trimers or tetramers observed in our data (Extended Data Fig. 9c,d), thus suggesting why homodimer formation was favored for the T4L-fused receptor.
The trimeric interface is characterized by a buried surface area of ~1,150 Å2 and is primarily mediated by crisscrossing of TM6 and TM1 of neighboring protomers near the midpoint of the membrane (Fig. 6a). The diagonal orientation of TM6 results in interprotomer contacts with the cytoplasmic half of TM7. TM1 of the neighboring protomer makes additional interprotomer contacts with cytoplasmic end of TM5 and the extracellular tip of TM7. As expected from interactions between TM helices, most of the residues involved are hydrophobic. As noted above, the tetramer interface is similar to the trimer interface (Fig. 6b). However, close inspection revealed a remarkable difference in the tetramer: a sterol-shaped density that we tentatively built as cholesterol present at the cytoplasmic half of the bilayer sandwiched between TM5 and TM6 of one protomer and TM1 and TM7 of its neighbor (Fig. 6b,c). To make space for sterol binding at the tetrameric interface, the intracellular portion of TM6 splays away from the interface and TM1 of the neighboring protomer rotates relative to their conformations in the trimeric interface (Fig. 6d). The TM1 rotation is concurrent with the rotation of the entire CXCR4 protomer, which in turn allows space for the additional subunit present in the tetrameric assembly (Fig. 5h). Our structures, therefore, imply that the absence or presence of lipid at the CXCR4 interprotomer interface may drive the assembly of trimers and tetramers, respectively. These findings provide a structural example supporting the idea that cholesterol regulates chemokine receptor oligomerization62.

a, Interprotomer interface of CXCR4 trimer. Interface residues are shown in stick representation and labeled. b, Interprotomer interface of CXCR4 tetramer. Interface residues and modeled cholesterol are shown in stick representation. The density corresponding to cholesterol is shown as a transparent gray surface. c, Bottom-up view showing position of cholesterol at the tetramer interface. d, Structural alignment of TM6 and TM1 at the trimer (gray) and tetramer (blue and magenta, with cholesterol (yellow) shown in stick representation). e,f, Side (e) and bottom-up (f) views of protomeric structures of trimeric (cyan) and tetrameric (magenta) CXCR4. Binding of the Gαi α5 helix (gray) is prevented by steric clash. g, Structural alignment of trimeric CXCR4 protomer (cyan) and active CXCR4 protomer (yellow). Red asterisks highlight the distinct positions of ICL3 and TM7. h, Structural alignment of tetrameric CXCR4 protomer (magenta) and inactive CXCR4 (pink).
A super-resolution microscopy study reported that the simultaneous introduction of three substitutions (K239E, V242A and L246A) within TM6, located at the oligomerization interface observed in our structures, resulted in reduced higher-order oligomerization of CXCR4 (ref. 21). We used FSEC to examine the effect of this triple mutant and other substitutions at the oligomeric interface on the oligomerization behavior of the detergent-extracted receptor, using CXCR4EM as the background construct (Extended Data Fig. 10a). The K239E;V242A;L246A and K239E;V242W;L246W triple mutants both showed a reduced propensity to form oligomers relative to monomers, determined from the FSEC peak–area ratio of oligomer to monomer for each mutant (Extended Data Fig. 10b). We found that the single mutant V242W showed similarly reduced oligomerization, likely by introducing steric hindrance at the oligomerization interface. On the other hand, L246W increased apparent oligomerization and reduced monomer levels, possibly by augmenting the hydrophobic interactions between subunits. A substitution at a TM1 residue (L58W) that faces TM5 of the neighboring subunit also showed a reduced oligomer-to-monomer ratio. Other TM1, TM6 and TM7 mutants showed no notable change in oligomer-to-monomer ratio (T51W) or did not have clearly interpretable FSEC chromatograms, presumably because of impacts on expression level or stability of the receptor in detergent. Overall, these biochemical data corroborate the oligomeric interface observed in our structural data.
We next examined the conformations of the individual protomers within the CXCR4 trimer and tetramer. As noted above, a striking difference is the kink at TM6 associated with sterol binding (Fig. 6e,f). TM6 of the trimeric protomer is kinked outward relative to that of the tetrameric protomer, suggesting a more active-like conformation. Indeed, the structure of the trimeric protomer matches closely with active CXCR4 in complex with REGN7663 Fab and Gi, while the tetrameric protomer aligns well with the inactive CXCR4–REGN7663 Fab complex in the absence of Gi (Fig. 6g,h). A noteworthy distinction between the trimeric CXCR4 protomer and active, Gi-bound CXCR4 is the conformation of ICL3, TM7 and H8; in the trimer, ICL3 is pushed away from the cytoplasmic-facing core, the C-terminal end of TM7 is tucked inward, effectively blocking Gi binding, and H8 is not visible in the cryo-EM map (Fig. 6f,g). Therefore, while trimeric CXCR4 is composed of protomers with an active-like conformation, they are not structurally competent for Gi coupling and, as such, cannot be deemed fully active. This structural observation agrees with FSEC data showing that the presence of Gi did not result in a shift of the oligomeric peak (Extended Data Fig. 1b). Overall, these oligomeric structures demonstrate that distinct protomeric conformations underpin the trimeric and tetrameric arrangements of CXCR4. Lipids found at the central axis and at the tetrameric interface appear to be important for oligomeric assembly.
Discussion
CXCR4 is a longstanding drug target for HIV, cancer and immune disorders and one of the most well-studied chemokine receptors; it was also the first to be crystallized. However, critical structures of CXCR4 remained missing. We present here a thorough investigation of the CXCR4 structure using cryo-EM. Our structure of active CXCR4 bound to CXCL12 shows how the chemokine N terminus buries deep into the orthosteric pocket to activate the receptor. Amino acid substitutions at the distal CXCL12 N terminus40 likely diminish its agonistic activity by disrupting the interactions between chemokine and receptor at the TM domain that are required for activation. Because of the flexibility of the complex, we were unable to resolve interactions between the receptor N terminus and chemokine (chemokine recognition site 1). Therefore, further studies are necessary to visualize this important determinant of CXCL12–CXCR4 affinity.
Like CXCL12, the FDA-approved drug AMD3100 uses electrostatic interactions (namely between its two positively charged lactam rings and acidic residues in the CXCR4 TM domain) to stabilize a diagonal binding mode. We also showed how a potent antibody inhibitor, REGN7663, blocks CXCL12 by binding across the extracellular face of CXCR4 and partially inserting its CDR-H3 loop into the orthosteric pocket. The structures of REGN7663–CXCR4 complexes do not provide a clear answer as to why this monoclonal antibody has apparent inverse-agonist activity in the setting of CXCR4 overexpression. Stable binding of REGN7663 to active-state CXCR4–Gi, which might be unexpected for an inverse-agonist monoclonal antibody, was possibly enabled by the constitutively active N119S substitution present in our construct that shifts the conformational equilibrium of the receptor. Indeed, the functional action (inverse agonism, antagonism or partial agonism) of REGN7663 on constitutively active mutants of CXCR4 is yet to be determined. While it is tempting to speculate that inverse agonism is related to interactions between REGN7663 and the CXCR4 TM domains, inverse-agonist antibodies raised against the MC4R N terminus have been reported63, suggesting that TM domain interactions are not a prerequisite for GPCR inverse-agonist monoclonal antibodies.
Although the functional relevance of chemokine receptor oligomerization in vivo awaits confirmation, CXCR4 oligomerization has been reported in various experimental settings, including crystal structures of parallel homodimers17. In this study, we observed that detergent-solubilized CXCR4 exists in various oligomeric states and determined structures of receptor trimers and tetramers. The parallel orientation of the protomers and the encapsulation of lipids at the central axis support the notion that these oligomeric species are present at the cell surface of insect cells overexpressing CXCR4 before detergent solubilization. Nonetheless, whether these species correspond to cell membrane oligomers observed previously16,55 or are representative of in vivo CXCR4 requires further investigation. Interestingly, super-resolution microscopy experiments implicated three TM6 residues (K239, V242 and L246) located at the oligomerization interface observed in our structures as being important for higher-order oligomerization but not dimerization of CXCR4 in Jurkat cells21. Furthermore, the oligomerization-defective K239E;V242A;L246A triple mutant showed decreased chemotaxis in response to CXCL12 in vitro21. These previously reported data provide a link between our oligomeric structures of detergent-solubilized receptor and CXCR4 function in T cells.
Lastly, we observed that oligomeric state and, specifically, the binding of lipid at the oligomeric interface are correlated with distinct conformations of CXCR4 protomers. While the individual protomers of trimeric CXCR4 exhibited an active-like conformation characterized by outward kinking of TM6, the positioning of intracellular-facing structural elements (ICL3, TM7 and H8) appears to preclude the docking of Gi. Therefore, additional conformational changes would be required for the oligomeric CXCR4 entities observed here to participate directly in G-protein-mediated cellular signaling. Further studies are needed to better understand how the underlying conformational dynamics of CXCR4 monomers, trimers and tetramers contribute to chemokine and drug action in cells.
Overall, our structures build on previous crystallographic studies19,20 to provide a foundation for understanding how peptides, small molecules, chemokines and antibody bind and affect the function of CXCR4 in diverse ways. Our data also provide a structural perspective on oligomerization as a potential mode of GPCR regulation, adding a layer of complexity to studies that have focused on monomers as the functional units in physiology and disease.
Methods
FSEC-based construct screening
Expression constructs (shown in Extended Data Fig. 1a) were codon-optimized, synthesized and cloned into pFastBac1 or pFastbac Dual vectors by Genscript. Second-generation baculoviruses (P1) encoding human CXCR4, CXCR4EM, Gαi or Gβ1–Gγ2 (expressed together using pFastBac Dual) were generated in ExpiSf9 cells (Thermo Fisher, A35243), titered and adjusted to approximately 2.5 × 108 ivp per ml. The titering assay was performed using flow cytometry to detect envelope protein gp64 displayed on the surface of infected cells. ExpiSf9 cells at ~5 × 106 cells per ml were infected with CXCR4 alone (1:11 viral dilution) or with Gαi (1:22 viral dilution) and Gβ1–Gγ2 (1:22 viral dilution). Cells were harvested by centrifugation after 72 h of growth (120 r.p.m. shaking, 27 °C, 125-ml flat-bottom flask, Innova 44 shaker). After freeze–thaw (−80 °C) cycles, cell pellets, each from 1 ml of culture, were resuspended in 200 µl of lysis buffer (25 mM Tris pH 7.5, 50 mM NaCl, 2 mM MgCl2, cOmplete (EDTA-free) protease inhibitor, 5 mM CaCl2 and 50 mU per ml of Apyrase) and rotated at 4 °C for 1 h. For the samples to which Gi was added, Gi-containing pellets were first suspended in 200 µl of lysis buffer. Then, 200 µl of Gi slurry was used to resuspend the receptor-containing pellets. After 1 h, 200 µl of solubilization buffer (25 mM Tris pH 7.5, 50 mM NaCl, 2 mM MgCl2, 5 mM CaCl2, ~2% LMNG, ~0.2% CHS, Roche cOmplete (EDTA-free) protease inhibitor (Sigma, 4693132001) and 50 mU per ml of Apyrase) was added and the mixture was rotated at 4 °C for an additional 1 h at 4 °C. Insoluble material was removed by centrifugation and each sample was subjected to FSEC (buffer: 25 mM Tris pH 7.5, 150 mM NaCl, 2 mM MgCl2, 0.01% LMNG and 0.001% CHS). A Zenix-C SEC-300 3-µm 300-Å 4.6 × 300-mm column (Sepax, 233300P-4630) at a flow rate of 0.35 ml min−1 was used for the data shown in Extended Data Fig. 1b. For the data shown in Extended Data Fig. 10, a Zenix-C SEC-300 3-µm 300-Å 7.8 × 300-mm column (Sepax, 233300-7830) at a flow rate of 0.75 ml min−1 was used and the baculovirus used was not titered. FSEC data were collected using a Shimadzu liquid chromatography system using LabSolutions version 5.111 software.
Expression and purification of CXCR4 and Gi proteins
ExpiSf9 cells at ~5 × 106 cells per ml were infected with P1 baculovirus encoding either CXCR4EM or GαI and Gβ1–Gγ2 as described above. Cells were harvested by centrifugation (3,000g, 10 min, 4 °C) after 72 h of growth (120 r.p.m. shaking, 27 °C, 2-L flat-bottom flask, Innova 44 shaker). Cell pellets were washed in ice-cold DPBS with cOmplete (EDTA-free) protease inhibitor, then subjected to freeze–thaw (−80 °C) cycles and resuspended in lysis buffer (25 mM Tris pH 7.5, 50 mM NaCl, 2 mM MgCl2, 1× Roche cOmplete (EDTA-free) protease inhibitor, 5 mM CaCl2 and 50 mU per ml of Apyrase). Crude lysates containing CXCR4EM and Gi were then combined and stirred at 4 °C. After 1 h, an equal volume (1 ml for every 1 ml of lysis buffer) of solubilization buffer (25 mM Tris pH 7.5, 50 mM NaCl, 2 mM MgCl2, 5 mM CaCl2, 2% LMNG and 0.2% CHS) was added to the slurry and the mixture was stirred at 4 °C for 1 h. Insoluble material was removed by centrifugation (100,000g, 4 °C, 30 min). Anti-FLAG M2 Affinity Gel (Sigma, A2220) was used to capture CXCR4EM-containing species. The protein-loaded resin was washed with SEC buffer (25 mM Tris pH 7.5, 150 mM NaCl, 2 mM MgCl2, 0.01% LMNG and 0.001% CHS) and protein was eluted in SEC buffer containing 0.15 mg ml−1 3xFLAG peptide. The eluate was concentrated to approximately 0.5 ml and subjected to SEC. A tandem column was used to improve the separation of different CXCR4EM species, whereby a Superose 6 Increase 10/300 GL column (Cytiva, 29-0915-96) was connected upstream of a Superdex 200 Increase 10/300 GL column (Cytiva, 28-9909-44). Fractions containing CXCR4EM–Gi protein complex were selected, pooled, concentrated and mixed with either Fab′, CXCL12 or AMD3100 before cryo-EM grid making.
A comparable procedure was used for the production of CXCR4EM to which Gi was not added. In this case, SEC peaks corresponding to oligomeric and monomeric CXCR4EM were separately isolated and were each mixed with Fab′ before cryo-EM grid making.
Fab′ production
REGN7663 IgG was diluted to 2 mg ml−1 in 20 mM HEPES pH 7.4 and 150 mM NaCl. IdeS, an IgG-specific protease, was added to cleave off the Fc region, thereby producing F(ab′)2. Then, 10 µg of concentrated IdeS per 1 mg of antibody (1:100) was added and the cleavage reaction was carried out at 37 °C for 30 min. F(ab′)2 was reduced using approximately 88 mM cysteamine hydrochloride at 37 °C for 10 min, in the presence of approximately 18 mM EDTA. Reduced Fab′ was dialyzed against 20 mM HEPES pH 7.4 and 150 mM NaCl overnight at 4 °C. Fab′ was further purified by negative passes through immobilized metal affinity chromatography and a CaptureSelect IgG-Fc (Multispecies) affinity matrix (ThermoFisher, 2942852050) to remove the His-tagged IdeS and Fc fragment, respectively. F(ab′) was treated with 20 mM iodoacetamide at room temperature, in the dark, for 30 min to alkylate the reduced hinge cysteines. Fab′ was purified further by SEC (HighLoad 16/600 Superdex 75-pg column (Cytiva 28989333) equilibrated to 25 mM Tris pH 7.5 and 150 mM NaCl) and concentrated before use.
CRE luciferase CXCR4 functional assay
HEK293 cell lines were generated to stably express full-length human CXCR4 (hCXCR4; amino acids 1–352, accession number NP_003458.1) along with a luciferase reporter CRE4×-luciferase-IRES-GFP. For the CXCR4 CRE luciferase assay, HEK293/CRE-Luc/hCXCR4 cells were plated in Opti-MEM media (Invitrogen, 31985-070) supplemented with 0.1% FBS (Seradigm, 1500-500) at 37 °C with 5% CO2 overnight. The cells were then incubated with 5 μM forskolin (Sigma, F6886) and serially diluted CXCL12 (Tocris, 350-NS) for activation of CXCR4 or preincubated with REGN7663 or control antibody for 30 min before adding 5 μM forskolin without or with 500 pM SDF for the inhibition of CXCR4 basal activity or SDF-induced CXCR4 activation. Cells were incubated for 5.5 h at 37 °C with 5% CO2. At the conclusion of the incubations, the luciferase activity was detected using OneGlo (Promega, E6130) and luminescence was recorded by an EnVision Plate reader using EnVision Manager version 1.14 (Perkin Elmer). Results were analyzed using nonlinear regression (four-parameter logistics) with Prism 6 software (GraphPad) to obtain EC50 and IC50 values.
Cryo-EM grid preparation and data collection
CXCR4EM (Gi-bound complex, monomer or oligomer) were concentrated to ~1–5 mg ml−1 and left as is (apo, Gi-bound complex sample) or mixed with 0.5 mg ml−1 CXCL12 (recombinant human CXCL12/SDF1α; R&D Systems, 350-NS-050/CF), 1 mM AMD3100 (AMD3100 octahydrochloride; R&D Systems, 3299) or ~1–1.5 mg ml−1 REGN7663 Fab and incubated on ice for ~1 h. Samples were pipetted onto freshly hydrogen–oxygen plasma-cleaned UltrAuFoil 0.6/1 300-mesh grids, blotted, then plunge-frozen into liquid ethane using a Vitrobot Mark IV and stored in liquid nitrogen before data collection.
Samples were inserted into a Titan Krios G3i (Thermo Fisher) microscope equipped with a BioQuantum K3 (Gatan) imaging system or a Glacios microscope equipped with a Falcon 4i camera and Selectris energy filter (Thermo Fisher). Data were collected at nominal magnifications of ×105,000 (0.85 Å per pixel) or ×165,000 (0.696 Å per pixel) and energy filters were inserted with slit widths of 20 eV and 10 eV on the Titan Krios and Glacios microscopes, respectively. Automated data collections were carried using EPU version 2.12 with an applied defocus range of −1.0 to −2.2 µm. A 40° stage tilt was applied during collection of the oligomeric CXCR4EM–REGN7663 Fab complex sample to overcome preferred particle orientations. Additional details regarding data collection are shown in Table 1.
Cryo-EM image processing
Cryo-EM data processing for apo CXCR4EM–Gi, CXCR4EM–Gi–AMD3100, CXCR4EM–Gi–REGN7663 Fab, CXCR4EM–REGN7663 Fab trimer and CXCR4EM–REGN7663 Fab tetramer was carried out within the cryoSPARC version 3.3.2 pipeline64. Patch motion correction and Patch contrast transfer function (CTF) estimation were used to align video frames and estimate CTF parameters, respectively. Particle images were picked using 2D template-based picker or TOPAZ version 0.2.5 (ref. 65), extracted and subjected to multiple rounds of 2D classification, ab initio reconstruction and heterogeneous refinement to obtain a homogenous subset of particles with well-resolved features corresponding to the target complex. Final map calculations were carried out using the local refinement job type. C3 and C4 symmetry were applied for refinement of the trimeric and tetrameric reconstructions of CXCR4EM–REGN7663 Fab, respectively. Refinements of oligomeric CXCR4 conducted without applied symmetry yielded similar structures to the symmetric refinements, but at lower resolution.
Initial processing steps for the CXCR4EM–Gi–CXCL12 and CXCR4EM–REGN7663 Fab monomeric complexes were carried out in RELION-3 (ref. 66). CTF parameters were calculated using gctf67 and CTFFIND4 (ref. 68). Particles were picked using TOPAZ65 and then sorted by 2D and 3D classification. Initial 3D refinements of the CXCR4EM–Gi–CXCL12 complex had very weak density for the ligand. To improve signal for the bound ligand, successive rounds of alignment-free focused 3D classification were conducted, applying a mask around CXCL12. Selected particle images were then subjected to Bayesian polishing and then imported into cryoSPARC for final map refinements. For the CXCR4EM–REGN7663 Fab complex, signal from the constant region of the Fab was subtracted before final local refinement in cryoSPARC. Additional data processing details are listed in Table 1.
Model building, structure refinement and visualization
Model building was initiated by docking starting models into the cryo-EM maps using the fit in map function in Chimera69, followed by rounds of manual adjustment in Coot 0.8.9 (ref. 70) and real-space refinement in PHENIX 1.19 (ref. 71). Published structures of CXCR4 (Protein Data Bank (PDB) 4RWS)20, Gi heterotrimer (PDB 7T2G) and an internal Fab structure were used as initial models to build the CXCR4EM–Gi–REGN7663 Fab complex. CXCR4EM and Gi from this structure was then used as starting models for the other structures in this study. A crystal structure of CXCL12 (PDB 3HP3)72 was used as an initial model for the chemokine. Side chains for CXCL12 residues 13–65 (except disulfide bonds) were truncated to Cβ because of weak density. The REGN7663 Fab constant regions were omitted from the CXCR4EM–REGN7663 Fab (without Gi), CXCR4EM–REGN7663 Fab trimer and CXCR4EM–REGN7663 Fab tetramer models because of weak density. The eLBOW program73 in PHENIX was used to generate ligand coordinates and restraints for AMD3100. Structures were validated using PHENIX and MolProbity74. Buried surface areas were calculated using PISA75. PyMOL version 2.5.4 (Schrödinger), Chimera version 1.16 (ref. 69) and ChimeraX version 1.2.5 (ref. 76) were used to visualize structural data and generate figures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Materials described in this manuscript were either purchased commercially (with the manufacturer indicated) or derived internally at Regeneron. Regeneron materials described in this manuscript may be made available to qualified, academic, noncommercial researchers through a materials transfer agreement upon request at https://regeneron.envisionpharma.com/vt_regeneron/. For questions about how Regeneron shares materials, use the email address preclinical.collaborations@regeneron.com. Atomic coordinates and cryo-EM maps were deposited to the PDB and EM Data Bank under accession codes 8U4N and EMD-41888 (Apo CXCR4EM–Gi), 8U4O and EMD-41889 (CXCR4EM–Gi–CXCL12), 8U4P and EMD-41890 (CXCR4EM–Gi–AMD3100), 8U4Q and EMD-41891 (CXCR4EM–Gi–REGN7663 Fab), 8U4R and EMD-41892 (CXCR4EM–REGN7663 Fab), 8U4S and EMD-41893 (CXCR4EM–REGN7663 Fab trimer) and 8U4T and EMD-41894 (CXCR4EM–REGN7663 Fab tetramer). PDB accession codes for previously reported structures used in this study include 3OE0, 4RWS, 6LFO, 8IC0, 7VL9, 7XA3, 7F1Q, 7SK3, 3GV3, 7T2G and 3HP3. Source data are provided with this paper.
References
Hughes, C. E. & Nibbs, R. J. B. A guide to chemokines and their receptors. FEBS J. 285, 2944–2971 (2018).
Busillo, J. M. & Benovic, J. L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 1768, 952–963 (2007).
Britton, C., Poznansky, M. C. & Reeves, P. Polyfunctionality of the CXCR4/CXCL12 axis in health and disease: implications for therapeutic interventions in cancer and immune-mediated diseases. FASEB J. 35, e21260 (2021).
Guo, F. et al. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 35, 816–826 (2016).
Feng, Y., Broder, C. C., Kennedy, P. E. & Berger, E. A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872–877 (1996).
Heusinkveld, L. E., Majumdar, S., Gao, J. L., McDermott, D. H. & Murphy, P. M. WHIM syndrome: from pathogenesis towards personalized medicine and cure. J. Clin. Immunol. 39, 532–556 (2019).
Caspar, B. et al. CXCR4 as a novel target in immunology: moving away from typical antagonists. Future Drug Discov. 4, FDD77 (2022).
Donzella, G. A. et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 4, 72–77 (1998).
DiPersio, J. F., Uy, G. L., Yasothan, U. & Kirkpatrick, P. Plerixafor. Nat. Rev. Drug Discov. 8, 105–106 (2009).
DiPersio, J. F. et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J. Clin. Oncol. 27, 4767–4773 (2009).
Ghobrial, I. M. et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin. Cancer Res. 26, 344–353 (2020).
Kashyap, M. K. et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF-06747143) in chronic lymphocytic leukemia. J. Hematol. Oncol. 10, 112 (2017).
Peng, S. B. et al. Inhibition of CXCR4 by LY2624587, a fully humanized anti-CXCR4 antibody induces apoptosis of hematologic malignancies. PLoS ONE 11, e0150585 (2016).
Gusach, A., Garcia-Nafria, J. & Tate, C. G. New insights into GPCR coupling and dimerisation from cryo-EM structures. Curr. Opin. Struct. Biol. 80, 102574 (2023).
Milligan, G., Ward, R. J. & Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 57, 40–47 (2019).
Ward, R. J. et al. Chemokine receptor CXCR4 oligomerization is disrupted selectively by the antagonist ligand IT1t. J. Biol. Chem. 296, 100139 (2021).
Munoz, L. M. et al. Receptor oligomerization: a pivotal mechanism for regulating chemokine function. Pharmacol. Ther. 131, 351–358 (2011).
Sohy, D. et al. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of ‘selective’ antagonists. J. Biol. Chem. 284, 31270–31279 (2009).
Wu, B. et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 (2010).
Qin, L. et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 (2015).
Martinez-Munoz, L. et al. Separating actin-dependent chemokine receptor nanoclustering from dimerization indicates a role for clustering in CXCR4 signaling and function. Mol. Cell 71, 873 (2018).
Guan, X. M., Kobilka, T. S. & Kobilka, B. K. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J. Biol. Chem. 267, 21995–21998 (1992).
Zhang, W. B. et al. A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J. Biol. Chem. 277, 24515–24521 (2002).
von Stetten, D., Noirclerc-Savoye, M., Goedhart, J., Gadella, T. W. Jr. & Royant, A. Structure of a fluorescent protein from Aequorea victoria bearing the obligate-monomer mutation A206K. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 68, 878–882 (2012).
Liang, Y. L. et al. Dominant negative G proteins enhance formation and purification of agonist–GPCR–G protein complexes for structure determination. ACS Pharm. Transl. Sci. 1, 12–20 (2018).
Zhang, X. et al. Evolving cryo-EM structural approaches for GPCR drug discovery. Structure 29, 963–974 (2021).
Kawate, T. & Gouaux, E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 (2006).
Stephens, B. S., Ngo, T., Kufareva, I. & Handel, T. M. Functional anatomy of the full-length CXCR4–CXCL12 complex systematically dissected by quantitative model-guided mutagenesis. Sci. Signal 13, eaay5024 (2020).
Ray, P. et al. Secreted CXCL12 (SDF-1) forms dimers under physiological conditions. Biochem. J. 442, 433–442 (2012).
Drury, L. J. et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc. Natl Acad. Sci. USA 108, 17655–17660 (2011).
Liu, K. et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140 (2020).
Shao, Z. et al. Molecular insights into ligand recognition and activation of chemokine receptors CCR2 and CCR3. Cell Discov. 8, 44 (2022).
Shao, Z. et al. Identification and mechanism of G protein-biased ligands for chemokine receptor CCR1. Nat. Chem. Biol. 18, 264–271 (2022).
Zhang, H. et al. Structural basis for chemokine recognition and receptor activation of chemokine receptor CCR5. Nat. Commun. 12, 4151 (2021).
Wasilko, D. J. et al. Structural basis for chemokine receptor CCR6 activation by the endogenous protein ligand CCL20. Nat. Commun. 11, 3031 (2020).
Ishimoto, N. et al. Structural basis of CXC chemokine receptor 1 ligand binding and activation. Nat. Commun. 14, 4107 (2023).
Yen, Y. C. et al. Structures of atypical chemokine receptor 3 reveal the basis for its promiscuity and signaling bias. Sci. Adv. 8, eabn8063 (2022).
Saaber, F. et al. ACKR3 regulation of neuronal migration requires ACKR3 phosphorylation, but not β-arrestin. Cell Rep. 26, 1473–1488 (2019).
Gustavsson, M., Dyer, D. P., Zhao, C. & Handel, T. M. Kinetics of CXCL12 binding to atypical chemokine receptor 3 reveal a role for the receptor N terminus in chemokine binding. Sci. Signal 12, eaaw3657 (2019).
Crump, M. P. et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16, 6996–7007 (1997).
Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of three dimensional models and computational probing of structure–function relations in G-protein coupled receptors. In Receptor Molecular Biology 1st edn, Vol. 25 (eds Conn, P. M. & Sealfon S. C.) (Academic Press, 1995).
Brelot, A., Heveker, N., Montes, M. & Alizon, M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 275, 23736–23744 (2000).
Wescott, M. P. et al. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc. Natl Acad. Sci. USA 113, 9928–9933 (2016).
Wang, J., Tannous, B. A., Poznansky, M. C. & Chen, H. CXCR4 antagonist AMD3100 (plerixafor): from an impurity to a therapeutic agent. Pharmacol. Res. 159, 105010 (2020).
Rosenkilde, M. M. et al. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: transfer of binding site to the CXCR3 receptor. J. Biol. Chem. 279, 3033–3041 (2004).
Bridger, G. J. et al. Synthesis and structure–activity relationships of phenylenebis(methylene)-linked bis-tetraazamacrocycles that inhibit HIV replication. Effects of macrocyclic ring size and substituents on the aromatic linker. J. Med. Chem. 38, 366–378 (1995).
Bobkov, V. et al. Antibodies targeting chemokine receptors CXCR4 and ACKR3. Mol. Pharmacol. 96, 753–764 (2019).
Hutchings, C. J., Koglin, M., Olson, W. C. & Marshall, F. H. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat. Rev. Drug Discov. 16, 787–810 (2017).
Murphy, A. J. et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc. Natl Acad. Sci. USA 111, 5153–5158 (2014).
Macdonald, L. E. et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc. Natl Acad. Sci. USA 111, 5147–5152 (2014).
Ma, Y. et al. Structure-guided discovery of a single-domain antibody agonist against human apelin receptor. Sci. Adv. 6, eaax7379 (2020).
Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 55, 114–120 (2019).
Hernanz-Falcon, P. et al. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 5, 216–223 (2004).
Percherancier, Y. et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J. Biol. Chem. 280, 9895–9903 (2005).
Hamatake, M. et al. Ligand-independent higher-order multimerization of CXCR4, a G-protein-coupled chemokine receptor involved in targeted metastasis. Cancer Sci. 100, 95–102 (2009).
Lao, J. et al. Single-molecule imaging demonstrates ligand regulation of the oligomeric status of CXCR4 in living cells. J. Phys. Chem. B 121, 1466–1474 (2017).
Stephens, B. & Handel, T. M. Chemokine receptor oligomerization and allostery. Prog. Mol. Biol. Transl. Sci. 115, 375–420 (2013).
Martinez-Munoz, L., Villares, R., Rodriguez-Fernandez, J. L., Rodriguez-Frade, J. M. & Mellado, M. Remodeling our concept of chemokine receptor function: from monomers to oligomers. J. Leukoc. Biol. 104, 323–331 (2018).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Tucker, K., Sridharan, S., Adesnik, H. & Brohawn, S. G. Cryo-EM structures of the channelrhodopsin ChRmine in lipid nanodiscs. Nat. Commun. 13, 4842 (2022).
Morizumi, T. et al. Structures of channelrhodopsin paralogs in peptidiscs explain their contrasting K+ and Na+ selectivities. Nat. Commun. 14, 4365 (2023).
Legler, D. F. et al. Modulation of chemokine receptor function by cholesterol: new prospects for pharmacological intervention. Mol. Pharmacol. 91, 331–338 (2017).
Peter, J. C. et al. Antibodies against the melanocortin-4 receptor act as inverse agonists in vitro and in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R2151–R2158 (2007).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zhang, K. gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Murphy, J. W., Yuan, H., Kong, Y., Xiong, Y. & Lolis, E. J. Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins 78, 1331–1337 (2010).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 (2009).
Prisant, M. G., Williams, C. J., Chen, V. B., Richardson, J. S. & Richardson, D. C. New tools in MolProbity validation: CaBLAM for CryoEM backbone, UnDowser to rethink ‘waters’, and NGL Viewer to recapture online 3D graphics. Protein Sci. 29, 315–329 (2020).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
This study was funded by Regeneron Pharmaceuticals. We thank various Regeneron scientists including Y. Zhou, M. Rapp and A. Murphy for discussions, L. Molla and S. Chandwani for project management and the Regeneron cloud and HPC teams for supporting cryo-EM data storage and processing.
Author information
Authors and Affiliations
Contributions
K.S., L.L.M., J.H., M.M., J.H.K. and M.C.F. conceptualized the studies. L.L.M. and T.R. expressed and purified proteins for cryo-EM. L.L.M., T.R. and K.S. conducted the FSEC experiments. K.S. conducted cryo-EM experiments and analyzed the structural data, with contributions from L.L.M. and M.C.F. J.H. and S.S. conducted the CRE luciferase assays. K.S., L.L.M., J.H., M.M., J.H.K., R.L., W.C.O. and M.C.F. analyzed the data and supervised the overall project. K.S. and L.L.M. drafted the manuscript with input from J.H. and M.C.F. The manuscript was finalized by all authors.
Corresponding authors
Ethics declarations
Competing interests
All authors are employees of Regeneron Pharmaceuticals and may own stock and/or stock options of the company. W.C.O. is an officer of Regeneron. All funding for this work was provided by Regeneron Pharmaceuticals. The anti-CXCR4 antibody described in this manuscript is a subject of international patent applications (PCT/US2024/044209 and PCT/US2024/044111).
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Katarzyna Ciazynska, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Protein constructs and FSEC-based protein screening.
a, Primary structures of protein constructs used in structural studies. b, Fluorescence-detection size exclusion chromatography (FSEC) screening of wild type CXCR4 (blue) and N119S-containing CXCR4 (referred to as CXCR4EM, red) in the presence (solid lines) or absence (dashed lines) of added Gi. The nominally wild type CXCR4 construct was identical to that shown in (a) without the N119S point mutation. Chromatograms are annotated with presumed peak positions of various species present in the samples.
Extended Data Fig. 2 Purification of CXCR4 complexes.
a, Size-exclusion chromatography (SEC) traces for a CXCR4EM purification with added Gi (blue), and for a purification prepared in the absence of exogenously added Gi (orange). CXCR4EM/Gi complex chromatogram is representative of four purifications performed each yielding similar results, whereas purification of CXCR4EM in the absence of added Gi was conducted once. b,c SDS-PAGE (4–20% Tris-Glycine) showing the subunit content and purity of prepared cryoEM samples for CXCR4EM/Gi complex (b, representative of four preparations yielding similar results) and CXCR4EM prepared in the absence of added Gi (c, prepared once). 2% (v/v) 2-Mercaptoetanol was present in the SDS-PAGE samples prior to loading. d, Blue native (BN) PAGE (4–16% Bis-Tris) of SEC-purified CXCR4EM monomer and oligomer samples prepared in the absence of added Gi. BN PAGE analysis was conducted on one sample.
Extended Data Fig. 3 CryoEM reconstruction of Apo CXCR4EM/Gi, CXCL12-bound CXCR4EM/Gi, AMD3100-bound CXCR4EM/Gi.
a-d, FSC curve (a), particle angular distribution plot (b), local resolution map calculated in cryoSPARC (c), and map/model fits of selected regions (d) for Apo CXCR4EM/Gi. e-h, FSC curve (e), particle angular distribution plot (f), local resolution map calculated in cryoSPARC (g), and map/model fits of selected regions (h) for CXCL12-bound CXCR4EM/Gi. i, two views showing putative fit of a CXCL12 dimer (arranged on the basis of PDB 3GV3) into cryoEM map, shown at 4 sigma in PyMOL. j-m, FSC curve (j), particle angular distribution plot (k), local resolution map calculated in cryoSPARC (l), and map/model fits of selected regions (m) for AMD3100-bound CXCR4EM/Gi.
Extended Data Fig. 4 Structural comparisons of chemokine receptor structures.
a, structural alignment of active CXCR4 (yellow, this study, CXCL12/Gi-bound complex) and inactive, antagonist-bound CXCR4 (cyan, PDB 3OE0). Magenta block arrows depict movement of TM6. b, Alignment of CXCR4/CXCL12 complex (yellow) with CXCR2/CXCL8 complex (brown, PDB 6LFO), and CCR2/CCL2 complex (pink, PDB 7XA3). G protein models are omitted for clarity. c, Receptor-based alignment of CXCR4/CXCL12 (yellow/pink) with ACKR3/CXCL12 (gray/black, PDB 7SK3). Arrow highlights different docking orientations of CXCL12 onto the two receptors. d, expanded views of showing different binding modes of CXCL12 N-termini (pink in CXCR4 complex and black in ACKR3 complex) in CXCR4 and ACKR3. e, alignment of CXCR4/CXCL12 complex (yellow/pink) and CXCR4/vMIP-II (green/blue). Inset shows expanded views of chemokine N-terminal positions within orthosteric pocket, highlighting the positions of toggle switch residue W252 in sticks.
Extended Data Fig. 5 CryoEM reconstruction of REGN7663 Fab/CXCR4EM/Gi and REGN7663 Fab/CXCR4EM.
a-d, FSC curve (a), particle angular distribution plot (b), local resolution map calculated in cryoSPARC (c), and map/model fits of selected regions (d) for REGN7663 Fab/CXCR4EM/Gi. e, f, Expanded view of contacts between REGN7663 Fab (light chain in cyan, heavy chain in blue) and CXCR4 ECL2 (e) and N-term (f). Epitope and paratope residues are shown as sticks and labeled, and apparent salt bridges/hydrogen bonds between mAb and receptor are shown as dashed lines. g, structural alignment of CXCR4 bound to CXCL12 and REGN7663 Fab. N-term. and ECL2 are colored green (CXCL12-bound) or magenta (REGN7663 Fab-bound) to highlight their different positions. h-k, FSC curve (h), particle angular distribution plot (i), local resolution map calculated in cryoSPARC (j), and map/model fits of TM helices (k) for REGN7663 Fab/CXCR4EM without Gi. l, aligned structures of CXCR4/REGN7663 Fab complex in the inactive (pink) and active (yellow, Gi-bound) conformations. Note the REGN7663 Fab variable region and cytoplasmic half of the CXCR4 domain are mostly superimposable.
Extended Data Fig. 6 Structural features of Gi-bound chemokine receptors.
a, alignment of CXCR4/Gi complexes bound to CXCL12 (yellow), REGN7663 Fab (magenta), AMD3100 (green) or in the absence of ligand (apo, gray). Inset shows expanded view around E288 residue. Bound CXCL12 is shown as yellow transparent surface and sticks, highlighting how it enforces a rotameric change of E288 and slight shift of TM7 in the CXCL12-bound complex. b, receptor-based alignment showing architecture of various chemokine/chemokine receptor/Gi complexes. c, expanded view showing docking of Gαi α5 helix into cytoplasmic pocket of chemokine receptors. Note that the Gαi α5 helix is positioned closer to ICL2 in CC chemokine receptor complexes, while it is closer to ICL3 in CXC chemokine receptor complexes.
Extended Data Fig. 7 CryoEM of CXCR4 oligomers.
a-e, example 2D class averages obtained from tilted data collection (a), FSC curves (b), angular distribution plot (c), local resolution map (d) and model/map fits of TM helices (e) of trimeric CXCR4EM/REGN7663 Fab complex. f-j, example 2D class averages obtained from tilted data collection (f), FSC curves (g), angular distribution plot (h), local resolution map (i) and model/map fits of TM helices (j) of tetrameric CXCR4EM/REGN7663 Fab complex. k, Output maps from ab initio reconstruction conducted on the oligomeric CXCR4EM/REGN7663 Fab particles. Particles belonging to classes of tetramer with 4 fabs bound or trimer with 3 fabs bound were selected for further processing. l, An example 2D class average showing an anti-parallel dimer of CXCR4EM/Gi.
Extended Data Fig. 8 Lipids resolved in oligomeric structures of CXCR4.
a-c, side (a), top-down (b) and bottom-up (c) views of trimeric CXCR4, highlighting positions of built lipid molecules. Cholesterols (chol.) are shown as yellow sticks and phosphatidic acid (PA) are shown as green sticks. d, fit of lipid molecules (shown as sticks) to map (transparent blue surface) in trimeric CXCR4. Chol. 1 and chol 2. refer to lipids labeled in a-c. e-g, side (e), top-down (f) and bottom-up (g) views of tetrameric CXCR4, highlighting positions of built lipid molecules. h, fit of lipid molecules (shown as sticks) to map (transparent blue surface) in tetrameric CXCR4.
Extended Data Fig. 9 Structural analysis of dimeric and oligomeric structures of CXCR4.
a,b, top-down view of hypothetical models of four CXCR4 trimers (a) or five CXCR4 tetramers (b) clustered via dimeric interfaces (red ovals) observed in crystal structures. c,d, a single subunit from dimeric x-ray structure of CXCR4 (receptor in yellow, ICL3-fused T4 lysozyme (T4L) in gray) aligned to trimeric CXCR4 (c) or tetrameric CXCR4 (d). Side and top views are shown. Red symbols indicate steric clash between T4L and neighboring protomers that would prevent trimer or tetramer assembly. The steric hindrance caused by fused T4L may explain why dimeric CXCR4 was favored over trimeric or tetrameric CXCR4 in previous crystallographic studies.
Extended Data Fig. 10 FSEC analysis of CXCR4 oligomerization interface mutants.
a, FSEC chromatograms of control construct CXCR4EM (black trace) and its mutants (colored traces) tracking GFP fluorescence. The same chromatogram for the control are shown in each for comparison. Gray shaded regions indicate elution times corresponding to CXCR4 oligomer (7.8-8.8 min) and monomer (9.6-10 min). b, ratio of oligomer to monomer peak areas, calculated according to shaded regions in a. horizontal dotted line corresponds to mean value for CXCR4EM. Column heights and error bars show mean and standard deviations calculated from n = 3 (mutant constructs) or n = 4 (CXCR4EM control) FSEC experiments using two independently generated baculoviruses for each construct. Note that several mutants (I257W, I300W, F276A/W283A, F276A, W283A) showed poor chromatographic behavior overall, presumably due to poor expression or stability in detergent.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2.
Source data
Source Data Fig. 3
Source data for Fig. 3a,b.
Source Data Extended Data Fig. 1
Source Data for Extended Data Fig. 1b.
Source Data Extended Data Fig. 2
Source Data for Extended Data Fig. 2a.
Source Data Extended Data Fig. 2
Uncropped gels for Extended Data Fig. 2b–d.
Source Data Extended Data Fig. 10
Source data for Extended Data Fig. 10a,b.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saotome, K., McGoldrick, L.L., Ho, JH. et al. Structural insights into CXCR4 modulation and oligomerization. Nat Struct Mol Biol (2024). https://doi.org/10.1038/s41594-024-01397-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41594-024-01397-1
