
Abstract
The efficacy of convalescent plasma for coronavirus disease 2019 (COVID-19) is unclear. Although most randomized controlled trials have shown negative results, uncontrolled studies have suggested that the antibody content could influence patient outcomes. We conducted an open-label, randomized controlled trial of convalescent plasma for adults with COVID-19 receiving oxygen within 12 d of respiratory symptom onset (NCT04348656). Patients were allocated 2:1 to 500 ml of convalescent plasma or standard of care. The composite primary outcome was intubation or death by 30 d. Exploratory analyses of the effect of convalescent plasma antibodies on the primary outcome was assessed by logistic regression. The trial was terminated at 78% of planned enrollment after meeting stopping criteria for futility. In total, 940 patients were randomized, and 921 patients were included in the intention-to-treat analysis. Intubation or death occurred in 199/614 (32.4%) patients in the convalescent plasma arm and 86/307 (28.0%) patients in the standard of care arm—relative risk (RR) = 1.16 (95% confidence interval (CI) 0.94–1.43, P = 0.18). Patients in the convalescent plasma arm had more serious adverse events (33.4% versus 26.4%; RR = 1.27, 95% CI 1.02–1.57, P = 0.034). The antibody content significantly modulated the therapeutic effect of convalescent plasma. In multivariate analysis, each standardized log increase in neutralization or antibody-dependent cellular cytotoxicity independently reduced the potential harmful effect of plasma (odds ratio (OR) = 0.74, 95% CI 0.57–0.95 and OR = 0.66, 95% CI 0.50–0.87, respectively), whereas IgG against the full transmembrane spike protein increased it (OR = 1.53, 95% CI 1.14–2.05). Convalescent plasma did not reduce the risk of intubation or death at 30 d in hospitalized patients with COVID-19. Transfusion of convalescent plasma with unfavorable antibody profiles could be associated with worse clinical outcomes compared to standard care.
Similar content being viewed by others
Main
The immune response after severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection results in the formation of antibodies that can interfere with viral replication and infection of host cells in over 95% of patients1. Based on previous experience in other viral infections2, the use of convalescent plasma has been proposed as a therapeutic form of passive immunization for patients with acute COVID-19 (refs. 3,4). Early in the pandemic, several small randomized trials found no difference in clinical outcomes5,6,7,8. In the United States, an Extended Access Program outside of a controlled trial led to the use of convalescent plasma in over half a million patients. Data from these patients showed that the transfusion of plasma with high anti-SARS-CoV-2 antibody levels was associated with a lower risk of death in non-intubated patients compared to lower antibody levels; however, this study lacked a control group9. The RECOVERY trial was a large randomized trial in 11,558 hospitalized patients that found that the risk of death after the administration of high-titer plasma was not different from standard of care10.
The Convalescent Plasma for COVID-19 Respiratory Illness (CONCOR-1) trial was a multi-center, international, open-label, randomized controlled trial designed to assess the effectiveness and safety of COVID-19 convalescent plasma in hospitalized patients. The trial used plasma collected from four blood suppliers with a range of anti-SARS-CoV-2 antibody levels. The variability in antibody titers allowed for a characterization of the effect-modifying role of functional and quantitative antibodies on the primary outcome (intubation or death at 30 d).
Results
Patients
This trial was stopped at the planned interim analysis because the conditional power estimate was 1.6% (below the stopping criterion of 20%). Between 14 May 2020 and 29 January 2021, 940 patients were randomized (2:1) to convalescent plasma or standard of care in 72 hospital sites in Canada, the United States and Brazil (Fig. 1 and Supplementary Table 1). Two patients randomized to plasma withdrew consent before treatment. Demographics of the baseline study population (n = 938) were balanced between groups for all study populations (Table 1 and Supplementary Tables 2 and 3). The median age was 69 years, with 59% male and 41% female, and the median time from the onset of any COVID-19 symptom was 8 d (interquartile range (IQR), 5–10 d). Most patients (n = 766, 81.7%) were receiving systemic corticosteroids at the time of enrolment. Seventeen patients were lost to follow-up between discharge and day 30, precluding assessment of the primary outcome.

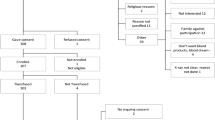
Patient flow in the CONCOR-1 study detailing the intention-to-treat population, per-protocol analysis population and excluded patients. Othera, n = 26: <16 years of age (n = 13), <18 years of age (n = 5), ABO-compatible plasma unavailable (n = 5) and other (n = 3). bIncludes not receiving supplemental oxygen at the time of randomization (but on oxygen at screening) and any symptom onset >12 d before randomization for protocol version 5.0 or earlier.
Primary outcome
In the intention-to-treat population (n = 921), intubation or death occurred in 199 (32.4%) of 614 patients in the convalescent plasma group and 86 (28.0%) of 307 patients in the standard of care group (RR = 1.16, 95% CI 0.94–1.43, P = 0.18) (Fig. 2a). The time to intubation or death was not significantly different between groups (Fig. 2b). In the per-protocol analysis (n = 851), intubation or death occurred in 167 (30.5%) of 548 patients in the convalescent plasma group and 85 (28.1%) of 303 patients in the standard of care group (RR = 1.09, 95% CI 0.87–1.35, P = 0.46) (Supplementary Table 4).

a, Patient outcomes for the primary and secondary endpoints. b, Cumulative incidence functions of the primary outcome (intubation or death) by day 30 and of in-hospital death by day 90. aRR and 95% CI; hazard ratio ((HR), 95% CI); and mean difference ((MD), with 95% CI based on robust bootstrap standard errors). bSeventeen patients were discharged before day 30 and were lost to follow-up at 30 d, and two withdrew consent before day 30; thus, outcomes collected at day 30 (primary outcome and some other secondary outcomes for day 30) were missing. cExcluding 11 patients on chronic kidney replacement therapy at baseline. dIntention-to-treat survival analyses were based on the complete baseline population (940 randomized patients minus two patients who withdrew consent).
Secondary efficacy outcomes and subgroup analyses
Secondary outcomes for the intention-to-treat population are shown in Fig. 2a. There were no differences in mortality or intubation or other secondary efficacy outcomes. Similarly, in the per-protocol analysis, there were no differences in the secondary efficacy outcomes (Supplementary Table 4 and Extended Data Figs. 1–3). No significant differences were observed in most subgroups, including time from diagnosis to randomization <3 d for both the intention-to-treat (Fig. 3) and per-protocol (Extended Data Fig. 4) populations. The subgroup of patients served by blood supplier 3 (Fig. 3) and the post hoc subgroup of patients who were not receiving corticosteroids (Extended Data Figs. 5 and 6) had worse outcomes with convalescent plasma compared to standard of care.

Forest plots are presented for the subgroup analyses for the intention-to-treat population. P values for RR and homogeneity are two sided without adjustment for multiple comparisons. BMI, body mass index.
Safety
Serious adverse events occurred in 205 (33.4%) of 614 patients in the convalescent plasma arm compared to 81 (26.4%) of 307 patients in the standard of care arm for the intention-to-treat population (RR = 1.27, 95% CI 1.02–1.57, P = 0.034; Fig. 2a and Supplementary Tables 4–6). Most of these events were worsening hypoxemia and respiratory failure. Transfusion-related complications were recorded in 35 (5.7%) of 614 patients in the convalescent plasma group (Supplementary Tables 7 and 8). Of the 35 reactions, four were life-threatening (two transfusion-associated circulatory overload, one possible transfusion-related acute lung injury and one transfusion-associated dyspnea), and none was fatal. Thirteen of the 35 reactions were classified as transfusion-associated dyspnea. Two patients underwent serological investigation for transfusion-related acute lung injury (both negative).
Effect-modifying role of antibodies in convalescent plasma
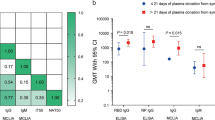
The distributions of antibodies in convalescent plasma units varied by blood supplier (Fig. 4, Supplementary Table 9 and Extended Data Fig. 7); therefore, antibody analyses controlled for supplier to address possible confounding. Transfusion of convalescent plasma with average (log-transformed) levels of antibody-dependent cellular cytotoxicity (ADCC) yielded an OR of 1.16 (95% CI 0.85–1.57) for the primary outcome relative to standard of care. Each one-unit increase in the standardized log-transformed ADCC was associated with a 24% reduction in the OR of the treatment effect (OR = 0.76, 95% CI 0.62–0.92) (Fig. 4 and Supplementary Table 10). This effect-modifying role was also significant for the neutralization test (OR = 0.77, 95% CI 0.63–0.94) but not for anti-receptor-binding domain (RBD) enzyme-linked immunosorbent assay (ELISA) (OR = 0.84, 95% CI 0.69–1.03) or IgG against the full transmembrane spike (OR = 1.01, 95% CI 0.82–1.23).

a, Absolute antibody amounts transfused per patient (n = 597) in the CCP arm for each marker, expressed as the product of volume and concentration. Center line: median; box limits: 25th and 75th percentiles; whiskers: 1.5× IQR; points: outliers. b, Effect-modifying role of CCP content for the primary outcome for each marker. The top row presents the trends in CCP effect compared to SOC as a function of the marker value, along with 95% CIs. Marker values are expressed as standard deviations of log values centered around the mean (standardized log). The horizontal dotted line represents CCP with no effect (OR = 1). The P values (two-sided test for trend without adjustment for multiple comparisons) refer to the effect modification observed with each marker (Supplementary Table 10). The histograms present the frequency distribution by marker. c,d, Contour plots of the OR for the primary outcome as a function of marker combinations. Overlaid data points indicate the value of the two markers for each CCP transfusion. Mfi, mean fluorescence intensity; OD, optical density; S, SARS-CoV-2 spike protein; SOC, standard of care.
When all four serologic markers were included in the multivariate model, each one-unit increase in the standardized log-transformed anti-spike IgG marker was associated with a 53% increase in the OR for the deleterious effect of convalescent plasma on the primary outcome (OR = 1.53, 95% CI 1.14–2.05); increases in ADCC and neutralization independently improved the effect of CCP (OR = 0.66, 95% CI 0.50–0.87 and OR = 0.74, 95% CI 0.57–0.95, respectively), whereas levels of anti-RBD antibodies had no effect-modifying role (OR = 1.02, 95% CI 0.76–1.38) (Supplementary Table 10). There was no evidence of significant interaction among the four serologic measures in the general additive model (Fig. 4 and Extended Data Fig. 8).
Meta-analysis
Of the 15 other reported randomized trials, 11 used only high-titer plasma5,7,10,11,12,13,14,15,16,17,18, and four applied less stringent plasma selection criteria, allowing for variable plasma titers6,19,20,21. Including the results from CONCOR-1, a total of 15,301 patients participated in trials using high-titer plasma, and 968 participated in trials applying less stringent criteria. The summary estimates for the RR of mortality in high-titer plasma trials was 0.97 (95% CI 0.92–1.02) compared to 1.25 (95% CI 0.92–1.69) in trials using unselected convalescent plasma (Fig. 5).

a, Meta-analysis of trials that used high-titer plasma. b, Meta-analysis of trials that used a mix of low-, medium- and high-titer plasma. df, degrees of freedom.
Discussion
The CONCOR-1 trial found that the use of convalescent plasma for the treatment of hospitalized patients with COVID-19 did not reduce the risk of intubation or death at 30 d. Patients in the convalescent plasma arm experienced more serious adverse events. Convalescent plasma was not associated with an improvement in any of the secondary efficacy outcomes or in any of the subgroup analyses. These results are consistent with the RECOVERY trial and a recent Cochrane meta-analysis8. A major additional contribution of our study comes from the study of immunologic markers, which suggest that the antibody profile significantly modified the effect of convalescent plasma compared to standard of care.
The RECOVERY trial showed that transfusion of high-titer plasma was no better than standard of care in the prevention of key outcomes. The U.S. National Registry report showed that high antibody level plasma was associated with a 34% RR reduction in mortality compared to low antibody level plasma9. Our assessment of the role of antibody profile on the clinical effect relative to standard of care is aligned with both of these conclusions. In the RECOVERY trial, plasma with a commercial ELISA cutoff corresponding to a neutralizing antibody titer of 100 or greater was used, and the mortality rate ratio compared to standard of care was 1.00 (95% CI 0.93–1.07). In our trial, plasma from one of the blood suppliers (blood supplier 1) that used a similar antibody threshold (neutralizing antibody titer of 160 of greater) was associated with a similar effect size (OR = 0.95 (95% CI 0.73–1.25)) (Fig. 4). In contrast, the U.S. National Registry study, which lacked a control group, reported that plasma containing high antibody levels (Ortho VITROS IgG anti-spike subunit 1, which contains the RBD, signal-to-cutoff ratio >18.45) was associated with a 34% reduction in mortality compared to plasma containing low antibody levels (signal-to-cutoff ratio <4.62)9. In our regression model (Supplementary Table 10), plasma with anti-RBD ELISA values corresponding to this low antibody cutoff (Fig. 4 and Extended Data Figure 9) would have a predicted OR of 1.49 compared to controls (95% CI 0.98–2.29), whereas plasma with the corresponding high antibody cutoff would have a predicted OR of 0.91 (95% CI 0.60–1.40), representing a 38% RR reduction. Thus, the 34% RR reduction observed by the U.S. National Registry9 could be explained by increased mortality with low antibody plasma rather than improved mortality with high antibody plasma.
This conclusion is corroborated by the meta-analysis of previous trials based on plasma selection strategy. Although the vast majority of patients included in convalescent plasma trials received high-titer plasma, most patients treated outside of clinical trials did not, including many of those who received plasma according to the current U.S. Food and Drug Administration (FDA) requirements (Ortho VITROS ≥9.5). Only 20% of convalescent plasma included in the U.S. National Registry was considered high-titer9. In our study, blood supplier 3 issued the same plasma as the one used in clinical practice as part of the emergency use authorization, and, in our subgroup analysis, convalescent plasma from this blood supplier was associated with worse clinical outcomes (OR = 1.89, 95% CI 1.05–3.43).
The antibody content is critical in determining the potency and potential harm of passive antibody therapy. Convalescent plasma demonstrating high levels of viral neutralization and high levels of Fc-mediated function were independently associated with a reduced risk for intubation or death. The importance of Fc-mediated function is in line with the known functional determinants of the anti-SARS-CoV-2 humoral response. In animal models of COVID-19, mutation of monoclonal antibodies leading to loss of Fc-mediated function, but sparing the neutralizing function, abrogated the protective effect of the antibody22,23,24,25. In cohort studies of severe COVID-19, low Fc-mediated function, but not neutralization, was associated with mortality26,27.
In contrast, high levels of IgG antibodies against the full transmembrane spike protein measured by flow cytometry (which is distinct from commercial assays for IgG against spike subunit 1) were associated with an increased risk of intubation or death after controlling for other antibody markers, suggesting that the transfusion of convalescent plasma containing non-functional anti-SARS-CoV-2 antibodies might be harmful. Antibody Fc-mediated function is dependent on the ability to aggregate and crosslink Fc receptors on target cells. This process can be disrupted by competition from other antibodies with low or absent Fc function28. Similar observations were made during HIV vaccine trials, where the development of IgA antibodies against the virus envelope paradoxically increased the risk of infection due to competition with IgG29,30, and in animal models of passive immunization where transfer of antibodies could be deleterious to the host31.
One positive clinical trial in mild disease (n = 160) found that high-titer convalescent plasma administered within 72 h of the onset of mild COVID-19 symptoms improved clinical outcomes compared to placebo in an elderly outpatient population13. Furthermore, in a Bayesian re-analysis of the RECOVERY trial, the subgroup of patients who had not yet developed anti-SARS-CoV-2 antibodies appeared to benefit from convalescent plasma32. The C3PO trial, which also assessed early treatment with high-titer plasma in high-risk patients, was stopped prematurely for futility after enrolling 511 of 900 planned participants (NCT04355767). In our trial, the median time from the onset of symptoms was 8 d; however, we did not observe a difference in the primary outcome in the subgroup of patients who were randomized within 3 d of diagnosis.
The frequency of serious adverse events was higher in the convalescent plasma group compared to the standard of care group (33.4% versus 26.4%; RR = 1.27, 95% CI 1.02–1.57). Most of these events were caused by worsening hypoxemia and respiratory failure occurring throughout the 30-d follow-up period. This frequency is consistent with the recent Cochrane review that reported an OR of 1.24 (95% CI 0.81–1.90) for serious adverse events8. The frequency of transfusion-associated dyspnea and transfusion-associated circulatory overload was 2.1% and 0.8%, respectively, which is similar to other studies of non-convalescent plasma33. The rates of transfusion reactions in CONCOR-1 were higher than what were reported in the RECOVERY trial, where transfusion reactions were reported in 13 of 5,795 (0.22%) patients. CONCOR-1 site investigators included many transfusion medicine specialists, and the open-label design might have encouraged reporting. However, the rate of serious transfusion-related adverse events was low (4/614 (0.65%) patients treated with convalescent plasma) and, thus, does not explain the difference in serious adverse events between groups.
CONCOR-1 was a randomized trial designed to examine the effect of convalescent plasma versus standard of care for the primary composite outcome of intubation or death, with a capacity to explore the immunological profile of convalescent plasma and its impact on the effect of convalescent plasma. The trial involved four blood suppliers that provided local convalescent plasma units based on different antibody criteria. As a result, plasma units with a wide distribution of antibody content were included, and comprehensive antibody testing using both quantitative and functional assays provided a detailed description of the plasma product. The open-label design represents a limitation of this study, as knowledge of the treatment group could influence the decision to intubate, report adverse events or administer other treatments. The antibody profile of recipients was unavailable at the time of this analysis. In future work, we will investigate the value of convalescent plasma in patients without a detectable humoral immune response. In addition, other antibody isotypes (IgM and IgA) and IgG subclasses should be evaluated in future studies to determine their effect on clinical outcomes. Additional randomized trials are warranted to assess the early use of high-titer convalescent plasma units in immunocompromised patients with COVID-19 who are unable to mount an efficient anti-SARS-CoV-2 antibody response.
In summary, the CONCOR-1 trial did not demonstrate a difference in the frequency of intubation or death at 30 d with convalescent plasma or standard of care in hospitalized patients with COVID-19 respiratory illness. The antibody content had a significant effect-modifying role for the effect of convalescent plasma on the primary outcome. The lack of benefit and the potential concern of harm caution against the unrestricted use of convalescent plasma for hospitalized patients with COVID-19.
Methods
Trial design and oversight
CONCOR-1 was an investigator-initiated, multi-center, open-label, randomized controlled trial conducted at 72 hospital sites in Canada, the United States and Brazil34. Eligible patients were randomly assigned to receive either convalescent plasma or standard of care. The study was approved by Clinical Trials Ontario (research ethics board of record: Sunnybrook Health Sciences Centre), project no. 2159; the Quebec Ministry of Health and Social Services multicenter ethics review (research ethics board of record: Comité d’éthique de la recherche du CHU Sainte-Justine), project no. MP-21-2020-2863; the Weil Cornell Medicine General Institutional Review Board, protocol no. 20-04021981; the Comissão Nacional de Ética em Pesquisa, approval no. 4.305.792; the Héma-Québec Research Ethics Board; the Canadian Blood Services Research Ethics Board; Research Ethics BC (research ethics board of record: the University of British Columbia Clinical Research Ethics Board); the Conjoint Health Research Ethics Board; the University of Alberta Health Research Ethics Board (Biomedical Committee); the Saskatchewan Health Authority Research Ethics Board; the University of Saskatchewan Biomedical Research Ethics Board; the University of Manitoba Biomedical Research Board; the Queensway Carleton Hospital Research Ethics Board; the Scarborough Health Network Research Ethics Board; the Windsor Regional Hospital Research Ethics Board; and the Bureau de l’Éthique of Vitalité Health Network. Regulatory authorization was obtained from Health Canada (control no. 238201) and the U.S. FDA (IND 22075). The trial was registered at ClinicalTrials.gov (NCT04348656). An independent data safety monitoring committee performed trial oversight and made recommendations after review of safety reports planned at every 100 patients and at the planned interim analysis based on the first 600 patients. External monitoring was performed at all sites to assess protocol adherence, reporting of adverse events and accuracy of data entry. Full details of the study design, conduct, oversight and analyses are provided in the protocol and statistical analysis plan, which are available online.
Participants
Eligible participants were (1) ≥16 years of age in Canada or ≥18 years of age in the United States and Brazil; (2) admitted to the hospital ward with confirmed COVID-19; (3) required supplemental oxygen; and (4) a 500-ml of ABO-compatible COVID-19 convalescent plasma (CCP) was available. The availability of ABO-compatible convalescent plasma from donors who had recovered from COVID-19 infection was an eligibility requirement. Exclusion criteria were (1) more than 12 d from the onset of respiratory symptoms; (2) imminent or current intubation; (3) a contraindication to plasma transfusion; or (4) a plan for no active treatment. Consent was obtained from all donors and participants (or their legally authorized representative).
Randomization and intervention
Patients were randomized in a 2:1 ratio to receive convalescent plasma or standard of care using a secure, concealed, computer-generated, web-accessed randomization sequence (REDCap v11.0.1)35. Randomization was stratified by site and age (<60 and ≥60 years) with allocation made with permuted blocks of size 3 or 6. Patients randomized to convalescent plasma received one or two units of apheresis plasma amounting to approximately 500 ml from one or two donors. The plasma was stored frozen and was thawed as per standard blood bank procedures and infused within 24 h of randomization. Patients were monitored by clinical staff for transfusion-related adverse events as per local procedures. Individuals assigned to standard of care received usual medical care as per routine practices at each site. The investigational product was prepared by Canadian Blood Services and Héma-Québec (Canada), the New York Blood Center (United States)36 and Hemorio (Brazil). Each supplier had different criteria for qualifying convalescent plasma units that were based on the presence of either viral neutralizing antibodies, measured by the plaque reduction neutralization assay and expressed as the concentration of serum that reduced the number of virus-induced plaques by 50% (PRNT50)37,38 using a threshold titer of >1:160 or antibodies against the RBD of the SARS-CoV-2 spike protein using a threshold titer of >1:100. Female donors with previous pregnancies were excluded from donation, unless they tested negative for HLA antibodies. In addition, a retained sample from every plasma donation was tested at reference laboratories after the transfusion for (1) anti-RBD antibodies (IgM, IgA and IgG) by ELISA;39,40 (2) viral neutralization by the PRNT50 assay using live virus;37,38 (3) IgG antibodies binding to the full-length trimeric transmembrane SARS-CoV-2 spike protein expressed on 293T cells by flow cytometry;41 and (4) Fc-mediated function by ADCC assay against the full spike protein expressed on CEM.NKr cells (see supplement for complete description)42,43. For each plasma unit, the absolute antibody content was defined as the product of the unit volume and the concentration of the antibody (or functional capacity) in the plasma. These calculations were used to estimate the total antibody content from the transfusion of two units.
Trial outcomes
The primary outcome was the composite of intubation or death by day 30. Secondary outcomes were: time to intubation or death; ventilator-free days by day 30; in-hospital death by day 90; time to in-hospital death; death by day 30; length of stay in critical care and hospital; need for extracorporeal membrane oxygenation; need for renal replacement therapy; convalescent plasma-associated adverse events; and occurrence of ≥3 grade adverse events by day 30 (classification of adverse events was performed using MedDRA (https://www.meddra.org/) and was graded by Common Terminology Criteria for Adverse Events, v4.03). All transfusion-related adverse events were classified and graded by International Society for Blood Transfusion (www.isbtweb.org) definitions. All patients were followed to day 30, including a 30-d telephone visit for patients who were discharged from hospital. Patients who were in hospital beyond day 30 were followed until discharge for the purpose of determining in-hospital mortality up to day 90.
Statistical analysis
The primary analysis was based on the intention-to-treat population, which included all individuals who were randomized and for whom primary outcome data were available. The per-protocol population was comprised of eligible patients who were treated according to the randomized allocation of the intervention and received two units (or equivalent) of convalescent plasma within 24 h of randomization.
The effect of convalescent plasma on the composite primary outcome of intubation or death by day 30 was assessed by testing the null hypothesis that the composite event rate was the same under convalescent plasma and standard of care. The RR for the primary outcome (convalescent plasma versus standard of care) was computed with a 95% CI. Secondary outcomes were analyzed as described in the statistical analysis plan (Appendix B in the (Supplementary Information). No multiplicity adjustments were implemented for the secondary analyses. Procedures planned for addressing missing data and subgroup analyses are described in the statistical analysis plan. Forest plots were used to display point estimates, and CIs across subgroups with interaction tests were used to assess effect modification.
The effect-modifying role of antibody content on the primary outcome was assessed via logistic regression controlling for the blood supplier, treatment and the antibody marker. Antibody markers were log-transformed, centered and then divided by the corresponding standard deviation before being entered into logistic regression models (see statistical analysis plan, Appendix B in the Supplementary Information). A multivariate logistic regression model was then fitted adjusting for all four markers. Generalized additive models were used to examine the joint effect of each pair of serologic markers on the primary outcome44.
The results from CONCOR-1 were subsequently included in a meta-analysis based on the 20 May 2021 update of the Cochrane systematic review8 and known randomized trials published since comparing convalescent plasma to placebo or standard care in patients with COVID-19. These were divided based on whether they used plasma with high antibody titer or not. For each trial, we compared the observed number of deaths at 30 d (or closest available time point before a crossover, if applicable) of patients allocated to convalescent plasma or the control group. Summary estimates for RR with 95% CI were calculated using random effects meta-analysis to account for variation in effect size among studies. Heterogeneity was quantified using inconsistency index (I2) and P values from the chi-square test for homogeneity.
With a 2:1 randomization ratio, 1,200 patients (800 in the convalescent plasma group and 400 in the standard of care group) were needed to provide 80% power to detect an RR reduction of 25% with convalescent plasma for the primary outcome with a 30% event rate under standard of care, based on a two-sided test at the 5% significance level. An interim analysis by a biostatistician unblinded to the allocation of the intervention was planned for when the primary outcome was available for 50% of the target sample. An O’Brien–Fleming stopping rule was employed45 to control the overall type I error rate at 5%. Conditional power was used to guide futility decisions with the nominal threshold of 20% to justify early stopping.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
De-identified individual patient data with the data dictionary that underlie the reported results will be made available upon request if the intended use is concordant with existing research ethics board approvals (requests will be reviewed by the CONCOR-1 Steering Committee within 3 months). Proposals for access should be sent to arnold@mcmaster.ca. The protocol and statistical analysis plan are available in the online supplement.
Change history
12 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41591-021-01667-1
References
Seow, J. et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat. Microbiol. 5, 1598–1607 (2020).
Devasenapathy, N. et al. Efficacy and safety of convalescent plasma for severe COVID-19 based on evidence in other severe respiratory viral infections: a systematic review and meta-analysis. CMAJ 192, E745–E755 (2020).
Wood, E. M., Estcourt, L. J. & McQuilten, Z. K. How should we use convalescent plasma therapies for the management of COVID-19? Blood 137, 1573–1581 (2021).
Blackall, D. et al. Rapid establishment of a COVID-19 convalescent plasma program in a regional health care delivery network. Transfusion 60, 2203–2209 (2020).
Simonovich, V. A. et al. A randomized trial of convalescent plasma in Covid-19 severe pneumonia. N. Engl. J. Med. 384, 619–629 (2021).
Agarwal, A. et al. Convalescent plasma in the management of moderate covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial). Brit. Med. J. 371, m3939 (2020).
Li, L. et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA 324, 460–470 (2020).
Piechotta, V. et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a living systematic review. Cochrane Database Syst. Rev. 5, CD013600 (2021).
Joyner, M. J. et al. Convalescent plasma antibody levels and the risk of death from Covid-19. N. Engl. J. Med. 384, 1015–1027 (2021).
The RECOVERY Collaborative Group. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised controlled, open-label, platform trial. Lancet 397, 2049–2059 (2021).
Ray, Y. et al. Clinical and immunological benefits of convalescent plasma therapy in severe COVID-19: insights from a single center open label randomised control trial. Preprint at https://www.medrxiv.org/content/10.1101/2020.11.25.20237883v1 (2020).
Gharbharan, A. et al. Effects of potent neutralizing antibodies from convalescent plasma in patients hospitalized for severe SARS-CoV-2 infection. Nat. Commun. 12, 3189 (2021).
Libster, R. et al. Early high-titer plasma therapy to prevent severe Covid-19 in older adults. N. Engl. J. Med. 384, 610–618 (2021).
O’Donnell, M. R. et al. A randomized double-blind controlled trial of convalescent plasma in adults with severe COVID-19. J. Clin. Invest. 131, e150646 (2021).
Avendaño-Solà, C. et al. Convalescent plasma for COVID-19: a multicenter, randomized clinical trial. Preprint at https://www.medrxiv.org/content/10.1101/2020.08.26.20182444v3 (2020).
Estcourt, L. J. Convalescent plasma in critically ill patients with Covid-19. Preprint at https://www.medrxiv.org/content/10.1101/2021.06.11.21258760v1 (2021).
Bennett-Guerrero, E. et al. Severe acute respiratory syndrome coronavirus 2 convalescent plasma versus standard plasma in Coronavirus Disease 2019 infected hospitalized patients in New York: a double-blind randomized trial. Crit. Care Med. 49, 1015–1025 (2021).
Körper, S. et al. High dose convalescent plasma in COVID-19: results from the randomized trial CAPSID. Preprint at https://www.medrxiv.org/content/10.1101/2021.05.10.21256192v1 (2021).
AlQahtani, M. et al. Randomized controlled trial of convalescent plasma therapy against standard therapy in patients with severe COVID-19 disease. Sci. Rep. 11, 9927 (2021).
Hamdy Salman, O. & Ail Mohamed, H. S. Efficacy and safety of transfusing plasma from COVID-19 survivors to COVID-19 victims with severe illness. A double-blinded controlled preliminary study. Egypt. J. Anaesth. 36, 264–272 (2020).
Bajpai, M. et al. efficacy of convalescent plasma therapy compared to fresh frozen plasma in severely ill COVID-19 patients: a pilot randomized controlled trial. Preprint at https://www.medrxiv.org/content/10.1101/2020.10.25.20219337v1 (2020).
Winkler, E. S. et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 184, 1804–1820 (2021).
Suryadevara, N. et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 184, 2316–2331 (2021).
Ullah, I. et al. Live imaging of SARS-CoV-2 infection in mice reveals neutralizing antibodies require Fc function for optimal efficacy. Preprint at https://www.biorxiv.org/content/10.1101/2021.03.22.436337v1.full (2021).
Schafer, A. et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 218, e20201993 (2021).
Brunet-Ratnasingham, E. A. S. et al. Integrated immunovirological profiling validates plasma SARS-CoV-2 RNA as an early predictor of COVID-19 mortality. Preprint at https://www.medrxiv.org/content/10.1101/2021.03.18.21253907v1 (2021).
Zohar, T. et al. Compromised humoral functional evolution tracks with SARS-CoV-2 mortality. Cell 183, 1508–1519 (2020).
Casadevall, A., Joyner, M. J. & Pirofski, L. A. Neutralizing antibody LY-CoV555 for outpatient Covid-19. N. Engl. J. Med. 384, 189 (2021).
Haynes, B. F. et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366, 1275–1286 (2012).
Tomaras, G. D. et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl Acad. Sci. USA 110, 9019–9024 (2013).
Taborda, C. P., Rivera, J., Zaragoza, O. & Casadevall, A. More is not necessarily better: prozone-like effects in passive immunization with IgG. J. Immunol. 170, 3621–3630 (2003).
Hamilton, F. W., Lee, T., Arnold, D. T., Lilford, R. & Hemming, K. Is convalescent plasma futile in COVID-19? A Bayesian re-analysis of the RECOVERY randomized controlled trial. Int. J. Infect. Dis. 109, 114–117 (2021).
Narick, C., Triulzi, D. J. & Yazer, M. H. Transfusion-associated circulatory overload after plasma transfusion. Transfusion 52, 160–165 (2012).
Begin, P. et al. Convalescent plasma for adults with acute COVID-19 respiratory illness (CONCOR-1): study protocol for an international, multicentre, randomized, open-label trial. Trials 22, 323 (2021).
Harris, P. A. et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 42, 377–381 (2009).
Budhai, A. et al. How did we rapidly implement a convalescent plasma program? Transfusion 60, 1348–1355 (2020).
Abe, K. T. et al. A simple protein-based surrogate neutralization assay for SARS-CoV-2. JCI Insight. 5, e142362 (2020).
Mendoza, E. J., Manguiat, K., Wood, H. & Drebot, M. Two detailed plaque assay protocols for the quantification of infectious SARS-CoV-2. Curr. Protoc. Microbiol. 57, ecpmc105 (2020).
Beaudoin-Bussieres, G. et al. Decline of humoral responses against SARS-CoV-2 spike in convalescent individuals. Preprint at https://www.biorxiv.org/content/10.1101/2020.07.09.194639v1 (2020).
Perreault, J. et al. Waning of SARS-CoV-2 RBD antibodies in longitudinal convalescent plasma samples within 4 months after symptom onset. Blood 136, 2588–2591 (2020).
Anand, S. P. et al. High-throughput detection of antibodies targeting the SARS-CoV-2 spike in longitudinal convalescent plasma samples. Transfusion 61, 1377–1382 (2021).
Tauzin, A. N. M. et al. A single BNT162b2 mRNA dose elicits antibodies with Fc-mediated effector functions and boost pre-existing humoral and T cell responses. Preprint at https://www.biorxiv.org/content/10.1101/2021.03.18.435972v1 (2021).
Anand, S. P. et al. Longitudinal analysis of humoral immunity against SARS-CoV-2 spike in convalescent individuals up to eight months post-symptom onset. Cell Rep. Med. 2, 100290 (2021).
Wood, S. N. Generalized Additive Models: An Introduction with R, Second Edition (Taylor and Francis, 2017).
O’Brien, P. C. & Fleming, T. R. A multiple testing procedure for clinical trials. Biometrics 35, 549–556 (1979).
R Development Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2010).
Wood, S. N. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J. R. Stat. Soc. Ser. B (Methodol.) 73, 3–36 (2011).
Acknowledgements
The authors acknowledge the recovered patients who donated their plasma; the staff involved in the recruitment, collection, qualification and distribution of COVID-19 convalescent plasma; and the staff of the COVID-19 wards who made the study possible. The authors would also like to thank the members of the independent data safety monitoring committee: K. Karkouti, R. Fowler, M. Delaney, G. Tomlinson, D. Davis and B. Juelg. Funding for the study was provided by Canadian Institutes of Health Research COVID-19 May 2020 Rapid Research Funding Opportunity, operating grant no. 447352 (D.A., P.B. and J.C.); Ontario COVID-19 Rapid Research Fund (D.A.); Toronto COVID-19 Action Initiative 2020 (University of Toronto) (J.C.); Fondation du CHU Ste-Justine (P.B.); Ministère de l’Économie et de l’Innovation du Québec, no. 2020-2021-COVID-19-PSOv2a-51169 (P.B.); Fonds de Recherche du Québec, santé no. 281662 (P.B.); University Health Network Emergent Access Innovation Fund (N.S.); University Health Academic Health Science Centre Alternative Funding Plan (Sunnybrook Health Sciences Centre) (N.D.); Saskatchewan Ministry of Health (O.P.T.); University of Alberta Hospital Foundation (S.N.); Alberta Health Services COVID-19 Foundation Competition (S.N.); Sunnybrook Health Sciences Centre Foundation (J.C.); Fondation du CHUM (A.F.); Ottawa Hospital Academic Medical Organization (A.T.); Ottawa Hospital Foundation COVID-19 Research Fund (A.T.); Sinai Health System Foundation (N.S.); and McMaster University (D.A.). These organizations and institutions did not have any role in the writing of the manuscript or the decision to submit it for publication. The authors did not receive payments from any pharmaceutical company or other agency to write this article. All authors had full access to the full data in the study and accept responsibility to submit for publication.
Author information
Authors and Affiliations
Consortia
Contributions
P.B., J.C., E.J., R. Cook, N.M.H., A.T., M.P.Z., G.B.B., R.B., K.C.L., R. Carl, M.C., N.D., D.V.D., D.A.F., A.M., N.R., D.C.S., L.S., N.S., A.F.T., R.Z., A.F. and D.M.A. were involved in study conceptualization, funding acquisition and methodology. P.B., J.C., R. Cook, G.B.B., R.B., H.W., A.F., N.L., Y.L. and D.M.A. developed the methodology for antibody analyses. P.B., J.C., N.M.H., A.T., M.P.Z., G.B.B., L.A., R.B., M.C., M.M.C., N.D., D.V.D., J.D., D.A.F., C.G., M.J.G., A.M., N.R., B.S.S., D.C.S., L.S., N.S., A.F.T., H.W., R.Z., A.F. and D.M.A. participated in the investigations. P.B., J.C., E.J., N.M.H., A.T., M.P.Z., L.A., R.B., M.M.C., D.V.D., C.G., M.J.G., N.R., B.S.S. and D.M.A. were responsible for project administration. P.B., J.C., E.J., R.C., G.B.B., R.B., D.V.D., N.L., Y.L., H.W. and D.M.A. were involved in data curation. P.B., J.C., R.C., N.L., Y.L. and D.M.A. were responsible for formal analyses and data visualization. P.B., J.C., Y.L. and D.M.A. verified the underlying data. P.B., J.C., R.C. and D.M.A. drafted the original manuscript. All members of the writing committee reviewed and edited the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Medicine thanks Nicole Bouvier, Ventura Simonovich and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Jennifer Sargent was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Cumulative incidence functions of intubation or in-hospital death by day 30.
Panel A presents the intention-to-treat population and panel B presents the per protocol population.
Extended Data Fig. 2 Cumulative incidence functions of in-hospital death by day 90.
Panel A presents the intention-to-treat population and panel B presents the per protocol population.
Extended Data Fig. 3 Kaplan-Meier estimate of distribution of length of stay in hospital by day 90.
Panel A presents the intention-to-treat population and panel B presents the per protocol population.
Extended Data Fig. 4 Subgroup analysis for the per-protocol population.
P-values for relative risk and homogeneity are two-sided without adjustment for multiple comparisons. BMI: Body mass index.
Extended Data Fig. 5 Post-hoc subgroup analyses for the intention-to-treat population.
Subgroups based on corticosteroid use and location at time of randomizations were added post-hoc at time of review. P-values for relative risk and homogeneity are two-sided without adjustment for multiple comparisons.
Extended Data Fig. 6 Post-hoc subgroup analyses for the per-protocol population.
Subgroups based on corticosteroid use and location at time of randomizations were added post-hoc at time of review. P-values for relative risk and homogeneity are two-sided without adjustment for multiple comparisons.
Extended Data Fig. 7 Pairwise scatter plots of plasma antibody markers and empirical distribution functions.
Markers (log transformed and standardized) include antibody (IgM,IgA,IgG) against the receptor binding domain (anti-RBD) by ELISA, plaque reduction neutralization test, IgG antibody against the full transmembrane Spike protein (anti-S IgG) by flow cytometry and the antibody-dependent cellular cytotoxicity (ADCC) assay.corr: Pearson correlation coefficients of pair of antibody markers.
Extended Data Fig. 8 Contour plots of the joint effect-modifying role of antibody markers for convalescent plasma versus standard of care on the composite endpoint of intubation or death.
The contours convey pairwise combinations of antibody markers yielding similar odds ratios for the CCP effect with the black line corresponding to an odds ratio of 1 (that is no effect of CCP). Data points for individual patients are overlaid with colours denoting the blood supply centre. Contours were obtained from fitting generalized additive logistic regression models for the primary outcome adjusting for blood supply center, treatment and the log transformed and standardized biomarkers - smoothing splines were used to relax linearity assumptions. The contour lines with positive slope suggest combinations of high (or low) values for both markers yield similar effects of CCP; the contour lines with negative slopes suggest high values of both markers yield strong CCP effects. For the combination of anti-S IgG with ADCC or anti-S IgG with anti-RBD, the general additive logistic regression models led to a complex equation that was not statistically significant nor clinical interpretable. These combinations were therefore excluded from this figure.
Extended Data Fig. 9 Comparison of in-house ELISA to commercial assays.
Values from the Héma-Québec in-house ELISA measuring antibody (IgM, IgA, IgG) binding the receptor binding domain of SARS-CoV-2 Spike protein (used in the current study) are compared to results from Euroimmun (Panel A) and Ortho Vitros (Panel B) commercial assays measuring IgG binding to subunit 1 of the SARS-CoV-2 Spike protein, which contains the receptor binding domain and which were used to qualify convalescent plasma in previous clinical trials. Each sample was tested with the commercial assays twice.
Supplementary information
Supplementary Information
List of CONCOR-1 investigators, Supplementary Methods, Supplementary Tables 1–10, Study Protocol and Statistical Analysis Plan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bégin, P., Callum, J., Jamula, E. et al. Convalescent plasma for hospitalized patients with COVID-19: an open-label, randomized controlled trial. Nat Med 27, 2012–2024 (2021). https://doi.org/10.1038/s41591-021-01488-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01488-2
This article is cited by
-
Does haste make waste? Prevalence and types of errors reported after publication of studies of COVID-19 therapeutics
Systematic Reviews (2023)
-
The prevention and treatment of COVID-19 in patients treated with hemodialysis
European Journal of Medical Research (2023)
-
Robust induction of functional humoral response by a plant-derived Coronavirus-like particle vaccine candidate for COVID-19
npj Vaccines (2023)
-
Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection
Nature Reviews Immunology (2023)
-
SARS-CoV-2 vaccination elicits broad and potent antibody effector functions to variants of concern in vulnerable populations
Nature Communications (2023)
