
Abstract
The highly pathogenic avian influenza (HPAI) H5N1 virus clade 2.3.4.4b has caused the death of millions of domestic birds and thousands of wild birds in the USA since January 2022 (refs. 1,2,3,4). Throughout this outbreak, spillovers to mammals have been frequently documented5,6,7,8,9,10,11,12. Here we report spillover of the HPAI H5N1 virus to dairy cattle across several states in the USA. The affected cows displayed clinical signs encompassing decreased feed intake, altered faecal consistency, respiratory distress and decreased milk production with abnormal milk. Infectious virus and viral RNA were consistently detected in milk from affected cows. Viral distribution in tissues via immunohistochemistry and in situ hybridization revealed a distinct tropism of the virus for the epithelial cells lining the alveoli of the mammary gland in cows. Whole viral genome sequences recovered from dairy cows, birds, domestic cats and a raccoon from affected farms indicated multidirectional interspecies transmissions. Epidemiological and genomic data revealed efficient cow-to-cow transmission after apparently healthy cows from an affected farm were transported to a premise in a different state. These results demonstrate the transmission of the HPAI H5N1 clade 2.3.4.4b virus at a non-traditional interface, underscoring the ability of the virus to cross species barriers.
Similar content being viewed by others

Main
The HPAI virus H5Nx goose/Guangdong lineage, an influenza A virus (IAV) from the family Orthomyxoviridae, emerged in China in 1996. This viral lineage was initially detected only in poultry, with detections in wild birds occurring in 2002 (ref. 13). The virus has frequently reassorted with other influenza viruses, with the haemagglutinin gene remaining as the only gene that defines this genetic lineage of viruses. These frequent reassortments and ongoing antigenic changes led to complex classification of the viruses into multiple clades14. Over the past decade, the H5Nx goose/Guangdong lineage evolved into eight clades (2.3.4.4a–2.3.4.4h) with three main neuraminidase subtypes: N1, N8 and N6. The H5N1 clade 2.3.4.4b has caused global outbreaks in recent years15,16, infecting various avian species and showing potential to infect humans and other mammals5,6,7,11,12,17,18,19,20,21,22,23. The World Health Organization (WHO) has reported 860 human infections since 2003, with a fatality rate of approximately 52.8%24, although serological evidence has suggested less severe more widespread infection25. Risk of human-to-human transmission remains low26. Since 2016, the H5Nx clade 2.3.3.4b has circulated broadly in migratory wild bird populations across Europe, Africa and Asia, being first detected in North America (Canada) in December 2021 (ref. 27). By January 2022, it was found in wild birds in North and South Carolina in the USA1,28, and soon after in commercial poultry2. Since then, H5N1 caused high morbidity and mortality in poultry leading to the loss of over 100 million birds (as of 13 August 2024) in the USA alone29. The continuous and widespread circulation of this high-consequence panzootic pathogen is of major concern and poses a substantial threat to animal and public health.
In addition to devastating consequences to domestic and wild avian species, HPAI H5N1 virus clade 2.3.4.4b spillovers have been detected in several mammals9,10,29,30, including domestic and wild carnivorous species such as cats (Felis catus)17, red foxes (Vulpes vulpes)6, bears (Ursus americanus)11 and harbour seals (Phoca vitulina)5. The virus has even spread to polar regions, killing a polar bear (Ursus maritimus) in the Arctic, and elephant seals (Mirounga leonina), Antarctic fur seals (Arctocephalus gazella) and gentoo penguins (Pygoscelis papua) in Antarctica31. In the USA, a human infection in a poultry worker that resulted in mild symptoms and full recovery was reported in April 2022 (ref. 32). During 2023, two major outbreaks of H5N1 in harbour seals resulted in high mortality in Maine and Washington. In January 2023, two indoor–outdoor cats died from HPAI-induced encephalitis12. Acute death of a striped skunk (Mephitis mephitis) in Washington has been reported20, along with additional cases in this species in 2023 and 2024 (ref. 33). On 20 March 2024, the Minnesota board of animal health reported that a juvenile goat (Capra hircus) tested positive for HPAI, resulting in the first report of HPAI H5N1 infection in a ruminant species, following positive tests in backyard poultry on the same premises34.
Here we report the spillover of the HPAI H5N1 virus clade 2.3.4.4b into dairy cattle and describe the findings of a clinical, pathological and epidemiological investigation in nine affected farms (farms 1–9) across four states in the USA.
Clinicoepidemiological investigation
From late January to mid-March 2024, a morbidity event of unknown aetiology affecting dairy cattle was reported by field veterinarians in the Texas Panhandle and surrounding states (New Mexico and Kansas). The first farm known to be affected by the morbidity event (30 January 2024) was in Texas; however, no clinical samples were collected from affected animals in this farm. The disease was subsequently reported in additional farms in Texas and other states. We conducted a clinicoepidemiological investigation in nine farms located in Texas (farms 1, 2, 5, 6 and 7), New Mexico (farms 4 and 8), Kansas (farm 9) and Ohio (farm 3) that reported the morbidity event between 11 February and 19 March 2024. Farm 3 in Ohio was affected after apparently healthy lactating cattle were transported from farm 1 in Texas to this location (Extended Data Table 1). Affected dairy cattle presented with decreased feed intake, decreased rumination time, mild respiratory signs (clear nasal discharge, increased respiratory rate and laboured breathing), lethargy, dehydration, dry/tacky faeces or diarrhoea, and milk with abnormal yellowish colostrum-like colour, thick and sometimes curdled consistency. In addition, an abrupt drop in milk production ranging from 20% to 100% in individual affected animals was noted. Upon clinical examination, mammary gland involution was observed in several of the affected cows (Extended Data Fig. 1). The proportion of clinically affected animals ranged between 3% and 20%. Mortality above average (twofold higher) was noted in cows in farms 2 and 3 during the clinical event. Of note, several of the affected farms reported simultaneous mortality events in passerines (great-tailed grackles (Quiscalus mexicanus)), peridomestic birds (rock pigeons (Columba livia)), and in outdoor domestic (cats) and wild (raccoons (Procyon lotor) mammals; Extended Data Table 1). The clinical disease in dairy cattle lasted 5–14 days, with animals returning to pre-outbreak health status, rumination times and feed intake, but maintaining decreased milk production for at least 4 weeks.
Multispecies detection of the H5N1 virus
A diagnostic investigation was conducted in samples collected from farms 1 to 9. Initially, nasal swabs, serum and blood buffy coats from ten affected cows from farm 1 were subjected to viral metagenomic sequencing. IAV sequences were detected in one nasal swab and no other bovine respiratory viruses were detected. Real-time PCR with reverse transcription (rRT–PCR) targeting the IAV matrix and haemagglutinin 5 (H5) genes confirmed the presence of HPAI-H5 in nasal swabs of this cow. Of note, 8 of 10 milk samples collected from the same cows were positive for HPAI-H5 via rRT–PCR (Supplementary Table 1). In addition, oropharyngeal swabs from great-tailed grackles and rock pigeons, and lung and brain tissues from a cat found dead on farm 1 tested positive for HPAI-H5 (Supplementary Table 1). A similar epidemiological scenario involving mortality events in domestic and wild mammals was observed in farms 3–5 and 8. Six domestic cats died in farm 3 after the disease onset in dairy cows. Cats found dead on farms 4 and 5 and cats and a raccoon found dead on farm 8 tested positive for HPAI-H5.
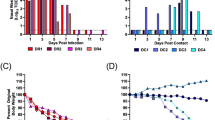
Testing of multiple sample types (n = 331) collected from cows from farms 1 to 9 by rRT–PCR showed sporadic viral RNA detection in nasal swabs (10 of 47), whole blood (3 of 25) and serum (1 of 15), and most frequent detection in milk (129 of 192). The milk samples consistently had the highest viral RNA loads of the samples tested (Fig. 1a and Supplementary Table 1). Results from rRT–PCR on tissues collected from three affected cows revealed the presence of viral RNA in the lungs, small intestines, supramammary lymph nodes and mammary glands. The highest viral RNA loads were detected in the mammary glands (Fig. 1b and Supplementary Table 1), corroborating high viral loads detected in milk. In addition, haemagglutination inhibition antibody testing in paired serum samples collected from animals in farm 2 (n = 19) confirmed H5N1 infection in affected dairy cows (Fig. 1c).

a, Viral RNA loads in nasal swab (n = 27), whole-blood (n = 25), serum (n = 15), urine (n = 4), faecal (n = 10) and milk (n = 167) samples collected from cattle from farms 1 to 9 quantified by rRT–PCR targeting the IAV matrix gene. b, Viral RNA loads in tissues of dairy cattle quantified by rRT–PCR targeting the IAV matrix gene. c, Serum antibody responses in affected cattle (n = 19) quantified by a haemagglutination inhibition (HI) assay. d, Cytopathic effect of the HPAI H5N1 virus isolated from milk in bovine uterine epithelial cells Cal-1. The photomicrographs shown are representative of two independent clinical samples. Scale bar, 50 µm. e, Detection of the infectious HPAI virus in Cal-1 cells by an immunofluorescence assay using a nucleoprotein-specific monoclonal antibody (red) counterstained with 4′,6-diamidino-2-phenylindole (DAPI, blue). The photomicrographs shown are representative of two independent clinical samples. Scale bar, 50 µm. f,g, Infectious HPAI H5N1 virus in milk (n = 69; f) and tissues (g) detected by virus titration. Virus titres were determined using end point dilutions and expressed as 50% tissue culture infectious dose (TCID50) per millilitre (TCID50 ml−1). The limit of detection for infectious virus titration was 101.05 TCID50 ml−1. Data are presented as mean ± s.e.m (a–c,f,g). All graphs and statistical analysis were generated using GraphPad Prism (v10).
Infectious virus shedding in dairy cows
Virus isolation and quantification were performed on milk samples from farms 1, 2 and 3. Infectious HPAI H5N1 virus was isolated from the pellet of pooled milk samples from 10 cows from farms 1 and 2 (Fig. 1d,e). Of note, virus titres in milk from affected animals ranged from 104.0 to 108.8 50% tissue culture infectious dose (TCID50) per millilitre (Fig. 1f), demonstrating high infectious viral loads in milk from infected animals. Consistent with this, high viral loads (107.3–107.8 TCID50 ml−1) were detected in mammary gland tissues (Fig. 1g).
Virus shedding was also investigated in samples (milk, nasal swabs, urine and faeces) collected from clinical and non-clinical animals from farm 3. Overall, virus shedding was detected more frequently in milk samples from clinical animals (24 of 25) with higher RNA viral loads than from non-clinical animals (1 of 15; Fig. 2a and Extended Data Table 2). Clinical animals shed virus at a low frequency in nasal swabs (6 of 25) and urine (2 of 15), and no viral RNA was detected in faeces (Fig. 2a and Extended Data Table 2). In non-clinical animals, viral RNA was detected in 6 of 19 nasal swabs and 4 of 8 urine samples (Fig. 2a and Extended Data Table 2), indicating subclinical infection.

a, Viral shedding and RNA load in milk (n = 41), nasal secretions (nasal swabs, n = 45), urine (n = 23) and faeces (n = 25) collected from clinical and non-clinical animals from farm 3. b, Viral RNA loads in milk samples collected from cattle from farm 3 on day 3 (n = 15), day 16 (n = 12) and day 31 (n = 9) post-clinical diagnosis quantified by rRT–PCR targeting the IAV matrix gene. c, Infectious HPAI virus in milk (n = 13 on day 3, n = 12 on day 16 and n = 9 on day 31) detected by virus titration. Virus titres were determined using end point dilutions and expressed as TCID50 ml−1. The limit of detection for infectious virus titration was 101.05 TCID50 ml−1. Data are presented as mean ± s.e.m. All graphs and statistical analysis were generated using GraphPad Prism (v10).
Duration of H5N1 virus shedding
Nasal swabs, whole blood, serum and milk samples were collected at approximately 3 (n = 15), 16 (n = 12) and 31 (n = 9) days post-clinical diagnosis of HPAI to assess the duration of virus shedding. On day 3, viral RNA was detected in nasal swabs from 2 of 15 animals, in whole blood of 1 of 15 animals, in serum of 1 of 15 animals, and in milk of 14 of 15 animals (Fig. 2b and Supplementary Table 2). Although no virus RNA was detected in nasal swabs, whole-blood or serum samples collected on days 16 and 31 post-clinical diagnosis, milk from 10 of 12 and 4 of 9 animals tested on days 16 and 31 post-clinical diagnosis, respectively, remained positive (Fig. 2b and Supplementary Table 2). Although high infectious viral loads were detected in milk on day 3 post-clinical diagnosis (104.05–108.80 TCID50 ml−1), no infectious virus was recovered from milk from days 16 and 31 post-clinical diagnosis (Fig. 2c).
Mammary gland tropism of the H5N1 virus
Histological examination of tissues from affected dairy cows revealed marked changes consisting of neutrophilic and lymphoplasmacytic mastitis with prominent effacement of tubuloacinar gland architecture, which were filled with neutrophils admixed with cellular debris in multiple lobules in the mammary gland (Fig. 3). The most pronounced histological changes in the cat tissues consisted of mild-to-moderate multifocal lymphohistiocytic meningoencephalitis with multifocal areas of parenchymal and neuronal necrosis (Extended Data Table 6 and Extended Data Fig. 2).

Haematoxylin and eosin (H&E) staining (left panels) showing intraluminal epithelial sloughing and cellular debris in mammary alveoli (Z1 and Z2), and normal mammary alveoli filled with milk and fat globules (Z3). In situ hybridization (ISH; middle panels) targeting the IAV (matrix gene) showing extensive viral RNA in milk-secreting epithelial cells in the alveoli and in intraluminal cellular debris (Z1 and Z2), and normal mammary alveoli showing no viral staining (Z3). Immunohistochemistry (IHC; right panels) targeting the IAV matrix gene showing intracytoplasmic immunolabelling of viral antigen in milk-secreting alveolar epithelial cells (Z1 and Z2), and normal mammary alveoli showing no viral staining (Z3). Scale bars, 500 µm (Z0) and 50 µm (Z1–Z3).
Pronounced viral RNA and antigen were detected via in situ hybridization (ISH) and immunohistochemistry (IHC) in the mammary gland of affected cows and in the brain (cerebrum, cerebellum and brain stem) of affected cats. In mammary glands, viral RNA and antigen were present in the alveolar milk-secreting epithelial cells and interacinar spaces. In the brain of an affected cat, viral RNA and antigen were detected in neuronal soma glial cells, endothelial cells lining the capillaries within the choroid plexus, and the Purkinje cells in the molecular layer of cerebellum (Fig. 3 and Extended Data Fig. 2). In addition, sparce viral RNA and antigen were detected in the lung, supramammary lymph nodes, spleen, heart, colon and liver from affected cows (Extended Data Table 3 and Extended Data Figs. 3 and 4). Virus-infected cells were detected in peripheral areas of germinal centres of lymph nodes and in cells surrounding blood vessels in the remaining tissues (Extended Data Table 3 and Extended Data Figs. 3 and 4). These results demonstrate a distinct tropism of HPAI H5N1 virus for the mammary tissue of cattle and the central nervous system tissue of cats with sporadic detection of virus-infected cells in other tissues.
Spillover of reassortant H5N1 virus
All HPAI H5N1 virus sequences obtained from the farms in our study (n = 91) were classified within a new reassortant B3.13 genotype (Extended Data Fig. 5), which comprises PA, HA, NA and matrix gene segments of an Eurasian wild bird ancestry (ea1), and NS, PB2, PB1, and NP gene segments from American bird lineages (am1.1, am2.2, am4 and am8, respectively; Extended Data Table 4). To identify potential parental genotypes and to define the most recent common ancestors leading to the emergence of genotype B3.13, we performed Bayesian evolutionary analysis sampling trees (BEAST) and TreeSort sorting using influenza A sequences obtained between 2020 and 2024. This analysis suggests that B3.13 genotype viruses acquired the PB2 and NP gene fragments before its initial detections in avian and mammalian species in January 2024 (Extended Data Table 4 and Extended Data Fig. 5). The first genome segment derived from the LPAI American bird lineage to be incorporated in HPAI H5N1 clade 2.3.4.4b was the NS gene (am1.1); the earliest evidence of its emergence derived from a reassortant genotype B3.2 virus obtained from chicken in British Columbia, Canada in June 2022 (A/chicken/BC/22-023547-001-original/2022(H5N1)); Extended Data Table 4). Incorporation of the PB1 (am4) and PB2 (am2.2) gene segments into HPAI H5N1 clade 2.3.4.4b was first detected in November 2023, in a genotype B3.9 virus sequence recovered from a tundra swan (Cygnus columbianus) from Minnesota (A/tundra_swan/Minnesota/23-037501-001-original/2023(H5N1)). The reassortant genotype B3.13 virus, which incorporated the am2.2 PB2 and am8 NP gene segments, was first detected on 26 November 2023 in Canada goose (Branta canadensis) in Colorado (A/goose/Colorado/23-038138-001/2023), then on 25 January 2024 in a Canada goose (Branta canadensis) in Wyoming (A/Canada_goose/Wyoming/24-003692-001-original/2024(H5N1)), followed by a detection in a peregrine falcon (Falco peregrinus) in California (A/peregrine_falcon/California/24-005915-001-original/2024(H5N1)) on 14 February 2024, and soon after in a skunk in New Mexico on 23 February 2024 (A/skunk/New_Mexico/24-006483-001-original/2024(H5N1); Extended Data Table 4 and Extended Data Fig. 5). The host species, in which the reassortment event culminating in the incorporation of the am8 NP segment and emergence of the HPAI H5N1 genotype B3.13 virus, remains unknown.
Phylogenomics of the H5N1 B3.13 genotype
Phylogenetic analysis based on concatenated whole genomes revealed that all sequences from farms 1 to 9, including sequences obtained from wild birds, cows and other mammals, formed a large monophyletic lineage (Fig. 4a and Extended Data Fig. 6). They were most closely related to a sequence obtained from the Canada Goose in November 2023 in Colorado (A/goose/Colorado/23-038138-001/2023) and a sequence from a skunk in New Mexico on 23 February 2024 (A/skunk/New_Mexico/24-006483-001-original/2024). The sequences obtained from the affected dairy farms characterized in the present study formed two large phylogenetic branches, with the largest one including three subclusters. Of note, these phylogenetic groups of closely related sequences were not always formed by sequences derived from the same farm (Fig. 4b). Phylogenetic branches formed by sequences obtained from cattle from farms 1 and 3, farms 1 and 8, and farms 7 and 9 suggested a close genetic relationship between the viruses in these farms (Fig. 4b), and potential transfer of the virus between farms. Similarly, sequences obtained from cattle from sites 1 and 2 of farm 2 (a multisite dairy operation), formed a monophyletic cluster, indicating co-circulation of the virus in these two sites (Fig. 4b).

a, Phylogeny of sequences derived from cattle, cats, raccoon and grackle sampled in the farms described in this study, and other sequences in closer ancestral branches, obtained from the GISAID database. Nodes are coloured by host species. b, Detailed view of the clade containing 91 sequences derived from animals sampled in the farms described in this study. Nodes are coloured by farm. All phylogenomic analyses were conducted with concatenated whole-genome sequences.
Next, the mutation profile of HPAI H5N1 clade 2.3.4.4b was investigated. Initially, we evaluated the occurrence of mutations with known functional relevance to IAV (for example, host adaptation, virulence and host specificity shift, among others) compared with the original H5N1 A/GsGd/1/1996 virus (Supplementary Table 3). Furthermore, we performed a detailed comparative genome analysis and mutational profiling using sequences obtained in the USA throughout the 2021–2024 HPAI outbreak (Extended Data Table 5). The first HPAI H5N1 clade 2.3.4.4b sequence A/chicken/NL/FAV-0033/2021 detected in Canada was used as a reference to identify mutations in different genome segments across affected species. Representative sequences from multiple genotypes (A1, A2, B1.3, B3.2, Minor01, B3.6, B3.9 and B3.13) were selected, including sequences from avian (chicken and great tailed grackle) and mammalian (skunk, red fox, harbour seals, human, goat, cat and cattle) hosts. A total of 132 amino acid substitutions were observed across the 8 genome segments, most of which are low-frequency mutations observed in a small proportion of cattle-derived viral sequences (Extended Data Table 5 and Supplementary Table 6). Fifteen mutations emerged in viruses of genotypes circulating in late 2023 (for example, A2 and B3.6) and were maintained in genotype B3.13 viruses in 2024, including mutations in PB2 (V109I, V139I, V495I and V649I), PB1 (E75D, M171V, R430K and A587P), PA (K113R), HA (T211I), NA (V67I, L269M, V321I and S339P), NP (S482N) and NS1 (C116S) genes. Seven additional mutations were detected exclusively in genotype B3.13 viruses, including five substitutions in PB2 (T58A, E362G, D441N, M631L and T676A), one in PA (L219I) and one in NS1 (S7L). When compared with the first reported B3.13 sequences (A/goose/Colorado/23-038138-001/2023[H5N1]), (A/Canada_goose/Wyoming/24-003692-001-original/2024(H5N1)), A/peregrine_falcon/California/24-005915-001-original/2024(H5N1) and A/skunk/New_Mexico/24-006483-001-original/2024(H5N1)), the cow HPAI H5N1 viral sequences presented five amino acid substitutions, including three in PB2 (E362G, D441N and M631L), one in PA (L219I) and one in NS (S7L; Extended Data Table 5), suggesting that these could have emerged following spillover in cattle.
H5N1 virus dispersal between farms
The HPAI H5N1 genotype B3.13 sequences obtained from farms presenting an epidemiological link (farm 2: separate production sites [site 1 and 2]; and farms 1 and 3: animals were transported from farm 1 to 3; Extended Data Table 1) or presenting closely related viral sequences (farms 7 and 9; Fig. 4b) were subjected to phylogeographical dispersal reconstructions (Fig. 5a). Haplotype network analysis of concatenated whole-genome sequences provided support for focusing the dispersal and phylogeographical inferences on farms 1 and 3, farm 2, and farms 7 and 9 (Fig. 5b). The phylogenetic relationship and dispersal pathways were inferred based on concatenated whole-genome sequences, the farm location and date of sample collection to reconstruct hypothetical dispersal trajectories of the HPAI virus between the farms. The viral sequences recovered from farm 2, which were collected from two separated production sites (1 and 2, approximately 50 km apart), formed two phylogenetic clusters, each comprising sequences from both sites, confirming the spread of the virus between these premises. Phylogeographical dispersal analysis of the HPAI H5N1 sequences recovered from farm 2 suggests site 1 as the likely source of the virus (Fig. 5c).

a, HPAI H5N1 virus dispersal in North America. Samples described in this study are coloured by farm, whereas locations in grey represent samples from closer ancestral branches obtained from the GISAID database. b, Haplotype network analysis of HPAI H5N1 viral sequences obtained from the farms described in this study. The different colours indicate different farms. The size of each vertex is relative to the number of samples, and the dashes on branches denote the number of mutations between nodes. c–e, Phylogenetic reconstruction and analysis of dispersal between sites 1 and 2 of farm 2 (c), farms 7 and 9 (d), and farms 1 and 3 (e). The directions of dispersal lines are counterclockwise. All phylogenomic and dispersal analyses were conducted with concatenated whole-genome sequences.
Viral sequences obtained from farms 7 and 9 (six sequences from farm 7 and 11 sequences from farm 9), which are approximately 280 km apart from each other, formed a monophyletic cluster, suggesting a link and potential bidirectional virus dispersal between these two farms (Fig. 5d). However, another hypothesis that cannot be formally excluded as it could not be resolved by our analysis is unidirectional dispersal of multiple viral lineages from farms 7 to 9 or vice versa. Given the close genetic relationship between the viruses in these farms, we conducted a broader phylogenetic analysis including other HPAI H5N1 B3.13 sequences available in GISIAD. This analysis revealed two additional H5N1 sequences recovered from blackbirds (unknown species) clustering with viral sequences from farms 7 and 9 (Extended Data Fig. 7). The blackbirds were collected at 8–12 km away from farm 7. Together, these results suggest both long-range and close-range lateral spread and transmission of the HPAI virus between farms.
Sequences obtained from farm 1 (Texas) and farm 3 (Ohio) branched interspersedly in two subclusters. Viral sequence recovered from animals from farm 1 were basal to all sequences from farm 3. Phylogeographical dispersal analysis revealed that the HPAI virus most likely spread from farm 1 (Texas) to farm 3 (Ohio; Fig. 5e). This is consistent with the epidemiological information revealing the transportation of 42 apparently healthy dairy cattle from farm 1 to farm 3 on 8 March 2024, 5 days before the first clinical signs were observed in animals in farm 1 and 12 days before the first clinical animal was identified in farm 3 (Extended Data Table 1). These results indicate transmission of HPAI H5N1 virus between subclinically infected cows.
Interspecies transmission of the H5N1 virus
Given that five of the nine farms included in our study (farms 1, 3, 4, 5 and 8) reported mortality events in wild (great-tailed grackles) and peri-domestic (pigeons) birds, and in wild (raccoon) and domestic (cats) mammals, we investigated potential HPAI infection in these species. Whole-genome sequencing of the samples from the grackles and a cat from farm 1 and a raccoon from farm 8 confirmed infection of these species with a HPAI H5N1 genotype B3.13 virus closely related to the viruses found in dairy cattle in these farms. The basal sequences for the viruses obtained from a cat in farm 1 and the raccoon in farm 8 were derived from dairy cattle, indicating cattle-to-cat and cattle-to-raccoon transmission (Extended Data Fig. 8). This is supported by epidemiological information revealing that feeding raw milk to farm cats was a common practice in these farms.
Discussion
Here we described the spillover of a new reassortant HPAI H5N1 virus clade 2.3.4.4b genotype B3.13 into dairy cattle and provide evidence of efficient transmission among cattle and between cattle and other species, highlighting the ability of the virus to cross species barriers. The farms that first reported and confirmed the HPAI H5N1 virus genotype B3.13 infection in cattle in Texas, New Mexico and Kansas are on the Central North American migratory bird flyway. The first reported genome sequence of the genotype B3.13 virus was obtained from a sample collected from a Canada goose in Colorado (26 November 2023) and then in a Canada goose in Wyoming (25 January 2024), within the same flyway. This was followed by detection in a peregrine falcon in California (14 February 2024) on the Pacific flyway, and then in a skunk in New Mexico (23 February 2024), again on the Central flyway. The lack of complete epidemiological information regarding the H5N1 genotype B3.13 sequence collected from the skunk in New Mexico precludes definitive conclusions on the link of this animal with affected dairy cattle farms in the region. However, these findings demonstrate the presence of the virus in wildlife in New Mexico around the same time (January–February 2024) that the first cases of sick cows presenting mild respiratory signs, drop in feed intake and milk production (which were later confirmed to be caused by HPAI H5N1 genotype B3.13) were reported35. Additional historical and prospective sequence data are needed for more detailed molecular epidemiological inferences.
Our results demonstrate a high tropism of HPAI H5N1 for the mammary gland tissue resulting in a viral-induced mastitis, which was confirmed by histological changes and direct viral detection in situ, demonstrating viral replication and defining the virus tropism for milk-secreting mammary epithelial cells lining the alveoli in the mammary gland. The tropism of HPAI H5N1 for milk-secreting epithelial cells is consistent with high expression of sialic acid receptors with an α2,3 (avian-like receptor) and α2,6 (human-like receptor) galactose linkage in these cells36. Although the tissue sample size included in our study was small, isolation of the virus in lung and suprammamary lymph nodes (which were also positive for viral RNA and antigen) suggests that other organs may also have a role in the virus infection dynamics and pathogenesis in dairy cattle. The initial site of virus replication remains unknown; however, it is possible that the virus infects through respiratory and/or oral routes replicating at low levels in the upper respiratory tract (for example, nasal turbinate, trachea and/or pharynx), from where it could disseminate to other organs via a short and low-level viraemia. The collected evidence suggests that the mammary gland is the main site of virus replication, resulting in substantial virus shedding in milk. Another possible transmission route includes direct infection of the mammary gland through the teat orifice and cisternae, which could occur through contaminated floors and bedding where animals lay in the farm or mechanically via the milking equipment during milking. This entry route could also lead to viraemia and subsequent virus dissemination/replication in other distant tissue sites. In the 1950s, several studies have shown that direct inoculation of virus into the udder of dairy cows and goats with the human PR8 strain of IAV led to infection and viral shedding37,38,39,40,41. These results suggest, considering the current outbreak, that mammary epithelial cells, which express α2,3 and α2,6 sialic acid36, may be generally susceptible to IAVs. There are a few studies that have suggested an association between IAV and clinical disease42,43,44,45,46,47, but, to our knowledge, there is no evidence yet of sustained transmission. The only published study of a goose/Guangdong lineage virus being inoculated intranasally into calves has shown limited viral replication with no clinical disease and no evidence of transmission48. Experimental infection studies using different inoculation routes (that is, intranasal versus intramammary) with the HPAI H5N1 genotype B3.13 virus and, perhaps, other contemporary viruses of the 2.3.4.4b lineage in dairy cattle with sequential and comprehensive sampling are critical to answer these important questions about the port of entry, infection dynamics and pathogenesis in this new host species.
Spillover of HPAI H5N1 clade 2.3.4.4b into mammalian species has been reported throughout the current global outbreak20,49; however, there was no evidence of sustained virus transmission in mammals. Our epidemiological investigation combined with genome sequence and geographical dispersal analysis provides evidence of efficient intraspecies and interspecies transmission of the HPAI H5N1 genotype B3.13. Soon after apparently healthy lactating cattle were moved from farm 1 to farm 3, resident animals in farm 3 developed clinical signs compatible with HPAI H5N1, providing evidence suggesting that non-clinical animals can spread the virus. Analysis of the genetic relationship between the viruses detected in farms 1 and 3 combined with phylogeographical modelling indicate that the viruses infecting cattle in these farms are closely related, supporting the direct epidemiological link and indicating long-range viral dispersal and efficient cattle-to-cattle transmission. The results from the phylogenomic and phylogeographical analyses in both sites of farm 2 and on farms 7 and 9 also indicate regional long-range farm-to-farm spread of the virus. In these cases, fomites such as shared farm equipment, vehicles or personnel may have a role in virus spread. The dispersal of virus between farms 7 and 9 could have been vectored by wild birds, as suggested by the fact that blackbirds found dead near farm 7 were infected with a virus closely related to the virus circulating in cattle in these farms. Alternatively, the birds at this premise could have been infected with virus shed by cattle. All affected farms from this study are large farms with cattle maintained in open-air pens, which facilitates access of wild birds or mammals to feed and water that could mediate indirect contact between cattle and potentially infected wild birds or mammals. Our phylogenomic analysis in sequences recovered from affected cats (farms 1, 2, 4 and 5) and a raccoon (farm 8) combined with epidemiological information revealing the practice of feeding raw milk to cats in these farms support cattle-to-cat and cattle-to-raccoon transmission. These observations highlight complex pathways underlying the introduction and spread of HPAI H5N1 in dairy farms (Fig. 6), underscoring the need for efficient biosecurity practices and enhanced surveillance efforts in affected and non-affected farms.

A reassortment event in an unknown host species led to the emergence of the H5N1 genotype B3.13, which circulated in wild birds and mammals before infecting dairy cattle. Following spillover of H5N1 into dairy cattle, the virus was able to establish infection and efficiently transmit from cow to cow (intraspecies transmission) and from cow to other species, including wild (great tailed grackles) and peridomestic (pigeons) birds and mammals (cats and raccoons; interspecies transmission). The spread of the virus between farms occurred by the movement of cattle between farms, and probably by movement of wild birds and fomites including personnel, shared farm equipment and trucks (feed, milk and/or animal trucks). The model was created using BioRender (https://biorender.com).
The spillover of HPAI H5N1 into dairy cattle and evidence for efficient and sustained mammal-to-mammal transmission are unprecedented. This efficient transmission is concerning as it can lead to the adaptation of the virus, potentially enhancing its infectivity and transmissibility in other species, including humans. Although none of the nine affected farms included in the present study reported cases of human HPAI H5N1 infection, there have been three confirmed human cases resulting in mild conjunctivitis and respiratory infection in other farms in Texas and Michigan50,51,52. These cases highlight the zoonotic potential of the virus, underscoring the need for robust measures to prevent and control the infection and further spread of HPAI H5N1 in dairy cattle. This would reduce the risk of the virus adapting in this new mammalian host species, thereby decreasing the pandemic risk to humans.
Methods
Inclusion and ethics statement
All authors of this study were committed to high standards of inclusion and ethics in research. Clinical samples used in the present study were collected as part of routine diagnostic procedures and the data used for research. The findings of this study are reported transparently, with a commitment to accuracy and integrity. All data and results are presented without manipulation, and any limitations of the study are clearly acknowledged.
Sample collection
Clinical samples used in the present study were collected by field veterinarians from nine clinically affected farms in Texas (farms 1, 2, 4, 5, 6 and 7), New Mexico (farm 8), Kansas (farm 9) or Ohio (farm 3). A total of 332 samples were collected from dairy cattle (n = 323), domestic cats (n = 4), great-tailed grackles (n = 3), a pigeon (n = 1) and a racoon (n = 1) in the affected farms. All samples including milk (n = 211), nasal swabs (n = 46), whole blood (n = 25), serum (n = 15), faeces (n = 10), urine (n = 4) and tissues (mammary gland (n = 4), lung (n = 1), lymph nodes (n = 3), small intestine (n = 3) and large intestine (n = 1)) from dairy cattle were submitted to the Cornell Animal Health Diagnostic Center (AHDC), Texas A&M Veterinary Medical Diagnostic Laboratory (TVMDL) or the Ohio Animal Disease Diagnostic Laboratory (OADDL) for diagnostic investigations. One domestic cat, two grackles and one pigeon (farm 1) were submitted to the AHDC, whereas three cats (farms 4 and 8), a racoon (farm 8) and four cows were submitted to the TVMDL for necropsy and testing (Supplementary Table 1).
Sequential samples (milk, nasal swabs and blood) collected from animals (n = 15) from farm 3 were used to investigate the duration of virus shedding (Supplementary Table 2). In addition, paired samples (milk, nasal sabs, urine and faeces) collected from animals presenting respiratory distress, drop in milk production and altered milk characteristics (clinical; n = 25) and from apparently healthy animals (non-clinical; n = 20) from farm 3 were used to compare virus shedding by clinical and non-clinical animals (Extended Data Table 2).
Clinical history and epidemiological information
Clinical history from all nine farms were obtained from the sample submission forms sent with the samples to the AHDC, TVMDL and OADDL. Additional relevant information from each farm were obtained from attending veterinarians through investigations conducted by laboratory diagnosticians.
rRT–PCR
Viral nucleic acid was extracted from milk, nasal swabs, whole blood, serum, faeces, urine and tissue homogenates. Of milk, nasal swabs, whole blood, serum and urine, 200 µl was used for extraction. Of raw milk samples, 200 µl was used directly or diluted at the ratio of 1:3 milk:PBS with 200 µl of the dilution used for nucleic acid extraction. Tissues and faeces were homogenized in PBS–BSA (1%; 10% w/v), cleared by centrifugation, and 200 µl of the supernatant was used for extraction. All RNA extractions were performed using the MagMAX Pathogen RNA/DNA Kit (Thermo Fisher) and the automated King Fisher Flex nucleic acid extractor (Thermo Fisher) following the manufacturer’s recommendations. The presence of IAV RNA was assessed using the VetMax-Gold AIV Detection Kit (Thermo Fisher) and the National Animal Laboratory Network (NAHLN) primers and probe targeting the conserved matrix gene or the H5 haemagglutinin gene53. Amplification and detection were performed using the Applied Biosystems 7500 Fast PCR Detection System (Thermo Fisher), under the following conditions: 10 min at 45 °C for reverse transcription, 10 min at 95 °C for polymerase activation, and 45 cycles of 15 s at 94 °C for denaturation and 30 s at 60 °C for annealing and extension. Relative viral loads were calculated and are expressed as 45 rRT–PCR cycles minus the actual CT value (45-Ct). Positive and negative amplification controls as well as internal inhibition controls were run side by side with test samples. Part of samples was also tested using 200 µl of undiluted milk and serum, and 100 µl of whole blood, targeting the matrix gene. These samples were extracted using the IndiMag Pathogen kit (INDICAL Bioscience) on the KingFisher Flex (Thermo Fisher), and rRT–PCR was performed using the Path-ID Multiplex One-Step RT-PCR Kit (Thermo Fisher) under the following conditions: 10 min at 48 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C for denaturation and 60 s at 60 °C.
Haemagglutination inhibition
Paired serum samples collected during the acute and convalescent phase of infection from animals (n = 20) from farm 2 were used to determine seroconversion to HPAI H5N1 virus using the haemagglutination inhibition test. Serum haemagglutination inhibition activity was determined using BPL-inactivated A/Tk/IN/3707/22 antigen (clade 2.3.4.4b), as previously described. Haemagglutination inhibition titres are expressed as log2 values, with 1 log2 being the minimum titre considered positive.
Virus isolation
Virus isolation was performed in pooled milk samples from farms 1 and 2. Approximately 5 ml of milk from individual animals was pooled, and a total of 50 ml of pooled milk was centrifuged at 1,700g for 10 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in 5 ml of sterile PBS–BSA (1%) followed by centrifugation at 1,700g for 10 min at 4 °C. The wash step was repeated one more time and the final pellet was resuspended in 1 ml PBS–BSA (1%). Virus isolation was conducted in bovine uterine epithelial cells (Cal-1; developed in-house at the Virology Laboratory at AHDC) cultured in minimal essential medium (MEM; Corning) supplemented with 10% FBS and penicillin–streptomycin (Thermo Fisher Scientific; 10 U ml–1 and 100 µg ml–1, respectively). Cells were cultured in T25 flasks and inoculated with 1 ml of the milk pellet resuspension from infected cows and incubated at 37 °C for 1 h (adsorption). The inoculum was then removed, and cells were washed once with PBS and replenished with 1 ml complete growth media (MEM 10% FBS). Cells were monitored daily for the development of cytopathic effects (CPEs), including cell swelling, rounding and detachment. When the CPE reached 70–80%, infected cells were harvested and cell suspensions were collected after three freeze–thaw cycles. The identity of the isolated virus was confirmed by rRT–PCR, an immunofluorescence assay using anti-nucleoprotein mouse monoclonal antibody (HB65, H16-L10-4R5, ATCC) and whole-genome sequencing. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 62248, Thermo Fisher Scientific).
Virus titrations
The infectious viral loads in milk and tissues of infected animals were quantified by viral titrations. For this, serial tenfold dilutions of rRT–PCR-positive milk samples and tissue homogenates were prepared in MEM and inoculated into Cal-1 cells in 96-well plates. Each dilution was inoculated in quadruplicate wells. At 48 h post-inoculation, culture supernatant was aspirated, and cells were fixed with 3.7% formaldehyde solution for 30 min at room temperature and subjected to an immunofluorescence assay using the anti-NP (HB65) mouse monoclonal antibody. Virus titres were determined using end-point dilutions and the Spearman and Karber’s method and expressed as TCID50 ml–1.
Microscopic changes, in situ hybridization and immunohistochemistry
A total of 25 tissue samples from 4 dairy cattle and 12 tissues from 1 domestic cat were collected and fixed in formalin. These formalin-fixed paraffin-embedded (FFPE) tissues were sectioned at 3 µm thickness, stained with haematoxylin and eosin, and examined for histological changes. To determine the virus tropism and tissue distribution in dairy cattle and cat affected with HPAI H5N1, we performed in situ hybridization (ISH) and immunohistochemistry (IHC) on FFPE tissues as previously described6. In brief, tissue sections were deparaffinized with xylene, washed with absolute ethanol, and blocked with peroxidase followed by antigen retrieval for 1 h. For the ISH, the V-InfluenzaA-H5N8-M2M1 probe (Advanced Cell Diagnostics), which targets H5Nx clade 2.3.4.4b viruses, and the RNAScope HD 2.5 assay were used as per the manufacturer’s instructions. ISH signals were amplified with multiple amplifiers conjugated with alkaline phosphatase enzymes, incubated with red substrate at room temperature for 10 min and counterstained with haematoxylin. IHC was performed at the University of Georgia Veterinary Diagnostic laboratory and the USDA-ARS Southeast Poultry Research Laboratory following standard diagnostic IHC procedure. Specifically, tissue sections were treated with proteinase K for 5 min for antigen retrieval and incubated with monoclonal antibody to IAV M-protein (C65331M, Meridian Bioscience) at 1:100 dilution (UGA and VDL) or monoclonal antibody to influenza A NP (clone 4F1; 10780-01, Southern Biotech) at 1:2,000 dilution (SEPRL) for 1 h. After washing the slides, the secondary antibody anti-mouse IgG (1070-04, Southern Biotech) at a 1:5,000 dilution was added and incubated with the slides for 1 h. All the slides were counterstained with haematoxylin, scanned at ×40 resolution and the digital slides were examined for virus tropism and tissue distribution.
Viral metagenomic sequencing
Sample collection and processing
Whole-blood nasal swab samples were obtained from ten cows from farm 1 in Texas. Samples were submitted to the AHDC at Cornell University on 16 March 2024. Upon receipt, metagenomic sequencing using the sequence-independent, single-primer amplification (SISPA) procedure, the Oxford Nanopore sequencing chemistry and GridION sequencing platform were performed as described below.
Nucleic acid extraction, library preparation and sequencing
Nucleic acid extraction was performed in 190 µl from each sample using the QIAamp MinElute Virus Spin Kit (Qiagen). Before nucleic acid extraction, samples were subjected to an enzymatic cocktail treatment composed of 10X DNase 1 buffer, DNAse 1, Turbo DNAse, RNase Cocktail (Thermo Fisher Scientific), Baseline ZERO DNAse (Lucigen), Benzonase (Sigma-Aldrich) and RNase ONE Ribonuclease (Promega) to deplete host and bacterial nucleic acid. Purified nucleic acid was subjected to SISPA, modified from a previously reported protocol54. In brief, 11 μl of nucleic acid was used in a reverse transcription reaction with 100 pmol of primer FR20RV-12N (5′-GCCGGAGCTCTGCAGATATCNNNNNNNNNNNN-3′) using SuperScript IV reverse transcriptase (Thermo Fisher Scientific), followed by second-strand synthesis using the Klenow Fragment of DNA polymerase (NEB) with primer FR20RV-12N at 10 pmol. After purification using Agencourt AMPure XP beads (Beckman Coulter), SISPA PCR amplification was conducted with TaKaRa Taq DNA Polymerase (Takara) using the primer FR20RV (5′-GCCGGAGCTCTGCAGATATC-3′) at 10 pmol. SISPA products were converted into sequencing libraries using the ligation sequencing kit (SQK-LSK109) and Native Barcoding Kit 96 V1 for multiplex sequencing. Sequencing was performed on the FLO-MIN106 MinION flow cell r9.4.1 using the GridION Sequencer (Oxford Nanopore Technologies). A 24-h sequencing run was conducted, with fastq generation performed by the GridION using high-accuracy base calling. Settings were adjusted to accommodate barcodes at both ends and filter mid-strand barcodes. Fastq reads were then filtered by size and quality using Nanofilt55 and classified using Kraken (v2.1.0)56 followed by Bracken57.
Targeted influenza A sequencing
Samples that tested positive for HPAI H5N1 and had Ct values of less than 30 were subjected to targeted influenza A sequencing at the Animal Health Diagnostic Center at Cornell University (Cornell AHDC) and the OADDL. The set of 107 samples included samples from farm 1 (n = 19), farm 2 (n = 33), farm 3 (n = 54) and farm 7 (n = 1). A complete metadata table with details on this set of samples is provided in Supplementary Table 1. Initial targeted sequencing attempts on milk samples at Cornell AHDC utilizing high-throughput diagnostic extraction methods6 were unsuccessful in obtaining whole influenza A genome sequences despite the utilization of samples with low Ct values. To overcome this limitation, up to 50 ml of each milk sample was pelleted at 1,770g for 15 min at 4 °C. The pellets were washed two times in PBS as described above and resuspended in 1 ml of PBS–BSA. The resuspended pellet was then diluted 1:5 or 1:10 in PBS, and 200 µl of this dilution was used for extraction with the Indical IndiMag Pathogen kit (INDICAL Bioscience) on the KingFisher Flex extractor (Thermo Fisher). Whole IAV genome sequences were generated using the MBTuni-12 and MBtuni-13 M-RT-PCR methods58. Sequencing libraries were generated using the Native Barcoding Kit EXP-NBD196 and the Ligation Sequencing Kit SQK-SQK109 (Oxford Nanopore Technologies), and sequenced on a FLO-MIN106 MinION flow cell r9.4.1 using the GriION platform.
In addition, 31 samples from farm 3 were subjected to target influenza A sequencing at the OADDL using the Illumina DNA Prep Kit and the Nextera DNA CD Indexes. Paired-end sequencing was performed on an Illumina MiSeq platform using the MiSeq Reagent Kit V3 (Illumina) with 2 × 250 base-pair chemistry.
Sequence analysis and mutational profiling
Sequencing data generated by the GridION platform underwent high-accuracy base calling and demultiplexing of barcodes. Settings were configured to require barcodes at both ends and to exclude reads with mid-read barcodes. The Nanofilt software (v2.8.0)55 was used to filter sequences based on quality thresholds. Reads with a quality score below 12 and those shorter than 600 base pairs were removed from further analysis. Filtered reads were aligned to a reference genome download from GenBank (A/Gallus/gallus_domesticus/Sonora/CPA-18486-23/2023/H5N1, NCBI accession numbers OR801090.1–OR801097.1) using Minialign software (v0.4.4; https://github.com/ocxtal/minialign). Consensus sequences were generated using Medaka software (v1.4.3) with medaka_haploid_variant and medaka_consensus programs for polishing (https://github.com/nanoporetech/medaka). Sequences with a read depth greater than 20 and a quality score exceeding 20 were retained. Analysis of Illumina MiSeq data was performed by trimming the reads with Trimmomatic (v0.39)59, and aligning, calling variants and generating consensus sequences with Snippy (v4.6.0; https://github.com/tseemann/snippy). Genome sequences were annotated using Prokka software (v1.14.5) to identify genetic features and functional elements60. The GenoFLU tool (v1.03) assessed potential reassortment events within the viral genome (https://github.com/USDA-VS/GenoFLU). Genome alignments, mutations, single-nucleotide polymorphisms and annotation data were visualized using Geneious Prime software (v2024.0). The FluSurver tool, available through GISAID EpiFlu, was utilized to interpret the effects of mutations identified in the sequences, leveraging previously published data (https://flusurver.bii.a-star.edu.sg/). Other mutation data were visualized using protein consensus alignments in Geneious Prime software.
Phylogenomic and phylogeographical analysis
The dataset consisted of HPAI H5N1 clade 2.3.4.4b genomes from samples collected between January 2023 and March 2024 in the American continent, downloaded from the GISAID Epiflu database33, and 91 complete genomes from the present study that include 50 genomes obtained from raw sequencing data, combined with another 41 complete genomes curated from the GISAID database that were obtained from the farms in our study (farm 1 (n = 11), farm 4 (n = 3), farm 5 (n = 1), farm 6 (n = 4), farm 7 (n = 5), farm 8 (n = 6) and farm 9 (n = 11)). The genomes generated in this study are deposited in the GISAID database (Supplementary Table 5), and raw reads are available in the Sequence Read Archive under BioProject accession number PRJNA1114404. Phylogenetic analyses were performed by using the Augur v21.0.1 tool kit61 procedures implemented in Nextstrain62. In brief, multiple sequence alignments were performed using MAFFT (v7.515)63; maximum likelihood trees were inferred using IQ-TREE (v1.6.12)64, and the initial tree was refined using sequence metadata through the augur refine subcommand. Discrete trait analysis was performed using TreeTime (v0.9.4)65. The resultant dataset was visualized through Auspice. Phylogenomic and phylogeographical analyses were also performed on complete genomes formed by concatenation of all gene segments. Analyses were performed using Nextstrain as described above, with the exception of the maximum-likelihood phylogenetic tree inferred using IQ-tree with an edge-linked partition model and 1,000 bootstrap replicates. The potential transmission networks between farms were inferred using the PB2 gene sequences in the PopART package (v1.7.2) using the median joining tree method with an epsilon of zero66.
Reassortment and MRCA identification
All the type A influenza sequences (n = 3,620, North America) from avian, dairy cattle and other mammals between January 2020 and May 2024 were downloaded from the Epiflu database in the GISAID33. Only complete gene fragments were used to infer maximum-likelihood phylogenetic trees for each fragment using IQ-TREE with generalized time-reversible nucleotide substitutions model64. The HA phylogeny was used to identify the reassortment events using TreeSort (v0.1.1; maximum molecular clock deviation parameter of 2.5; https://github.com/flu-crew/TreeSort). We implemented the Bayesian evolutionary analysis sampling tree (BEAST, v1.10) framework with BEAGLE library (v4.0.1) to estimate the most recent common ancestor (MRCA) for the individual gene fragments (generalized time-reversible gamma distributed site heterogeneity model, strict clock model, three independent Markov chain Monte Carlo sampling runs with 10 million iterations with sampling every 10,000 iterations)67. Tracer (v1.7.2)68 was used to analyse the results. The maximum clade credibility tree was generated using TreeAnnotator (v1.8.4) using median node heights and 10% burn-in67, and visualized with FigTree (v1.4.4; http://tree.bio.ed.ac.uk/software/figtree/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All HPAI H5N1 viral sequences generated in this study are deposited in the GISAID (https:www.gisaid.org/; accession numbers are available in Supplementary Table 5), and raw reads have been deposited in the NCBI’s Sequence Read Archive (BioProject number PRJNA1114404). All additional influenza sequences used in our analysis were obtained from the GISAID (accession numbers are available in Supplementary Table 4) or NCBI Nucleotide (https://www.ncbi.nlm.nih.gov/nucleotide/). Source data are provided with this paper.
References
Kandeil, A. et al. Rapid evolution of A(H5N1) influenza viruses after intercontinental spread to North America. Nat. Commun. 14, 3082 (2023).
Youk, S. et al. H5N1 highly pathogenic avian influenza clade 2.3.4.4b in wild and domestic birds: introductions into the United States and reassortments, December 2021–April 2022. Virology 587, 109860 (2023).
USDA–APHIS. Detections of highly pathogenic avian influenza in wild birds. USDA–APHIS https://www.aphis.usda.gov/livestock-poultry-disease/avian/avian-influenza/hpai-detections/wild-birds (2024).
USDA–APHIS. Confirmations of highly pathogenic avian influenza in commercial and backyard flocks. USDA–APHIS https://www.aphis.usda.gov/livestock-poultry-disease/avian/avian-influenza/hpai-detections/commercial-backyard-flocks (2024).
Puryear, W. et al. Highly pathogenic avian influenza A(H5N1) virus outbreak in New England seals, United States. Emerg. Infect. Dis. 29, 786–791 (2023).
Cronk, B. D. et al. Infection and tissue distribution of highly pathogenic avian influenza A type H5N1 (clade 2.3.4.4b) in red fox kits (Vulpes vulpes). Emerg. Microbes Infect. 12, 2249554 (2023).
Rijks, J. M. et al. Highly pathogenic avian influenza A(H5N1) virus in wild red foxes, the Netherlands, 2021. Emerg. Infect. Dis. 27, 2960–2962 (2021).
Bordes, L. et al. Highly pathogenic avian influenza H5N1 virus infections in wild red foxes (Vulpes vulpes) show neurotropism and adaptive virus mutations. Microbiol. Spectr. 11, e0286722 (2023).
Plaza, P. I., Gamarra-Toledo, V., Rodríguez Euguí, J., Rosciano, N. & Lambertucci, S. A. Pacific and Atlantic sea lion mortality caused by highly pathogenic avian influenza A(H5N1) in South America. Travel Med. Infect. Dis. 59, 102712 (2024).
Alkie, T. N. et al. Characterization of neurotropic HPAI H5N1 viruses with novel genome constellations and mammalian adaptive mutations in free-living mesocarnivores in Canada. Emerg. Microbes Infect. 12, 2186608 (2023).
Jakobek, B. T. et al. Influenza A(H5N1) virus infections in 2 free-ranging black bears (Ursus americanus), Quebec, Canada. Emerg. Infect. Dis. 29, 2145–2149 (2023).
Sillman, S. J., Drozd, M., Loy, D. & Harris, S. P. Naturally occurring highly pathogenic avian influenza virus H5N1 clade 2.3.4.4b infection in three domestic cats in North America during 2023. J. Comp. Pathol. 205, 17–23 (2023).
Ellis, T. M. et al. Investigation of outbreaks of highly pathogenic H5N1 avian influenza in waterfowl and wild birds in Hong Kong in late 2002. Avian Pathol. 33, 492–505 (2004).
Xu, X., Subbarao, K., Cox, N. J. & Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 261, 15–19 (1999).
Ali, M. et al. Genetic characterization of highly pathogenic avian influenza A(H5N8) virus in Pakistani live bird markets reveals rapid diversification of clade 2.3.4.4b viruses. Viruses 13, 1633 (2021).
de Vries, E. et al. Rapid emergence of highly pathogenic avian influenza subtypes from a subtype H5N1 hemagglutinin variant. Emerg. Infect. Dis. 21, 842–846 (2015).
Lee, K. et al. Characterization of highly pathogenic avian influenza A (H5N1) viruses isolated from cats in South Korea, 2023. Emerg. Microbes Infect. 13, 2290835 (2024).
Adlhoch, C. et al. Avian influenza overview March–April 2023. EFSA J. 21, e08039 (2023).
Harvey, J. A., Mullinax, J. M., Runge, M. C. & Prosser, D. J. The changing dynamics of highly pathogenic avian influenza H5N1: next steps for management & science in North America. Biol. Conserv. 282, 110041 (2023).
Elsmo, E. et al. Highly pathogenic avian influenza A(H5N1) virus clade 2.3.4.4b infections in wild terrestrial mammals, United States, 2022. Emerg. Infect. Dis. J. 29, 2451 (2023).
Tammiranta, N. et al. Highly pathogenic avian influenza A (H5N1) virus infections in wild carnivores connected to mass mortalities of pheasants in Finland. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 111, 105423 (2023).
Ulloa, M. et al. Mass mortality event in South American sea lions (Otaria flavescens) correlated to highly pathogenic avian influenza (HPAI) H5N1 outbreak in Chile. Vet. Q. 43, 1–10 (2023).
Agüero, M. et al. Highly pathogenic avian influenza A(H5N1) virus infection in farmed minks, Spain, October 2022. Eurosurveillance 28, 2300001 (2023).
WHO. Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO, 2003–2024, 28 March 2024. WHO https://www.who.int/publications/m/item/cumulative-number-of-confirmed-human-cases-for-avian-influenza-a(h5n1)-reported-to-who–2003-2024-28-march-2024 (2024).
de Jong, M. D. & Hien, T. T. Avian influenza A (H5N1). J. Clin. Virol. 35, 2–13 (2006).
Sutton, T. C. The pandemic threat of emerging H5 and H7 avian influenza viruses. Viruses 10, 461 (2018).
Caliendo, V. et al. Transatlantic spread of highly pathogenic avian influenza H5N1 by wild birds from Europe to North America in 2021. Sci. Rep. 12, 11729 (2022).
Bevins, S. et al. Intercontinental movement of highly pathogenic avian influenza A(H5N1) clade 2.3.4.4 virus to the United States, 2021. Emerg. Infect. Dis. J. 28, 1006–1011 (2022).
USDA–APHIS. Detections of highly pathogenic avian influenza. USDA–APHIS https://www.aphis.usda.gov/livestock-poultry-disease/avian/avian-influenza/hpai-detections (2024).
Plaza, P. I., Gamarra-Toledo, V., Euguí, J. R. & Lambertucci, S. A. Recent changes in patterns of mammal infection with highly pathogenic avian influenza A(H5N1) virus worldwide. Emerg. Infect. Dis. 30, 444–452 (2024).
Schnirring, L. Avian flu detected again in Antarctic penguins. CIDRAP https://www.cidrap.umn.edu/avian-influenza-bird-flu/avian-flu-detected-again-antarctic-penguins (2024).
U.S. case of human avian influenza A(H5) virus reported. CDC https://www.cdc.gov/media/releases/2022/s0428-avian-flu.html (2022).
Shu, Y. & McCauley, J. GISAID: global initiative on sharing all influenza data — from vision to reality. Euro Surveill. https://doi.org/10.2807/1560-7917.ES.2017.22.13.30494 (2017).
Schnirring, L. Avian flu detected for first time in US livestock. CIDRAP https://www.cidrap.umn.edu/avian-influenza-bird-flu/avian-flu-detected-first-time-us-livestock (2024).
Federal and state veterinary, public Health agencies share update on HPAI detection in Kansas, Texas dairy herds. APHIS https://www.aphis.usda.gov/news/agency-announcements/federal-state-veterinary-public-health-agencies-share-update-hpai (2024).
Kristensen, C., Larsen, L. E., Trebbien, R. & Jensen, H. E. The avian influenza A virus receptor SA-α2,3-gal is expressed in the porcine nasal mucosa sustaining the pig as a mixing vessel for new influenza viruses. Virus Res. 340, 199304 (2024).
Mitchell, C. A., Nordland, O. & Walker, R. V. L. Myxoviruses and their propagation in the mammary gland of ruminants. Can. J. Comp. Med. XXII, 154–156 (1958).
Mitchell, C. A., Walker, R. V. L. & Bannister, G. L. Further experiments relating to the propagation of virus in the bovine mammary gland. Can. J. Comp. Med. XVII, 218–222 (1953).
Mitchell, C. A., Walker, R. V. L. & Bannister, G. L. Persistence of neutralizing antibody in milk and blood of cows and goats following the instillation of virus into the mammary gland. Can. J. Comp. Med. XVIII, 426–430 (1954).
Mitchell, C. A., Walker, R. V. & Bannister, G. L. Preliminary experiments relating to the propagation of viruses in the bovine mammary gland. Can. J. Comp. Med. Vet. Sci. 17, 97–104 (1953).
Mitchell, C. A., Walker, R. V. & Bannister, G. L. Studies relating to the formation of neutralizing antibody following the propagation of influenza and Newcastle disease virus in the bovine mammary gland. Can. J. Microbiol. 2, 322–328 (1956).
Sreenivasan, C. C., Thomas, M., Kaushik, R. S., Wang, D. & Li, F. Influenza A in bovine species: a narrative literature review. Viruses 11, 561 (2019).
Campbell, C. H., Easterday, B. C. & Webster, R. G. Strains of Hong Kong influenza virus in calves. J. Infect. Dis. 135, 678–680 (1977).
Hu, X. et al. Highly pathogenic avian influenza A (H5N1) clade 2.3.4.4b virus detected in dairy cattle. Preprint at bioRxiv https://doi.org/10.1101/2024.04.16.588916 (2024).
Gunning, R. F., Brown, I. H. & Crawshaw, T. R. Evidence of influenza a virus infection in dairy cows with sporadic milk drop syndrome. Vet. Rec. 145, 556–557 (1999).
Brown, I. H., Crawshaw, T. R., Harris, P. A. & Alexander, D. J. Detection of antibodies to influenza A virus in cattle in association with respiratory disease and reduced milk yield. Vet. Rec. 143, 637–638 (1998).
Graham, D. A., Calvert, V. & McLaren, I. E. Retrospective analysis of serum and nasal mucus from cattle in Northern Ireland for evidence of infection with influenza A virus. Vet. Rec. 150, 201–204 (2002).
Kalthoff, D., Hoffmann, B., Harder, T., Durban, M. & Beer, M. Experimental infection of cattle with highly pathogenic avian influenza virus (H5N1). Emerg. Infect. Dis. 14, 1132–1134 (2008).
Arruda, B. et al. Divergent pathogenesis and transmission of highly pathogenic avian influenza A(H5N1) in swine. Emerg. Infect. Dis. 30, 738–751 (2024).
Uyeki, T. M. et al. Highly pathogenic avian influenza A(H5N1) virus infection in a dairy farm worker. N. Engl. J. Med. https://doi.org/10.1056/NEJMc2405371 (2024).
CDC. CDC confirms second human H5 bird flu case in Michigan, third case tied to dairy outbreak. CDC https://www.cdc.gov/media/releases/2024/p0530-h5-human-case-michigan.html (2024).
Garg, S. et al. Outbreak of highly pathogenic avian influenza A (H5N1) viruses in U. S. dairy cattle and detection of two human cases — United States, 2024. MMWR Morb. Mortal. Wkly Rep. 73, 501–505 (2024).
National Animal Health Laboratory Network. Standard operating procedure for real-time RT-PCR detection of influenza A and avian paramyxovirus type-1 (NVSL-SOP-0068). APHIS https://www.aphis.usda.gov/media/document/15399/file (2023).
Chrzastek, K. et al. Use of sequence-independent, single-primer-amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 509, 159–166 (2017).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Lu, J. et al. Metagenome analysis using the Kraken software suite. Nat. Protoc. 17, 2815–2839 (2022).
Mitchell, P. K. et al. Method comparison of targeted influenza A virus typing and whole-genome sequencing from respiratory specimens of companion animals. J. Vet. Diagnostic Investig. 33, 191–201 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Huddleston, J. et al. Augur: a bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 6, 2906 (2021).
Hadfield, J. et al. NextStrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Sagulenko, P., Puller, V. & Neher, R. A. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 4, vex042 (2018).
Leigh, J. W. & Bryant, D. POPART: full-feature software for haplotype network construction. Methods Ecol. Evol. https://doi.org/10.1111/2041-210X.12410 (2015).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Acknowledgements
We thank the producers and veterinarians who submitted samples and contributed to this investigation. The work was funded by the AHDC, OADDL and TVMDL. This work was supported in part by USDA-NIFA grant no. 2021-68014-33635 (to D.G.D. and K.M.D.) and APHIS NALHN Enhancement grant no. AP21VSD&B000C005 (to K.M.D. and D.G.D.). We acknowledge all data contributors including the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID initiative. The artwork in Fig. 6, Extended Data Fig. 5, the panels of Fig. 3 and Extended Data Figs. 2–4 were created with BioRender (https://biorender.com).
Author information
Authors and Affiliations
Contributions
D.G.D. conceptualized the study. E.A.F., S.L.B., M. Laverack, M.N., L.M.C., A.C.T., M.P.K., B.C., A.J., K.K., E.E.E., G.G., G.H., M.M., E.R.A.M. and T.H. were responsible for the methodology. L.C.C. and B.C. provided the software. S.L.B., M. Laverack, M.N., L.M.C., M.P.K., B.C. and A.J. validated the data. L.C.C., S.L.B., M. Laverack, B.C. and D.G.D. conducted the formal analysis. L.C.C., E.A.F., S.L.B., M. Laverack, L.M.C., A.C.T., M.P.K., B.C., A.J., D.R.K., M.M. and E.R.A.M. performed the investigation. E.A.F., A.C.T., D.L.S., M. Lejeune, J.S.B., A.K.S., F.E., K.M.D. and D.G.D. provided resources. L.C.C., E.A.F., S.L.B., M. Laverack, K.M.D. and D.G.D. curated the data. L.C.C., S.L.B., B.C. and D.G.D. wrote the original draft of the manuscript. L.C.C., E.A.F., S.L.B., M. Laverack, M.N., L.M.C., A.C.T., M.P.K., B.C., A.J., K.K., E.E.E., G.G., G.H., M.M., D.R.K., D.L.S., E.R.A.M., T.H., M. Lejeune, A.K.S., F.E., K.M.D. and D.G.D. reviewed and edited the manuscript. L.C.C., S.L.B., B.C. and D.G.D. performed the visualization. D.G.D. supervised the study. M.P.K., K.M.D. and D.G.D. provided project administration. A.K.S., F.E., K.M.D. and D.G.D. acquired funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Vijaykrishna Dhanasekaran, Philippe Lemey, Patricia Pesavento and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Clinical presentation of HPAI H5N1 infection in dairy cattle.
a, Clinically affected animals presenting clear nasal discharge and involution of the mammary gland/udder (gold arrowheads, top images) and depression (bottom images). b, Milk from HPAI H5N1 infected animals presenting yellowish colostrum-like color and appearance (top panels) or coloration varying from yellowish to pink/brown color. Curdling of milk visible in some samples.
Extended Data Fig. 2 Highly pathogenic avian influenza H5N1 virus detection in cat tissues.
Hematoxylin and eosin (H&E) staining (left panels) showing; a, multifocal area of perivascular cuffing, vascular congestion, and perivascular edema (Z0), neuronal swelling and neuronal necrosis and perivascular edema in brain (Z1, Z2 and Z3). b, pulmonary edema with strands of fibrin, thickened alveolar septa and intraepithelial lymphocytes, alveolar capillary congestion. c, single cell necrosis and hemorrhage in liver. In situ hybridization (ISH) (middle panels) targeting Influenza A virus (Matrix gene) showing (a) multifocal areas with extensive viral RNA (Z0), in neurons and glial cells within the granular layer and nuclear and intracytoplasmic viral RNA in neuronal soma, axon, and vascular endothelial cells in brain (Z1, Z2 and Z3), b, viral RNA in bronchiolar epithelial cells and type II pneumocytes, and c, viral RNA in resident sinusoidal Kupffer cells and vascular endothelial cells. Immunohistochemistry (IHC) (right panels) targeting Influenza A virus M gene showing immunolabeling of (a) multifocal areas of immunolabeling (Z0), intracytoplasmic immunolabeling of viral antigen in neuronal soma and axons within granular layer in brain (Z1, Z2 and Z3), b, bronchiolar epithelial cells and type II pneumocytes in lung, and c, vascular endothelial cells and resident sinusoidal Kupffer cells. Tissues from one cat were available and subjected to histological, ISH and IHC analysis.
Extended Data Fig. 3 Highly pathogenic avian influenza H5N1 virus RNA detection in cow tissues.
In situ hybridization of viral RNA in mononuclear cells of lymphoid follicles in lymph node, mononuclear cells of bronchial associated lymphoid tissue (BALT) in the lung, endothelial cells of blood vessels in the heart, endothelial cells of blood vessels in the colon, mononuclear cells in the spleen and endothelial cells and resident sinusoidal Kupffer cells in the liver. The zoom in (Z1) represents the demarcated area in the left panels (Z0). Tissues from three cows were available and subjected to histological, ISH and IHC analysis.
Extended Data Fig. 4 Highly pathogenic avian influenza H5N1 virus antigen detection in cow tissues.
Immunohistochemical staining of viral antigen in mononuclear cells of lymphoid follicles in lymph node, mononuclear cells of bronchial associated lymphoid tissue (BALT) in the lung, endothelial cells of blood vessels in the heart, endothelial cells of blood vessels in the colon, mononuclear cells in the spleen and endothelial cells and resident sinusoidal Kupffer cells in the liver. The zoom in (Z1) represents the demarcated area in the left panels (Z0). Tissues from three cows were available and subjected to histological, ISH and IHC analysis.
Extended Data Fig. 5 Bayesian analysis and estimation of reassortment events leading to emergence of HPAI H5N1 virus clade 2.3.4.4b genotype B3.13.
Estimation of tMRCA and immediate descendants of MRCA donors of PB2 (a), PB1 (b), NP (c) and NS (d) genes, respectively. e, Reassortment event of PB2 and NP which lead to emergence of genotype 3.13 in an unknown host before detection in skunk, avian species, and dairy cattle. The teal color of branches indicates the reassortment event.
Extended Data Fig. 6 Phylogenetic analysis of HPAI H5N1 viruses.
Phylogenetic trees constructed with each influenza A virus genome segment, comprising 91 sequences of samples described in this study and 648 sequences of samples collected throughout the American continent, collected between January 2023 and March 2024, available at the GISAID EpiFlu database.
Extended Data Fig. 7 Wild bird sequences HPAI H5N1 are related to sequences from cows in affected dairy farms.
a, Genetic relationship of HPAI H5N1 sequences recovered from blackbirds with sequences recovered from cattle in Farms 7 and 9. Nodes are colored by premise and all the samples collected in the referred farm are highlighted. b, Detailed/zoom in view of the sequence clusters containing samples from Farm 7, Farm 9 and sequences from blackbirds collected at 8–12 Km from Farm 7. Analysis was conducted based on whole concatenated genome sequences.
Extended Data Fig. 8 Evidence of interspecies transmission of HPAI H5N1.
a, Close phylogenetic relationship between HPAI H5N1 sequences recovered from dairy cows, great-tailed grackles, and cat in Farm 1. b, Close phylogenetic relationship between HPAI H5N1 sequences recovered from dairy cows and a racoon in Farm 8. Nodes are colored by host and all the samples collected in the specific farm are highlighted. Panels on the right are a detailed view of the clusters containing more than one host species. Analysis was conducted based on whole concatenated genome sequences.
Supplementary information
Supplementary Discussion
This file contains Supplementary discussion that complements the discussion section of the main text, and Supplementary References.
Supplementary Table 1
RT-PCR data in CSV format showing testing results for Influenza A Matrix and H5-specific RT-PCR on all samples tested in the present study.
Supplementary Table 2
RT-PCR data in CSV format showing testing results for Influenza A Matrix and H5-specific RT-PCR on serial samples collected on days 3, 16 and 31 post-clinical diagnosis (pcd).
Supplementary Table 3
Mutational analysis in CVS format showing all amino acid mutations identified in PB2, PB1, PA, HA, NA, M (M1 and M2), NS (NS1 and NS2) gene segments of the highly pathogenic influenza A H5N1 virus sequences obtained in the present study. Predicted phenotype changes as reported through FluServer (GISAID) are also included.
Supplementary Table 4
List of sequences including GISAID accession numbers in CVS format used in the phylogenomic and mutational analyses conducted in the present study.
Supplementary Table 5
List of highly pathogenic influenza A H5N1 virus sequences obtained in the present study including GISAID accession numbers in CVS format and sample metadata (H5 clade classification, genotype, collection date, state, host, sample type and premise).
Supplementary Table 6
Mutational analysis in CVS format showing amino acid mutations that accumulated over time and identified in PB2, PB1, PA, HA, NA, NP and NS1 genome segments of highly pathogenic influenza H5N1 virus and their frequency among the available H5N1 sequences derived from cattle.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Caserta, L.C., Frye, E.A., Butt, S.L. et al. Spillover of highly pathogenic avian influenza H5N1 virus to dairy cattle. Nature (2024). https://doi.org/10.1038/s41586-024-07849-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-024-07849-4
This article is cited by
-
Pathogenicity and transmissibility of bovine H5N1 influenza virus
Nature (2024)
-
The highly pathogenic H5N1 virus found in U.S. dairy cattle has some characteristics that could enhance infection and transmission among mammals
Lab Animal (2024)
-
Highly Pathogenic Avian Influenza A (H5N1) Virus Outbreak in Ecuador in 2022–2024
Current Infectious Disease Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
