
Abstract
Placebo effects are notable demonstrations of mind–body interactions1,2. During pain perception, in the absence of any treatment, an expectation of pain relief can reduce the experience of pain—a phenomenon known as placebo analgesia3,4,5,6. However, despite the strength of placebo effects and their impact on everyday human experience and the failure of clinical trials for new therapeutics7, the neural circuit basis of placebo effects has remained unclear. Here we show that analgesia from the expectation of pain relief is mediated by rostral anterior cingulate cortex (rACC) neurons that project to the pontine nucleus (rACC→Pn)—a precerebellar nucleus with no established function in pain. We created a behavioural assay that generates placebo-like anticipatory pain relief in mice. In vivo calcium imaging of neural activity and electrophysiological recordings in brain slices showed that expectations of pain relief boost the activity of rACC→Pn neurons and potentiate neurotransmission in this pathway. Transcriptomic studies of Pn neurons revealed an abundance of opioid receptors, further suggesting a role in pain modulation. Inhibition of the rACC→Pn pathway disrupted placebo analgesia and decreased pain thresholds, whereas activation elicited analgesia in the absence of placebo conditioning. Finally, Purkinje cells exhibited activity patterns resembling those of rACC→Pn neurons during pain-relief expectation, providing cellular-level evidence for a role of the cerebellum in cognitive pain modulation. These findings open the possibility of targeting this prefrontal cortico-ponto-cerebellar pathway with drugs or neurostimulation to treat pain.
Similar content being viewed by others


Main
Pain is a subjective experience during which mind–body interactions exert a powerful influence both on pain perception and on the success of pain treatment5,8. One notable example is placebo analgesia, a contextual, cue-based learning phenomenon in which an individual’s positive expectation suffices to reduce pain perception and pain-related behaviours in the absence of any analgesic drug or procedure5,6,8. Placebo analgesia has a prominent role in both medical practice and clinical trials9. Expectations of pain relief are induced during cognitive behavioural therapy to promote recovery in patients with postoperative and/or chronic pain, while strong analgesic responses in the placebo groups of clinical trials hinder the development of pain treatments. Notwithstanding the importance of this placebo effect, our understanding of its underlying biological mechanisms remains limited to human brain imaging data showing that activity in some brain regions, such as the anterior cingulate cortex (ACC), correlates with placebo analgesia10,11,12,13,14,15. Here we combined an advanced mouse behavioural assay of expectation-based pain relief, targeted recombination in active populations (TRAP) of neurons mediating pain-relief expectation, neural Ca2+ imaging in freely behaving mice, single-cell RNA sequencing (scRNA-seq), electrophysiological recordings and optogenetics to establish circuit, cellular and synaptic mechanisms through which positive expectations produce pain relief.
An animal model of placebo analgesia
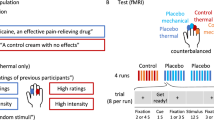
We first developed a 7-day placebo analgesia conditioning (PAC) assay that generates a placebo-like anticipatory pain-relief expectation in mice and permits evaluation of the resulting analgesic effect (Fig. 1a–f and Methods). The PAC apparatus consists of two chambers with distinct visual cues. The assay comprises three phases: habituation (days 1–3), conditioning (days 4–6) and post-conditioning analgesia testing (day 7; Fig. 1a). During the habituation phase, the floors of both chambers are set at 30 °C (innocuously warm) and the mice can freely explore both chambers. Mouse performance (for example, latency of border crossing, time spent in each chamber) on day 3 serves as the pre-conditioning baseline exploratory pattern. During the conditioning phase, the floor of chamber 1, on which the mouse begins the session, is set at 48 °C (noxiously hot), whereas the floor of chamber 2 remains at an innocuous 30 °C. This trains mice to expect pain relief when leaving chamber 1 and entering chamber 2. Finally, for the post-conditioning analgesia test (post-test), the floors of both chambers are set at 48 °C to evaluate any analgesic effect induced by the expectation of pain relief.

a, PAC assay and representative traces of mouse locomotion. Pre, pre-conditioning test; Post, post-conditioning test. b, The time spent in chamber 2 (F1,85 = 48.84, P = 3 × 10−8). c, The latency of the first crossing back (F1,85 = 38.45, P = 5 × 10−7). d, The latency preceding first paw licking (left; P = 0.001), rearing (middle; P = 0.028) and jumping (right; P = 0.013) after reaching chamber 2 during the post-test. e, Nocifensive behaviours after the first border crossing during the post-test. The grey highlighted region indicates the estimated duration (~45 s) of PAC-induced pain relief. f, The frequency of licking (left; P = 0.04) and rearing (middle; P = 0.06) and the duration of rearing (right; P = 0.007) during the first 45 s after the first border crossing on the post-test day in e. g, The strategy to label rACC neurons that are active during PAC. Scale bar, 2 mm. h, Image of rACC neuron cell bodies. i, The density of TRAPed ACC neurons (P = 2 × 10−5). n = 12 per group. Scale bars, 100 μm. j, Axon terminals of TRAPed rACC neurons. CPu, caudate putamen; MD, mediodorsal thalamic nucleus; Th, thalamus; VPM, ventral posteromedial thalamic nucleus; ZI, zona incerta. Scale bars, 100 μm. k, The axon terminal density in each area (F1,63 = 131.691, P = 2 × 10−16). n = 9 and 6 (CPu); 7 and 6 (ZI); 5 and 8 (MD); and 6 and 7 (VPM and Pn) in the control and conditioned groups, respectively. Statistical analysis was performed using two-way analysis of variance (ANOVA) with Tukey post hoc test (b, c and k), two-sided Wilcoxon rank-sum tests (d, f and i). For a–f, n = 9 (control) and 10 (conditioned). For the box plots, the centre lines show the median values, the box limits show the quartiles, and the whiskers show the most extreme datapoints ≤interquartile range from the box edges. For b and c, data are mean ± s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001.
Compared with the unconditioned control mice, conditioned mice progressively developed a significant preference for chamber 2 during the conditioning phase (days 4–6). Importantly, this preference persisted on the post-test day (Fig. 1b), despite that the floors of both chambers were set at the same temperature and should, in the absence of conditioning, elicit identical heat pain perception. Furthermore, conditioned mice exhibited increased latencies to revisit chamber 1 during both the conditioning phase and during the post-test (Fig. 1c). However, setting both chambers at 30 °C during the post-test diminished this preference (Extended Data Fig. 1a,b). Together, these results suggest that PAC generates an expectation of pain relief from chamber 2.
To determine whether this PAC-induced expectation of pain relief indeed recapitulates key features of human placebo analgesia, we compared the nocifensive behaviours displayed by control and conditioned mice during the post-test (Fig. 1d–f). PAC significantly prolonged the latency for paw licking, rearing and jumping (Fig. 1d). Moreover, mice subjected to PAC exhibited fewer overall nocifensive behaviours during the post-test (Fig. 1e,f). This PAC-induced analgesia persisted for at least a week after the conditioning phase (Extended Data Fig. 1c). Importantly, administration of the opioid receptor antagonist naloxone during the post-test, but not during the conditioning phase, abolished this analgesic effect (Extended Data Fig. 1d–i), consistent with the known involvement of endogenous opioid signalling in human placebo analgesia14,16. Furthermore, PAC-conditioned mice exhibited reduced sensitivity to chemical pain induced by formalin injection during a modified post-test in which both floors were set to 30 °C (Extended Data Fig. 1j–l). However, outside of the PAC apparatus, the mechanical, thermal and chemical pain sensitivities of PAC-conditioned mice on day 7 were comparable to those of unconditioned mice (Extended Data Fig. 1m,n). Finally, when we confined PAC-conditioned mice to either chamber 1 or chamber 2 during the post-test by blocking the opening between the two chambers, mice displayed similar latencies to initiate nocifensive behaviours regardless of the chamber in which they were confined (Extended Data Fig. 1o,p). Notably, consistent with human studies suggesting sex differences in placebo analgesia17, female mice showed behaviours similar to those of male mice during PAC (Extended Data Fig. 1q–s), but with different variability for licking (P = 0.017) and jumping (P < 0.001) nocifensive behaviours.
Taken together, these results show that PAC produces an expectation-based analgesic effect that shares key features of human placebo analgesia, enabling modelling and investigation of placebo analgesia in rodents.
rACC→Pn neurons encode pain-relief expectation
Human brain imaging studies suggest that the ACC, especially the rACC, contributes to placebo analgesia10,11,12,14. Notably, the ACC contains a wide variety of cell types, including projection neurons such as intratelencephalic (IT) and pyramidal tract (PT) pyramidal neurons located in distinct cortical layers and that have diverse intracortical and subcortical connections18. To identify rACC pathways that might contribute to placebo analgesia, we injected into the rACC of TRAP2 (FosCreERT2) mice an adeno-associated virus (AAV) that permits expression of synaptophysin–mRuby in a Cre-dependent manner; this approach enabled us to label the presynaptic terminals of rACC neurons that were active during PAC (Fig. 1g–i). This procedure revealed dense axonal projections from labelled layer 5 (L5) rACC neurons to three brain areas: the striatum (dorsal caudate nucleus and putamen), thalamic/subthalamic nuclei (ventral posteromedial thalamic nucleus, mediodorsal thalamic nucleus, zona incerta) and, notably, the Pn (Fig. 1j,k), a region of the pons that mediates cortico-cerebellar communication. The contributions of striatal and thalamic circuits to various sensory-discriminative and affective-motivational aspects of pain have been described previously19,20,21. However, the Pn has no established role in pain modulation, although previous studies have reported Pn activation during pain22,23. Notably, while relatively understudied in the pain field, the cerebellum, like the rACC, consistently shows increased activity during placebo analgesia10,11,13,15. Furthermore, patients who have experienced a cerebellar infarction exhibit impaired placebo analgesia24. We therefore next investigated the function of the rACC→Pn pathway in placebo analgesia.
To record the neural dynamics of rACC→Pn neurons in real time during placebo analgesia, we used a head-mounted miniature microscope to image single-cell somatic Ca2+ activity during the PAC assay. Viral anterograde and retrograde tracing, and whole-cell recording from Pn neurons during optogenetic stimulation of rACC neuron terminals confirmed monosynaptic glutamatergic connectivity between the rACC and Pn (Extended Data Fig. 2a–h). To express GCaMP7f selectively in rACC→Pn cells, we injected a Cre-encoding rAAV with retrograde transport properties into the Pn and injected a Cre-dependent GCaMP7f-encoding rAAV into the rACC (Fig. 2a–c). The numbers of cells that we detected from each mouse varied across different phases of PAC (Extended Data Fig. 3a–c). By aligning cell maps from day 3 (before conditioning), day 6 (conditioning) and day 7 (after conditioning), we aligned a total of 205 cells across days from 6 mice (34 ± 7 cells per mouse). Notably, intracranial virus injection, GRIN lens implantation and miniature microscope mounting had no significant effect on the measured performance metrics of mice during PAC, including total walking distance and average movement speed (Extended Data Fig. 3d).

a, The strategy to monitor rACC→Pn neuron activity. Scale bar, 2 mm. b, Maximal projection of a Ca2+ video with 82 rACC→Pn neurons. Scale bar, 50 μm. c, z-scored Ca2+ activity for the neurons numbered in b. d, rACC→Pn neuron activity during the first border crossing. e,f, rACC→Pn neuron activity averaged for individual neurons (e; F2,612 = 5.16, P = 0.006; n = 205) and individual mice (f; F2,15 = 15.6, P = 0.0002; n = 6). g, rACC→Pn neuron activity during border crossings on the post-test day. h,i, Averaged rACC→Pn neuron activity for individual neurons (h; F2,954 = 10.13, P = 4 × 10−5; n = 320) and individual mice (i; F2,15 = 16.8, P = 0.0002; n = 6). j, The latency preceding the first border crossing against the averaged signal of rACC→Pn neurons for each mouse, with the linear regression fit. k, Recording (rec.) configuration of rACC→Pn neurons. IN, local interneurons; PN, pyramidal neurons; II/III, layers II and III of ACC. l, EPSCs evoked at different holding potentials. The dot represents the peak AMPAR EPSC; the dashed line indicates the NMDAR EPSC amplitude. m, The AMPAR/NMDAR ratio (P = 0.03). n = 11 (Ctrl) and 10 (Cond.) neurons. n, TBS-induced EPSC amplitude changes. Inset: averaged EPSCs before and after TBS. n = 6 neurons per group. o, Recordings of isolated EPSCs and IPSCs. The arrows indicate the onset of electrical stimulation to onset of EPSCs and IPSCs. p, The EPSC–IPSC conductance ratio (P = 0.005). q, The EPSC–IPSC delay (P = 8 × 10−5). n = 8 (Ctrl) and 10 (Cond.) neurons. Statistical analysis was performed using one-way ANOVA with Tukey post hoc test (e, f, h and i), two-sided Wilcoxon rank-sum tests (m, p and q) and Pearson’s two-sided correlation tests (l). For d and g, neurons are ordered by mean Ca2+ activity for each condition. For the box plots, the centre lines show the median values, the box limits show the quartiles, and the whiskers show the most extreme datapoints ≤interquartile range from the box edges. For n, data are mean ± s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001.
We next examined Ca2+ signals in these neurons during border crossing, a timepoint around which mice should expect pain relief as a conditioned response to PAC training. We found that the Ca2+ activity of rACC→Pn neurons increased progressively during the conditioning phase (Extended Data Fig. 3e,f). On the post-test day, these neurons showed elevated Ca2+ activity, at the levels of individual neurons (Fig. 2d,e) and of individual mice (Fig. 2f). Among the cross-day-aligned rACC→Pn neurons, 58% exhibited greater activity during the post-test compared with the pre-conditioning baseline, while 25% showed progressively increased activity throughout all phases (Extended Data Fig. 3g,h). Furthermore, the discriminability index of rACC→Pn neurons between the first border crossing (with conditioned pain-relief expectation) and the first crossing back (without a conditioned expectation of pain relief) also increased after conditioning (Extended Data Fig. 3i). This higher discriminability index during the post-test suggests that the increased activity of rACC→Pn neurons is not due to an overall increase in neural activity after conditioning. To exclude the possibility that the biophysical properties of the Ca2+ indicator, especially its long decay dynamics, might explain the observed differences before and after PAC conditioning, we performed the same analysis using binary Ca2+ transient event data, which yielded similar results (Extended Data Fig. 3j–m). These increases in Ca2+ activity and discriminability index disappeared when we tested a shuffled control dataset with randomized crossing times (Extended Data Fig. 3n–s). Furthermore, we found no correlation between the activity of rACC→Pn neurons and mouse locomotor speed (Extended Data Fig. 3t–v), indicating that these cells are not merely responding to generic movement.
To further confirm that increased activity in rACC→Pn neurons corresponds with pain-relief expectation and not pain-associated aversion, we compared, on the post-test day, the activity of rACC→Pn neurons during the first border crossing (with conditioned pain-relief expectation), the first crossing back (without conditioned pain-relief expectation) and the last border crossing (reduced or no conditioned pain-relief expectation due to expectation violation25). rACC→Pn neurons showed no increased activity during the first crossing back and significantly reduced activity during the last border crossing, averaged across individual neurons (Fig. 2g,h) and across mice (Fig. 2i). Moreover, the elevated activity of rACC→Pn neurons during the post-test progressively decreased after arriving in chamber 2 (Extended Data Fig. 3w), aligning well with the violation of expectation.
Furthermore, we examined the relationship between the latency of the first border crossing and the activity of rACC→Pn neurons during that crossing for each mouse. We reasoned that, if rACC→Pn neurons encode the expectation of pain relief, then mice with the strongest expectation-based motivation to cross the border should show the greatest increase in rACC→Pn neuron activity. Consistent with this prediction, linear regression analysis revealed a negative correlation between the latency of the first border crossing and rACC→Pn neural activity (Fig. 2j), further evincing the involvement of the rACC→Pn pathway in pain-relief expectation.
To clarify whether the increase in activity after conditioning was specific to rACC→Pn neurons, or rather a general feature of all rACC output neurons, we recorded the Ca2+ dynamics of IT neurons, the other major type of deep-layer pyramidal neurons (rACC→Pn neurons are PT neurons), during PAC. IT neurons showed no significant change in Ca2+ activity during the first border crossing after conditioning (Extended Data Fig. 4a–n), suggesting a specific role for rACC→Pn neurons during pain-relief expectation.
Finally, in a separate experiment, we examined rACC→Pn neural activity during noxious thermal, noxious mechanical or innocuous mechanical stimulation (Extended Data Fig. 5a–j). None of these stimuli significantly affected rACC→Pn neural Ca2+ activity, in agreement with the lack of significant change in rACC→Pn neural activity during licking and rearing nocifensive behaviours on the post-test day (Extended Data Fig. 5k,l), arguing against a general role for these cells in nociception or mechanosensation.
PAC enhances rACC→Pn neuron excitability
Consistent with the learning-based nature of placebo analgesia6,26, the Ca2+ activity of rACC→Pn neurons increased progressively during PAC (Fig. 2d–f and Extended Data Fig. 3e,f). To examine the underlying synaptic mechanisms, we used brain-slice electrophysiology. First, we labelled rACC→Pn neurons by injecting a tdTomato-encoding AAV with retrograde transport properties into the Pn (Fig. 2k and Extended Data Fig. 6a). Mice were then subjected to PAC and euthanized immediately after the conditioning phase for electrophysiology recording (Extended Data Fig. 6a,b). Passive membrane properties of rACC→Pn neurons, such as the resting membrane potential, input resistance, amplitude and half-duration of the action potential, and action-potential firing frequency, remained unchanged after PAC (Extended Data Fig. 6c–f). However, rACC→Pn neurons from conditioned mice (Extended Data Fig. 6e,g), but not other L5 rACC neurons (Extended Data Fig. 6h,i), displayed more burst firing at the beginning of current injection than rACC→Pn neurons from control mice. Moreover, PAC significantly increased the amplitude, but not the frequency, of spontaneous excitatory postsynaptic currents (EPSCs; Extended Data Fig. 6j–l), suggesting a postsynaptic change in rACC→Pn neuron function. Consistent with this, the paired-pulse ratio (PPR), a measure of presynaptic function, was statistically indistinguishable between conditioned and control mice (Extended Data Fig. 6m,n), whereas the AMPAR/NMDAR ratio, a postsynaptic characteristic of synaptic transmission, increased significantly (Fig. 2l,m).
We next tested whether PAC alters long-term potentiation (LTP), a cellular process that underlies learning and memory27, in rACC→Pn neurons. We induced LTP using classical theta-burst stimulation (TBS). In rACC→Pn neurons from control mice, the amplitude of EPSCs increased after TBS, then quickly returned to the baseline levels. By contrast, rACC→Pn neurons from conditioned mice showed robust LTP that lasted for the entire recording period (40 min) after induction (Fig. 2n), indicating enhanced synaptic plasticity.
Cortical inhibitory interneurons control Ca2+ dynamics, burst firing, spontaneous release and synaptic plasticity of principal neurons through feedforward inhibition, facilitating learning28. To test whether PAC alters feedforward inhibition, we recorded evoked EPSCs or IPSCs (inhibitory postsynaptic currents) in isolation by holding the membrane potential of rACC→Pn neurons at −70 mV or +10 mV, respectively (Fig. 2o). In brain slices from control mice, monosynaptic EPSCs were followed by large disynaptic IPSCs, confirming strong feedforward inhibition in this circuit (Extended Data Fig. 6o). By contrast, rACC→Pn neurons from conditioned mice received significantly weaker feedforward inhibition (Fig. 2p). Furthermore, the delays between EPSCs and IPSCs were markedly prolonged after conditioning (Fig. 2q). Similarly, PAC decreased the amplitude and delayed the latency of IPSCs specifically from parvalbumin-positive (PV+) interneurons (Extended Data Fig. 6p–w), which critically contribute to feedforward inhibition in the cortex28,29. Taken together, these results demonstrate that PAC impairs both the efficacy and timing of feedforward inhibition of rACC→Pn neurons and enhances their excitability.
Adjusting placebo analgesia and pain thresholds
To test the function of the rACC→Pn pathway in placebo analgesia, we injected AAVs to express halorhodopsin (NpHR) or channelrhodopsin-2 (ChR2) in rACC neurons and implanted optic fibres bilaterally over the Pn (Fig. 3a and Extended Data Fig. 7a). We then photomanipulated rACC→Pn neuron terminals of conditioned mice during the PAC post-test, beginning when mice crossed from chamber 1 to chamber 2 (Extended Data Fig. 7b). We found that photoinhibition substantially reduced PAC-induced latency increases in paw licking, rearing and jumping (Fig. 3b). Conversely, optogenetically activating the rACC→Pn pathway during the post-test significantly prolonged the latency of mice to display paw licking, but not rearing and jumping behaviours (Extended Data Fig. 7c). These less-pronounced behavioural changes may indicate a ceiling effect, given that Pn neurons, especially their axon terminals, show high instantaneous firing frequencies (>700 Hz) while coding sensory information30. Neither photoinhibition nor photoexcitation produced detectable changes in motor coordination (Extended Data Fig. 7d). These results indicate that the rACC→Pn pathway mediates PAC-induced analgesia.

a, The strategy and experimental timeline to optogenetically manipulate the activity of the rACC→Pn pathway. Scale bar, 2 mm. b, The latency preceding first paw licking (left; P = 0.005), rearing (middle; P = 0.04) and jumping (right; P = 0.009) during the post-test. n = 10 mice per group. c, The strategy to measure thermal pain using a hot plate while optogenetically activating or inhibiting the rACC→Pn pathway. d, The latency preceding paw withdrawal on a 48 °C plate (left; F2,26 = 10.66, P < 0.001) or 52 °C plate (right; F2,26 = 7.38, P = 0.003). n = 10 (eYFP control), 10 (NpHR) and 9 (ChR2) mice. e, The strategy to measure the mechanical pain threshold with von Frey filaments while optogenetically activating or inhibiting the rACC→Pn pathway. f, Quantification of changes in paw withdrawal frequency in response to six different von Frey filaments induced by optogenetic manipulation of the rACC→Pn pathway (F2,156 = 62.965, P = 2 × 10−16). n = 10 (eYFP control), 10 (NpHR) and 9 (ChR2) mice. g, The pain threshold of mice with or without light stimulation (F2,26 = 25.98, P = 1.3 × 10−9). n = 10 (eYFP control), 10 (NpHR) and 9 (ChR2) mice. Statistical analysis was performed using two-sided Wilcoxon rank-sum tests (b), one-way ANOVA with Tukey post hoc test (d) and two-way ANOVA with Tukey post hoc test (f and g). For the box plots, the centre lines show the median values, the box limits show the quartiles, and the whiskers show the most extreme datapoints ≤interquartile range from the box edges. For f, data are mean ± s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001.
To test whether modulating the activity of the rACC→Pn pathway could alter pain, we subjected naive mice to commonly used thermal (hot plate) and mechanical (von Frey) sensitivity tests while optogenetically manipulating the rACC→Pn pathway (Fig. 3c–g). In the hotplate test, photoinhibition of the rACC→Pn pathway decreased paw withdrawal latency, while photoexcitation increased this latency compared with control mice (Fig. 3d). In the von Frey test, photoinhibition of the rACC→Pn pathway increased the paw withdrawal frequency, whereas photoexcitation decreased the paw withdrawal frequency compared with the control mice (Fig. 3f and Extended Data Fig. 7e). Furthermore, photoinhibition decreased the mechanical sensitivity threshold, whereas photoexcitation increased it (Fig. 3g). Together, these results indicate that the rACC→Pn pathway can be activated to generate analgesia.
Placebo analgesia requires Oprd1 + Pn neurons
We next sought to manipulate the function of Pn neurons during PAC. However, our understanding of the molecular identity of Pn neurons is limited. We therefore used single-cell transcriptomics to investigate the cellular composition of the Pn and identify marker genes to gain genetic access to and manipulate Pn neurons. We used both high-throughput/low-depth (10x Genomics) and low-throughput/high-depth (SMART-seq) scRNA-seq approaches to comprehensively characterize Pn neurons (Fig. 4a and Extended Data Fig. 8a).

a, The strategy to characterize gene expression in the Pn using single-cell transcriptomics. Scale bar, 1 mm. D, dorsal; L, lateral. b, Pn neurons in low-dimensional uniform manifold approximation and projection (UMAP) space, colour coded by cluster. E, excitatory; I, inhibitory. c, Opioid and cluster-specific gene expression. d, Euler diagram depicting Oprm1 and Oprd1 expression in Pn excitatory neurons. e, Fluorescence in situ hybridization verifying Oprd1 expression by excitatory Pn neurons. Scale bar, 50 µm. f, The strategy to optogenetically manipulate the activity of Oprd1+ neurons in the Pn. Scale bar, 2 mm. g, The latency preceding first paw licking (left; P = 0.01), rearing (middle; P = 0.07) and jumping (right; P = 0.01) during the post-test with photoinhibition of eYFP control versus NpHR mice. n = 8 (eYFP control) and 9 (NpHR) mice. h, The strategy to monitor Purkinje cell activity using a miniscope. Scale bar, 2 mm. i, The mean projection of a Ca2+ video with 89 Purkinje cells. In total, 40 cross-day-aligned Purkinje cells were classified into clusters 1 or 2. Scale bar, 100 μm. j, Ca2+ trace of 5 Purkinje cells in each cluster. k, Purkinje cell activity during the first border crossing. For each cluster, neurons are ordered by mean Ca2+ activity during the last day of conditioning. l,m, Cluster 1 Purkinje cell activity averaged for individual neurons (l; F2,171 = 23.63, P = 8.6 × 10−10; n = 58) and individual mice (m; F2,15 = 13.94, P = 0.0003; n = 6). Statistical analysis was performed using two-sided Wilcoxon rank-sum tests (g) and one-way ANOVA with Tukey post hoc test (l and m). For the box plots, the centre lines show the median values, the box limits show the quartiles, and the whiskers show the most extreme datapoints ≤interquartile range from the box edges. *P < 0.01 and ***P < 0.001.
Focusing our analysis on neurons, we detected ten transcriptionally distinct clusters (Fig. 4b,c). Five clusters were Slc17a7+ (encoding vesicular glutamate transporter 1) excitatory neurons, comprising 72% of all Pn neurons. The remaining clusters were largely Slc32a1+ (encoding vesicular inhibitory amino acid transporter) inhibitory neurons (Fig. 4c and Extended Data Fig. 8b). More than half of the excitatory Slc17a7+ Pn neurons coexpressed Slc17a6 (encoding vesicular glutamate transporter 2)—a rare feature for glutamatergic neurons throughout the nervous system (Fig. 4c and Extended Data Fig. 8c). After examining the expression of endogenous opioid peptides and receptors, which critically contribute to pain modulation and placebo analgesia, we determined that a very large proportion of Pn neurons expresses the δ- and/or μ-opioid receptors (Fig. 4c,d). Specifically, 54% and 26% of Pn neurons express Oprd1 and Oprm1 (encoding the δ- and μ-opioid receptor, respectively). In total, 81% of Oprd1+ Pn neurons coexpress excitatory neuron markers (Slc17a7, Slc17a6 or both; Extended Data Fig. 8c). We confirmed the presence of Slc17a6+Oprd1+ neurons in the Pn using fluorescence in situ hybridization (Fig. 4e). On the basis of these observations, we used an Oprd1cre mouse line to investigate the anatomy of Oprd1+ Pn neurons and their function in placebo analgesia.
To investigate whether Oprd1+ Pn neurons have a role in placebo analgesia and/or pain modulation, we first tested for a direct connection between the Oprd1+ Pn neurons and rACC projection neurons. Both AAV1-mediated anterograde transsynaptic tracing in WT mice and rabies-mediated retrograde transsynaptic tracing in Oprd1cre mice (Extended Data Fig. 8d–h) indicated a monosynaptic connection between rACC neurons and Oprd1+ Pn neurons. Moreover, anterograde transsynaptic tagging suggested that 53% of neurons in rACC-targeted subregions of the Pn receive monosynaptic inputs from the rACC (Extended Data Fig. 8f). Given that photoinhibition of rACC→Pn neuron terminals abolished placebo analgesia (Fig. 3b), we next tested whether postsynaptic manipulation of Oprd1+ Pn neurons could produce similar effects. We injected AAVs to express NpHR or eYFP (control) in Oprd1+ Pn neurons and implanted optic fibres bilaterally over the Pn (Fig. 4f). Photoinhibition of Oprd1+ Pn neurons during the post-test abolished the PAC-induced prolonged latency of mice to display first paw licking, rearing and jumping (Fig. 4g). A more-specific strategy targeting only the Oprd1+ Pn neurons receiving rACC inputs yielded similar results (Extended Data Fig. 8l–o). Consistent with these findings, systemic administration of selective agonists for either the µ- or δ-opioid receptor also diminished the analgesic effects induced by PAC (Extended Data Fig. 8p,q). Moreover, photoinhibition of Oprd1+ Pn neurons significantly increased mechanical and thermal sensitivity in the von Frey and hotplate tests, respectively (Extended Data Fig. 8r,s). Photoinhibition of Oprd1+ Pn neurons produced no detectable change in locomotion or motor coordination (Extended Data Fig. 8t,u). Lastly, photoinhibition of Oprd1+ Pn neurons during the conditioning phase of PAC showed a trend toward attenuating PAC-induced analgesia on the post-test day (Extended Data Fig. 8i–k). Together, these results indicate that L5 rACC neurons projecting onto Oprd1+ Pn neurons critically contribute to both placebo analgesia and pain processing.
Purkinje cells encode pain-relief expectation
To gain further evidence that the rACC→Pn pathway mediates pain-relief expectation, we examined the primary target of Pn neurons—the cerebellum. To label the projections of Oprd1+ Pn neurons in the cerebellum, we injected into the Pn of Oprd1cre mice an AAV encoding mGFP and synaptophysin–mRuby in a Cre-recombinase-dependent manner. The resulting tracing data showed that Oprd1+ Pn neurons mainly project to cerebellar lobules VI, Crus I and Crus II (Extended Data Fig. 9a–c), which support the cognitive functions of the cerebellum31.
We then used a head-mounted miniature microscope to image the dendritic Ca2+ activity of Purkinje cells32, the principal neurons of the cerebellar cortex, in lobule VI of the cerebellar vermis in freely behaving mice during PAC (Fig. 4h–m). Cerebellar Purkinje cells receive excitatory input from a single climbing fibre (CF) originating in the inferior olive and from around 200,000 parallel fibres (PFs) that relay information sent disynaptically from the Pn through cerebellar granule cells. Spontaneous CF activity (1–2 Hz) triggers dendritic Ca2+ spikes that can pervade the entire dendritic tree, whereas PF inputs, depending on their activity level, can evoke smaller to moderate dendritic spikes33,34,35. Furthermore, when near coincident in time with CF input, PF activity can lead to supralinear Ca2+ excitation33,35. Thus, if the Pn relays pain-relief expectation from the rACC to the cerebellum through PFs, then PAC should increase the amplitudes of Purkinje cell dendritic Ca2+ spikes and the occurrence frequency of dendritic Ca2+ spikes that are large enough to be detected by Ca2+ imaging.
To test these predictions, we analysed Ca2+ imaging recordings from 276 cross-day-aligned Purkinje cells (Fig. 4h–j) during PAC. To identify Purkinje cells that might encode pain-relief expectation, we performed a classification analysis (Extended Data Fig. 9e–g). On the basis of Ca2+ activity during the first border crossing (with conditioned pain-relief expectation), the first crossing back (without conditioned pain-relief expectation) and the last border crossing (reduced or no conditioned pain-relief expectation due to expectation violation), the classification algorithm resolved two main classes of Purkinje cells (clusters 1 and 2). Notably, during first crossing back and last border crossing, the average Ca2+ activity of Purkinje cells in cluster 1 declined substantially, whereas the activity of Purkinje cells in cluster 2 increased modestly but significantly (Extended Data Fig. 9f,g). Furthermore, the Ca2+ activity of Purkinje cells in cluster 1, but not cluster 2 (Extended Data Fig. 9h,i), progressively increased across the before, during and after conditioning phases, as seen in statistical analyses of individual neurons (Fig. 4k,l) and individual mice (Fig. 4m).
To disentangle the distinct contributions of PF and CF inputs to the elevated Ca2+ activity of Purkinje cells in cluster 1 during PAC, we examined the amplitudes and occurrence frequency of Purkinje cell dendritic Ca2+ spikes. Ca2+ spikes in Purkinje cells of cluster 1 occurred at 1.4 Hz during border crossing on the pre-test day, consistent with the spontaneous firing rate of CFs36,37. However, Ca2+ spiking increased to 2.6 Hz on the last day of the conditioning phase and to 2.5 Hz on the post-test day (Extended Data Fig. 9j). By contrast, the Ca2+ spiking of Purkinje cells in cluster 2 was not significantly altered during PAC (Extended Data Fig. 9k). Moreover, Purkinje cells of cluster 1 (Extended Data Fig. 9l), but not cluster 2 (Extended Data Fig. 9m), displayed more Ca2+ spikes with large amplitudes after PAC. Furthermore, when considering only Ca2+ spikes with amplitudes exceeding 3 z-scored ΔF/F, suggestive of supralinear Ca2+ signals, both the number and amplitude of these spikes were increased for Purkinje cells in cluster 1 (Extended Data Fig. 9l,n), but not in cluster 2 (Extended Data Fig. 9m,n).
Moreover, linear regression analysis showed that, across days of PAC, increased net activity levels of Purkinje cells in cluster 1, but not in cluster 2, were associated with shorter latencies for the first border crossing (Extended Data Fig. 9o,p). Both Purkinje cells in cluster 1 and the entire set of recorded Purkinje cells displayed elevated Ca2+ activity during first border crossing after conditioning, but not during the first crossing back or the last border crossing during the post-test (Extended Data Fig. 9q–s). This effect was absent in a shuffled dataset with randomized crossing times (Extended Data Fig. 9t). Together, these findings support the idea that elevated PF inputs during the PAC assay drive the increased Ca2+ activity of Purkinje cells in cluster 1 and thereby promote behavioural changes. This similar response of cerebellar Purkinje cells to that of rACC→Pn neurons during PAC directly demonstrates the cerebellum’s involvement in pain-relief expectation and the critical role of the rACC–ponto–cerebellar pathway in placebo analgesia.
Discussion
Pain is a complex experience with sensory-discriminative, affective-motivational and cognitive-evaluative dimensions38. Placebo analgesia epitomizes the cognitive-evaluative dimension of pain by demonstrating how cognitive factors such as expectations alter the perception of noxious events or injuries to shape pain subjectivity. To investigate the neural basis of placebo analgesia in rodents, we developed the PAC assay. This paradigm effectively establishes an expectation of pain relief in mice, resulting in reduced pain (Fig. 1). Although PAC-induced analgesia may not capture all of the complexities of human placebo analgesia, this assay replicates several key features of the phenomenon, including reliance on the endogenous opioid system (Extended Data Figs. 1d and 8q), the recall of pain-relief expectations over time (Extended Data Fig. 1c) and the inherent variability observed in human placebo analgesia (Fig. 1b–d and Extended Data Fig. 1q–s). Thus, building on previous approaches to model placebo analgesia in rodents39, PAC offers a simple, practical and non-pharmacological method to investigate the biology of expectation-induced analgesia at several levels.
At the circuit level, our data suggest that the rACC mediates placebo analgesia by engaging the cerebellum through L5 PT projections to the Pn. Among all Pn neurons, we found that 65% express Oprd1 and/or Oprm1 (Fig. 4c,d), suggesting that the activity of Pn neurons is probably modulated by the endogenous opioid system. Notably, naloxone injections during the conditioning phase prevent the attenuation of PAC-induced analgesia caused by three consecutive days of injections (Extended Data Fig. 1g–i), and optogenetically inhibiting Oprd1+ Pn neurons during the same phase reduces PAC-induced analgesia (Extended Data Fig. 8i–k). Although these observations suggest that associative learning requires the activation of Oprd1+ Pn neurons, aligning with the Ca2+ imaging data, the contribution of opioid signalling in this process needs to be further confirmed. Similarly, although our data suggest the involvement of both the µ- and δ-opioid receptors in PAC-induced placebo analgesia (Extended Data Fig. 8q), the specific opioid peptides and receptors within the rACC-Pn-cerebellar pathway, and potentially in other regions, that collectively mediate this phenomenon remain to be established. Moreover, by imaging the dendritic activity of cerebellar Purkinje cells, the sole outputs of the cerebellar cortex, during pain-relief expectation (Fig. 4h–m and Extended Data Fig. 9), our study provides direct, cellular-level evidence for the cerebellum’s contribution to placebo analgesia. The mechanisms by which the cerebellum modulates placebo analgesia remain to be explored. Based on its connectivity with numerous brain regions40,41,42,43,44, the cerebellum could modulate pain perception through ascending and/or descending pathways. For example, the rACC may recruit the cerebellum to indirectly modulate the descending pain modulation pathway, especially the periaqueductal grey45,46, to produce analgesia; this could explain the enhanced coactivation between the rACC and periaqueductal grey during placebo analgesia reported in several human studies13,14.
At the cellular level, our Ca2+ imaging and electrophysiological data show that PAC specifically increases the activity of Pn-projecting L5 rACC neurons (Extended Data Fig. 6e,f,h,i). While previous studies suggest that PT neurons targeting specific subcortical areas tend to be homogeneous, both genetically47 and in terms of morphology and function48, we cannot exclude the possibility that only a subset of rACC→Pn neurons drives placebo analgesia. Notably, PT (rACC→Pn) and IT neurons display opposite activity during PAC: our electrophysiological data show reduced activity in non-PT L5 rACC neurons after conditioning (Extended Data Fig. 6h,i), and our Ca2+ imaging data indicate that IT neurons are more active, whereas PT neurons less active, during the last border crossing in the PAC post-test (Extended Data Fig. 4k,l,n). The opposite activity of rACC PT and IT neurons during placebo analgesia is an interesting subject for future study.
At the synaptic level, our electrophysiology data show that PAC increases the synaptic plasticity and impairs the feedforward inhibition of rACC→Pn neurons (Fig. 2n–q). Feedforward inhibition contributes to burst firing, shapes network representations of behavioural events and modulates ensemble Ca2+ signalling during learning28,49. Thus, the diminished feedforward inhibition of rACC→Pn neurons probably underlies their increased burst firing after conditioning and progressively enhanced activity during PAC (Fig. 2f and Extended Data Fig. 6g). Notably, in the cerebellar cortex, feedforward inhibition has also been shown to gate supralinear Ca2+ signalling in Purkinje cell dendrites50. Given that Purkinje cells display an increased number of supralinear Ca2+ spikes after PAC (Extended Data Fig. 9n), suppressed feedforward inhibition may serve as a common synaptic mechanism of pain-relief expectation in both cerebral and cerebellar cortices.
In conclusion, this study reveals circuit, cellular and synaptic mechanisms that underlie placebo analgesia and, more broadly, the cognitive-evaluative dimension of pain, bridging the gap with our more advanced understanding of the sensory-discriminative and affective-motivational dimensions of pain20,51,52,53,54,55,56,57,58,59. Crucially, we provide evidence that this rACC–ponto–cerebellar pathway could be engaged by analgesic drugs, neurostimulation protocols and/or cognitive behavioural therapies to produce pain relief.
Methods
Animals
All of the procedures were performed according to animal care guidelines approved by the Administrative Panel on Laboratory Animal Care (APLAC) of Stanford University, by the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill and by the International Association for the Study of Pain. Mice were housed at a maximum of 5 mice per cage and maintained under a 12 h–12 h light–dark cycle in a temperature-controlled environment with ad libitum access to food and water. Male or female mice with an age range of 8–12 weeks were used for the experiments. C57BL/6 wild-type (000664), TRAP2 (FosCreERT2, 030323) and Pvalbcre (017320) mice were purchased from Jackson Laboratory. Oprd1cre mice were generated at the Stanford Transgenic Research Center using standard gene targeting procedures. In brief, an IRES-cre cassette was introduced immediately following the Oprd1 stop codon through homologous recombination. After electroporation of the targeting construct into 129Sv/SvJ-derived ES cells, neomycin-resistant ES cell colonies were screened for IRES-cre insertion using long-range PCR and Southern blotting. Flp transfection was then performed to remove the FRT-flanked neomycin-resistance gene. Confirmed neomycin-excised colonies were then injected into C57BL/6 blastocysts to generate chimeric males, which were bred to C57BL/6 females to generate founders. Sample sizes for mouse behaviour and electrophysiology experiments were determined using the power analysis (‘pwr’ R package). Specifically, the function ‘pwr.t.test’ was used, with a significance level of 0.05, a power of 0.80, and effect sizes estimated from pilot experiments and/or previous studies using similar methods. For the histology experiment, samples were allocated by randomly selecting brain slices containing regions of interest. For all other experiments, samples and animals were randomized into groups. Experimenters were blinded to experimental groups before and during all mouse behavior experiments. Calcium imaging data were analyzed by two independent researchers using two different analysis pipelines at two universities.
Drugs
4-OHT (H6278, Sigma-Aldrich) was prepared in absolute ethanol and Kolliphor EL (C5135, Sigma-Aldrich) and administered intraperitoneally (50 mg per kg). Naloxone (N7758, Sigma-Aldrich) was prepared in saline and administered intraperitoneally (5 mg per kg).
Viral reagents
To express GCaMP7f60 in rACC→Pn projection neurons for Ca2+ imaging, we intracranially injected 400 nl of rAAV2-retro-hSyn-Cre-WPRE-hGH (105553-AAVrg; Addgene; titre: 7 × 1012 viral genomes (vg) per ml) into the right Pn at the coordinates anteroposterior (AP): −4.0 mm, mediolateral (ML): +0.4 mm, dorsoventral (DV): −5.4/−5.8 mm and 400 nl of AAV1-syn-FLEX-jGCaMP7f-WPRE (104492-AAV1; Addgene; titre: 1 × 1013 vg per ml) into the right ACC (AP: +0.75 mm, ML: +0.5 mm, DV: −1.75 mm). To express GCaMP6s in ACC IT neurons, we injected 400 nl of AAVretro-EF1a-Flpo (55637-AAVrg; Addgene; titre: 2.2 × 1013 vg per ml) into the left dorsomedial striatum at the coordinates AP: +0.2 mm, ML: −2.0 mm, DV: −4.1 mm and 400 nl of AAV8-EF1a-fDIO-GCaMP6s (105714-AAV8; Addgene; titre: 1.8 × 1013 vg per ml) into the right ACC. To express GCaMP8m in cerebellar Purkinje cells for Ca2+ imaging, we intracranially injected 200 nl of AAV1-CamKIIa-JGCaMP8m (176751-AAV1; Addgene; titre: 1.8 × 1013 vg per ml) into lobule VI of the vermis at the coordinates AP: −7.0 mm, ML: +0.0 mm, DV: −360 and −200 µm.
To trace the output of rACC neurons active during PAC, we intracranially injected 400 nl of AAV-DJ-hSyn-FLEX-mGFP-2A-Synaptophysin-mRuby (Stanford Virus Core; titre: 1.2 × 1013 vg per ml) into the rACC of TRAP2 (FosCreERT2) mice at the coordinates AP: +0.75 mm, ML: +0.5 mm, DV: −1.75 mm.
To optogenetically manipulate the activity of the rACC→Pn pathway, we intracranially injected 400 nl of either AAV5-hSyn-eNpHR-eYFP (UNC Vector Core; titre: 4.4 × 1012 vg per ml) or AAV5-hSyn-ChR2-eYFP (UNC Vector Core; titre: 5.3 × 1012 vg per ml) into the ACC of WT mice, and 400 nl of AAV5-EF1a-DIO-eNpHR-eYFP (UNC Vector Core; titre: 4.5 × 1012 vg per ml) into the Pn of Oprd1cre mice.
To optogenetically manipulate the activity of the Oprd1+ Pn neurons receiving monosynaptic inputs from rACC neurons, we intracranially injected in Oprd1cre mice 400 nl of AAV1-EF1a-Flpo (55637-AAV; Addgene; titre: 2.3 × 1013) into the rACC bilaterally, and then 400 nl AAV8-nEF-Con/Fon-NpHR-EYFP (137152-AAV8; Addgene; titre: 2.6 × 1013) into the Pn bilaterally.
To trace the output of ACC and secondary motor cortex projections to the Pn, we intracranially injected 400 nl of AAV5-hSyn-eGFP (UNC Vector Core; titre: 4 × 1012 vg per ml) into the ACC (AP: +0.75 mm, ML: +0.5 mm, DV: −1.75 mm) and 400 nl of rAAV5-hsyn-chrimsonr-tdT (UNC Vector Core; titre: 4.6 × 1012 vg per ml) into the secondary motor cortex (AP: +1.41 mm, ML: +0.75 mm, DV: −1.5 mm).
To label rACC→Pn neurons for electrophysiological recording, we intracranially injected 400 nl of AAVretro-CAG-tdTomato (59462-AAVrg; titre: 1.2 × 1012 vg per ml) into the right Pn at the coordinates listed above. To measure the feedforward inhibition of rACC→Pn neurons by PV+ interneurons, in addition to the AAVretro-CAG-tdTomato virus injected into the Pn, we injected 400 nl of AAV5-DIO-EF1a-ChR2-eYFP (UNC Vector Core; titre: 4.0 × 1012 vg per ml) into the rACC to express ChR2 in PV+ interneurons.
To trace the output of Pn neurons that receive inputs from the ACC, we injected 300 nl of the anterograde transsynaptic virus AAV1-hSyn-cre-WPRE-hGH (Addgene; titre: 1 × 1013 vg per ml) into the ACC61, then injected 400 nl of AAV-DJ-hSyn-FLEX-mGFP-2A-Synaptophysin-mRuby (Stanford Virus Core; titre: 1.2 × 1013 vg per ml) into the Pn. The coordinates for the ACC and Pn injections were the same as for the Ca2+ imaging experiment.
To trace brain areas forming monosynaptic connections with Oprd1+ Pn neurons, we injected the AAV helper viruses AAV-FLEx-TVA-Mkate and AAV-FLEx-G into the Pn of Oprd1cre mice. Then, 3 weeks later, a recombinant rabies virus (RVdG) was injected into the Pn. Next, 1 week after injection of the rabies virus, the brains were collected.
The labelling efficiency of viruses is governed by a multifaceted interplay of factors, including the virus’s serotype and titre, the selected promoter, and the targeted cell types. Thus, the labelling efficiency of the virus used in this study cannot be consistently quantified.
Stereotaxic injection and surgical procedures
All surgeries were performed under aseptic conditions. Animals were anaesthetized using isoflurane (07-893-8441, Patterson Veterinary). After anaesthesia induction with 4% isoflurane in a chamber, animals were transferred to a small-animal digital stereotaxic instrument (David Kopf Instruments) and anaesthesia was maintained using 2% isoflurane. Injections were performed using a calibrated microcapillary tube (P0549, Sigma-Aldrich) and pulled with a P-97 micropipette puller (Sutter Instruments). The viral reagents were aspirated into the tube using negative pressure and delivered at a rate of around 50 nl min−1 using positive pressure. After injection, the tube was raised ~100 µm and held stationary for an additional 10 min to allow diffusion of the virus, and then slowly withdrawn at a rate of 0.05 mm s−1. After surgery, the mice were transferred to a warm chamber until they had fully recovered, and were then returned to their home cage.
Microendoscope or optical cannula implantation
Microendoscope implantation in the ACC was performed as described previously56,61. In brief, we stereotaxically implanted a stainless steel cannula 1 week after viral injection. The cannula was fabricated with 18-G 304 S/S Hypodermic Tubing, custom cut to pieces 4.3 mm in length (Ziggy’s Tubes and Wires) and attached at one end to a Schott Glass 2 mm in diameter and 0.1 mm thick (TTL) using an optical adhesive (Norland Optical Adhesive #81, Thermo Fisher Scientific). After grinding away the excess glass using a polisher, the cannulas were carefully stored until use in implantation surgeries.
For cannula implantation surgeries, mice were anaesthetized with isoflurane (4% for induction and 2% for maintenance) while the body temperature was maintained using a heating pad. After cranial hair removal, skin sterilization and scalp incision, we performed small craniotomies in three locations (AP: +5.10 mm, −3.56 mm, −3.56 mm; ML: −0.77mm, +2.06 mm, −3.01 mm) and then screwed three stainless steel screws (MX-000120-01SF, Component Supply Company) down to the dura of the skull to stabilize the implantation. We then performed a fourth craniotomy using a drill (Model EXL-M40, Osada) and a 1.4 mm round drill burr (19007-14, FST) at the coordinates AP: +0.75 mm, ML: +1.25 mm. The bone fragments and other detritus were cleared away from the opening using sterilized forceps. To prevent any increase in intracranial pressure and improve image quality, we aspirated away the overlying tissue down to approximately DV: −1.0 mm at an angle of 18°.
The custom cannula was then attached to a holder (David Kopf Instruments) and lowered to AP: +0.75 mm, ML: +1.25 mm, DV: −1.8 mm at 18°. Blood and additional debris around the craniotomy were quickly removed and adhesive cement (S380 Metabond Quick Adhesive Cement System, C&B) was applied to seal the gap between the cannula and the skull. A custom-designed laser-cut headbar (18–24 G stainless steel, LaserAlliance) was placed over the left posterior skull screw, then layers of dental cement (Lang Dental) were applied to affix both the cannula and headbar to the skull. After the cement dried (7–10 min), we transferred the animal to a heated pad for recovery. After full recovery, the mice were returned to their home cage.
Placement of the cranial window
After cranial hair removal, skin sterilization and scalp incision, we performed small craniotomies in two locations (AP: +5.10 mm, −3.56 mm; ML: +0.77 mm, −2.89 mm) and then screwed two stainless steel screws (MX-000120-01SF, Component Supply Company) down to the dura of the skull to stabilize the implantation. We then opened an approximately 4-mm-diameter craniotomy above lobule VI of cerebellar vermis (7.0 mm posterior to bregma, 0.0 mm lateral). We first injected a virus expressing GCaMP8m into the cerebellum (DV: −360 to 200 µm). After virus injection, we gently removed the dura with fine forceps (91197-00, FST). Next, we applied Kwik-Sil (World Precision Instruments) to the border of the craniotomy. We then covered the brain with a 3-mm-diameter coverslip that we attached beneath a 5-mm-diameter coverslip before the experiment using ultraviolet-light-activated epoxy (Norland Optical Adhesive #81, Thermo Fisher Scientific). We fixed the 5-mm-diameter coverslip to the cranium with adhesive cement (S380 Metabond Quick Adhesive Cement System, C&B) and dental cement (Lang Dental). After the cement dried (7–10 min), we transferred the animal to a heated pad for recovery. After full recovery, the mice were returned to their home cage.
Verification of microendoscope implantation and GCaMP expression in awake, behaving mice
Three weeks after the cannula implantation, we verified the GCaMP7f/8m fluorescence and Ca2+ activity in awake mice on a custom-designed apparatus to avoid using any general anaesthetics56. Mice were head-fixed by clamping (CC-1, Siskiyou) their headbar and were allowed to run on a freely rotating wheel (InnoWheel, Thermo Fisher Scientific). For imaging rACC→Pn neurons, a naked 1.0-mm-diameter gradient refractive index (GRIN) lens probe (1050-004598, Inscopix) was lowered into the implanted cannula using forceps. A miniature microscope (nVoke, Inscopix) was attached to a holder (1050-002199, Inscopix) connected to a goniometer (GN1, Thorlabs) for x–y and y–z plane tilting. The holder was connected to a three-axis micromanipulator for lowering the miniature microscope to the optimal focal plane. Image acquisition software (nVoke, Inscopix) was used to display the incoming image frames in units of relative fluorescence changes (ΔF/F), enabling observation of Ca2+ activity in awake, behaving mice. If we observed Ca2+ transients, we proceeded by mounting the miniature microscope baseplate.
Miniature microscope baseplate mounting
Mice were anaesthetized with isoflurane (4% for induction and 2% for maintenance) and placed onto the stereotaxic instrument. The GRIN lens probe was fixed in place with ultraviolet-light-curable epoxy (Loctite Light-Activated Adhesive, 4305). The miniature microscope with baseplate attached was stereotaxically lowered toward the top of the GRIN lens probe or coverslips until the brain tissue was in focus. We then adjusted the orientation of the miniature microscope until it was parallel to the surface of the GRIN lens probe. The baseplate was then fixed onto the skull with dental cement. To prevent external light from contaminating the imaging field of view during recording, the outer layer was coated with black nail polish (Black Onyx NL T02, OPI). After attaching the baseplate cover (1050-004639, Inscopix), the mice were transferred to a heated pad for recovery. The mice were then returned to their home cages and then housed individually.
Ca2+ imaging video recording, cell extraction and estimation of firing rates
To perform Ca2+ imaging in mice during PAC, we used an implanted GRIN lens for rACC→Pn neurons or a cranial window for cerebellar Purkinje cells and a miniaturized microscope (nVista, Inscopix). Miniature microscopes were mounted onto the head of the mouse before each behavioural experiment using a custom mounting station. Images were acquired using the Inscopix Data Acquisition Software (IDAS; Inscopix) at a frame rate of 20 Hz. The light-emitting diode intensity was set at 1.5 mW for imaging rACC→Pn neurons or 1 mW for imaging cerebellar Purkinje cell dendrites. A gain of 2 was used for all mice.
Before performing cell extraction, we first corrected for brain motion in the videos using the TurboReg62 motion-correction algorithm. Later, we extracted both the spatial filters and the robust time traces for the regions of interest (ROIs) using EXTRACT63, a robust cell extraction routine. We matched the resulting ROIs across days based on Tanimoto similarity, defined as \(T({\bf{x}},{\bf{y}})=\frac{{\bf{x}}\cdot {\bf{y}}}{| | \,{\bf{x}}\,| {| }^{2}+| | \,{\bf{y}}\,| {| }^{2}-{\bf{x}}\cdot {\bf{y}}}\), where x and y are vectors corresponding to flattened spatial filters. We adjusted the cut-off for the Tanimoto similarity to each mouse for best results evaluated by visual inspection, then reinitialized EXTRACT with the global cell map. This process allows EXTRACT to find cells that may be missed on different days. We performed this routine of cell extraction, across-day registration and reinitialization for five iterations. We next visually inspected all videos and discarded spurious, duplicate and dendritic ROIs. Lastly, using the verified ROIs that correspond to cells, we performed a final robust regression via EXTRACT to obtain the final traces. We took the z-score of the traces, subtracting the mean and dividing by the s.d., to standardize the trace units across days.
To obtain a discrete approximation of the firing rates, we first thresholded the raw traces by a constant value, chosen as 0.5× the maximum value of the trace. The thresholded traces were searched for peak points that were at least 3 frames apart from each other, and then binarized at these peaks (Extended Data Fig. 3c). The firing rates were obtained by convolving the binarized peaks through a Gaussian kernel with an s.d. of 500 ms. As we thresholded the traces at the beginning and binarized the peaks at the end before convolution, the resulting firing rates did not necessarily contain all of the event times, but should be considered a discrete approximation using the binarized versions of large Ca2+ events. We chose this approximation of firing rates for its robustness against day-to-day variations in the cell baselines and/or absolute ΔF/F values.
Analysis of neural responses during crossing times across days
For each condition, the mean activity of each neuron during border crossing times was calculated by averaging the z-scored neural activities (firing rates for Extended Data Fig. 3j–m) within a time window beginning 2 s before and ending 2 s after the crossing event. We chose the averaged activities inside the time window instead of performing a point estimation of neural activity. This choice was motivated by our desire to mitigate the measurement errors in crossing times from the behavioural videos and to remain agnostic to the fine structure of the neural code in short time durations, as our interest is in understanding whether the neural population reacts differently, corresponding to the level of pain relief expectation during crossing and crossing back, during each phase of PAC. To determine the extent to which the neural responses of individual cells may differ between first border crossing and crossing back, we computed a discriminability index (d′)2 for each neuron using the equation \({({d}^{{\prime} })}^{2}=\frac{{\mu }_{{\rm{forward}}}-{\mu }_{{\rm{backward}}}}{{\sigma }_{{\rm{pooled}}}}\,\), where μforward and μbackward are the mean activity of the neuron calculated during first crossing and crossing back, respectively. σpooled is the pooled s.d. from both conditions. To perform randomized controls for all analyses, we averaged the values of interest (discriminability index, z-scored traces and/or firing rates) with randomly selected crossing times for each neuron 100 times and created a null distribution over the neural population.
Classification of Purkinje cells
We conducted k-means clustering analysis to categorize Purkinje cells based on their activity during the first border crossing, first crossing back and the last crossing on the post-test day. We extracted the Ca2+ activity of each Purkinje cell during these events within the 4 s border crossing period for each condition and then concatenated them, resulting in a Ca2+ activity trace for each Purkinje cell with a total duration of 12 s. Subsequently, we performed silhouette analysis64 to determine the optimal number of clusters for this dataset, which we found to be two. Accordingly, we then applied k-means clustering to classify all cross-day-aligned Purkinje cells into two clusters.
Purkinje cell Ca2+ spike frequency and amplitude analysis
To find Ca2+ events in Purkinje cells, we first applied a threshold to the raw traces using a constant value set at 0.1 times the maximum amplitude of the trace. The thresholded traces were then scanned for peaks that were at least 3 frames apart from each other and binarized at these peaks. For frequency analysis, we counted the number of Ca2+ events in the binarized traces of each cell during border crossing and calculated their firing frequency during this 4 s border-crossing period (Extended Data Fig. 9j,l). For Ca2+ spike amplitude analysis, we extracted the value from the z-scored Ca2+ traces of each cell at the timepoint of detected Ca2+ events as the amplitude. We then selected these amplitudes during the 4 s border-crossing time for comparison (Extended Data Fig. 9l,m). For Ca2+ spike waveform analysis, we used the z-scored trace of each Purkinje cell to identify the start, peak and end points of each spike that had an amplitude larger than 3 z-scored ΔF/F (Extended Data Fig. 9n).
Histology
Tissue collection and processing
Mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 4% formaldehyde in PBS. Brains were then dissected, post-fixed in 4% formaldehyde, and cryoprotected in 30% sucrose. Tissues were then frozen in Optimum Cutting Temperature compound (OCT; 4583, Tissue Tek) and sectioned using a cryostat (Leica). The brains were sectioned at 40 μm and stored in PBS at 4 °C if used immediately. For longer storage, tissue sections were placed in glycerol-based cryoprotectant solution and stored at −20 °C. For in situ hybridization, tissues were sectioned at 14 μm, collected on Superfrost Plus slides (22-037-246, Thermo Fisher Scientific) and stored at −80 °C.
Immunohistochemistry
Tissues were incubated for 1 h and blocked in 0.1 M PBS with 0.3% Triton X-100 (Sigma-Aldrich) plus 5% normal donkey serum. Primary and secondary antibodies were diluted in 0.1 M PBS with 0.3% Triton X-100 plus 1% normal donkey serum. The sections were then incubated overnight at 4 °C in primary antibody solution, washed in 0.1 M PBS with 0.3% Triton X-100 for 40 min, incubated for 2 h in secondary antibody at room temperature and washed in 0.1 M PBS for 40 min. The sections were then mounted using Fluoromount-G (00-4958-02, Thermo Fisher Scientific). Images were acquired on the Zeiss LSM 780 confocal microscope using Zeiss Zen software, running on a Windows PC, in the UNC Neuroscience Microscopy Core. A streptavidin conjugate (Alexa Fluor 594 conjugate, Thermo Fisher Scientific; 1:1,000) was used to visualize biocytin.
In situ hybridization
For in situ hybridization experiments, we used Advanced Cell Diagnostics RNAscope Technology (ACD Bioscience). In brief, wild-type mice 5–8 weeks old were deeply anaesthetized with 0.1 ml of Euthasol (NDC-051311-050-01, Virbac) and perfused transcardially with 0.1 M PBS followed by 4% formaldehyde solution in PBS. Brains were dissected, cryoprotected in 30% sucrose overnight and then frozen in OCT. Frozen tissue was cut at 20 μm onto Superfrost Plus slides and stored at −80 °C. Tissue was thawed from −80 °C, washed with PBS at room temperature and subsequently processed according to the protocol provided by the manufacturer. We first pretreated the tissue with solutions from the pretreatment kit to permeabilize the tissue, then incubated with protease for 30 min and then with the hybridization probe(s) for another 2 h at 40 °C.
Data analysis for the output of TRAPed rACC neurons
To quantify the output of neurons in the rACC TRAPed after PAC, we analysed the expression of mRuby in putative presynaptic axonal terminals. First, background subtraction was performed on the mRuby channels of each image using the rolling-ball algorithm in ImageJ. Subsequently, the images were thresholded to a value 4 times the mean background intensity, converting them into binary format. The pixel densities of these binary images were then calculated and normalized to the size of the specific regions displaying mRuby expression.
Electrophysiology
ACC slice preparation
We began the PAC paradigm 3–4 weeks after virus injection. Mice were euthanized immediately after the conditioning phase of PAC. After decapitation, the brain was rapidly collected and immersed in ice-cold slicing solution containing 87 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM d-glucose, 75 mM sucrose, 0.5 mM CaCl2 and 7 mM MgCl2 (pH 7.4 in 95% O2 and 5% CO2, 325 mOsm). Coronal brain slices 300 μm thick and containing the rACC were cut using a VT1200 vibratome (Leica Microsystems). After around 20 min incubation at 35 °C, the slices were stored at room temperature. Slices were then transferred to the chamber for electrophysiological recording. Slices were used for a maximum of 5 h after dissection. The experiments were performed at 21–24 °C.
During the experiment, slices were superfused with a physiological extracellular solution containing 125 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, 25 mM d-glucose, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4 in 95% O2 and 5% CO2, ~325 mOsm). Whole-cell patch recording of rACC→Pn neurons was performed as described previously65. The pipettes (1B150F-4, WPI) were formed using a P-97 puller (Sutter Instruments). The resistance was 3–5 MΩ.
Measuring action potential properties, spontaneous release and LTP induction
The intracellular solution used for testing the action potential firing properties, spontaneous release and LTP induction of rACC→Pn neurons contained 135 mM K-gluconate, 20 mM KCl, 0.1 mM EGTA, 2 mM MgCl2, 2 mM Na2ATP, 10 mM HEPES and 0.3 mM Na3GTP (pH adjusted to 7.28 with KOH, ~310 mOsm); in a subset of recordings, 0.2% biocytin was added. To measure membrane properties and evoke action potential firing of rACC→Pn neurons, a 1 s step current (−50, 0, 50, 100, 150, 200, 250, 300 pA) was injected into the cell through the recording pipette. Spontaneous EPSCs were recorded while holding the rACC→Pn neurons at −70 mV.
For LTP induction, biphasic electrical stimulations (5–8 V, 100 ms) were delivered by placing a borosilicate theta glass (2.0 mm, Warner Instruments) in layer II/III of the rACC. The glass was pulled using a vertical pipette puller and filled with perfusion solution. The fibre was stimulated using the DS4 Bi-Phasic Current Stimulator (Digitimer) at 0.02 Hz to measure evoked EPSCs of the rACC→Pn neurons for 6 min as the baseline. TBS (5 trains of burst with 4 pulses at 100 Hz, at 200 ms intervals, repeated 4 times at intervals of 10 s) was then administered to induce LTP. After LTP induction, evoked EPSCs were recorded for another 30 min to compare against the baseline. No blocker was used to block inhibitory synaptic inputs.
Measuring AMPA/NMDA ratio, PPR and feedforward inhibition
The Cs+-based intracellular solution used to measure the AMPA/NMDA ratio and PPR contained 130 mM Cs-methanesulfonate, 2 mM KCl, 10 mM EGTA, 2 mM MgCl2, 2 mM Na2ATP, 10 mM HEPES and 5 mM QX-314 (pH adjusted to 7.28 with CsOH, ~310 mOsm). To evoke synaptic response of rACC→Pn neurons, electrical stimulation (50–80 μA, 100 μs) was delivered by placing a concentric bipolar electrode (FHC) in layer II/III of the rACC. The selective GABAA receptor antagonist SR-95531 (10 μM; Sigma-Aldrich) was used to block IPSCs. To record EPSCs mediated by both AMPA and NMDA receptors, membrane potentials were held at voltages increasing from −80 mV to +60 mV. To measure the PPR, two electrical stimulations at different time intervals (20, 50, 100, 200, 500 ms) were used to evoke synaptic transmission. The membrane potential was set to −30 mV to record both EPSCs and IPSCs in the same trace (Extended Data Fig. 6o) or to either −70 mV or +10 mV to examine EPSCs or IPSCs in isolation (Fig. 2o).
Measuring inhibitory input from PV+ interneurons
Slices from Pvalbcre mice were prepared as described above. An intracellular solution containing high chloride concentration (140 mM KCl, 10 mM EGTA, 2 mM MgCl2, 2 mM ATP, 10 mM HEPES and 2 mM QX-314; pH adjusted to 7.28 with KOH; 313 mOsm) was used for postsynaptic recordings of rACC→Pn neurons, which were conducted in the voltage-clamp configuration with a holding potential of −70 mV.
For all voltage-clamp recordings, we applied hyperpolarizing test pulses (5 mV, 100 ms) to monitor series and input resistance throughout the entire experiment. Data from experiments in which series resistance changed more than 15% were discarded.
Data acquisition and analysis
Electrophysiological data were acquired using the Multiclamp 700b amplifier (Axon Instruments), low-pass filtered at 10 kHz, and sampled at 20 or 50 kHz using the Digidata 1440A low-noise digitizer (Axon Instruments). Stimulation and data acquisition were performed using Clampfit 10 software (Axon Instruments). Data were analysed using Stimfit v.0.14.9 (https://github.com/neurodroid/stimfit), Clampfit v.11.2 (Molecular Devices) and R v.4.0.3 (The R Project for Statistical Computing). sEPSCs were detected using a template-matching algorithm and verified by visual inspection66. The location at which the peak EPSC was recorded while holding the membrane potential at −80 mV was used to measure the amplitude of AMPAR EPSCs. The amplitude of NMDAR EPSCs was measured 50 ms after the electrical stimulation. The synaptic latency of monosynaptic EPSCs or IPSCs was measured from the onset of the electrical stimulus to the onset of the EPSC or IPSC. The disynaptic IPSC delay (Fig. 2o) was measured from the onset of the EPSC at −70 mV to the onset of the IPSC at +10 mV.
Cannula implantation and optogenetic manipulations
For fibreoptic cannula implantation surgeries, mice were anaesthetized with isoflurane (4% for induction and 2% for maintenance) while the body temperature was maintained using a heating pad. After cranial hair removal, skin sterilization and scalp incision, we bilaterally injected a virus encoding inhibitory or excitatory opsin into the ACC using the coordinates described above for manipulating ACC terminals in the Pn. To manipulate the activity of Oprd1+ cells in the Pn, we bilaterally injected a virus encoding an inhibitory opsin into the Pn at the coordinates AP: −4.0 mm, ML: ±0.4 mm, DV: −5.4/−5.8 mm. To manipulate the activity of Oprd1+ cells in the Pn that receive rACC inputs, we bilaterally injected the AAV1-Flpo virus into the rACC, then another virus into the Pn to express an inhibitory opsin in a Cre- and Flp-dependent manner. After virus injection, we performed small craniotomies in three locations (AP: +5.10, −1.06, −3.56 mm; ML: −0.77, +2.87, −3.13 mm). Next, to stabilize the implantation, three stainless steel screws (MX-000120-01SF, Component Supply Company) were drilled into the dura of the skull. We then performed two additional craniotomies at the coordinates AP: −4.0 mm, ML: ±1.2 mm. The cannula (CFMLC12L05, Thorlabs or RWD) was then attached to a holder (David Kopf Instruments) and lowered at 10° to the coordinates AP: +4.0 mm, ML: ±1.2 mm, DV: −4.9 mm. Blood and debris around the craniotomy were quickly removed and adhesive cement (S380 Metabond Quick Adhesive Cement System, C&B) was used to seal the gap between the cannula and skull. A custom-designed laser-cut headbar (18–24 G stainless steel, LaserAlliance) was placed over the left posterior skull screw, then layers of dental cement were applied (Lang Dental) to affix both the cannula and headbar to the skull. After the cement dried (7–10 min), we transferred the animal to a heated pad until full recovery, then to their home cage.
For optogenetic photostimulation of inhibitory (eNpHR3.0) or excitatory (ChR2) opsins, ferrules were connected to a 561 nm (yellow) laser diode (MGL-FN-561, Opto Engine) or 494 nm (blue) laser diode (MBL-III-473, Opto Engine LLC) using a FC/PC adaptor and a fibreoptic rotary joint (Thorlabs). The laser output was controlled using a shutter controller (SR470, Stanford Research System), which delivered yellow light continuously for the inhibitory opsin and 4 ms blue light pulses at 20 Hz for the excitatory opsin. Light output through the optical fibres was adjusted to ~5 mW at the tip of the optical fibre for inhibition and ~10 mW at the tip of the optical fibre for excitation.
Behavioural tests
For all behavioural assays described below, mice were acclimatized to the researcher and testing environment for at least 30 min before testing.
PAC assay to induce and evaluate placebo analgesia
The PAC apparatus consists of two adjacent and visually distinct chambers, using two separate thermal plates (BIOSEB) as the floor. PAC is a 7 day behavioural assay consisting of three phases: habituation (days 1–2) and pre-test (day 3), conditioning (days 4–6), and post-test (day 7; Fig. 1a). During the habituation and pre-test phases, the floors of both chambers are set at 30 °C and the mice are free to explore both compartments for 3 min; their performance on the pre-test day is compared with their performance on the post-test day. During the conditioning phase, the floor of the chamber on which the mouse begins the session (chamber 1) is set at 48 °C. Mice progressively learn that chamber 1 is painful and to associate chamber 2, which remains at 30 °C, with pain relief. On the post-test day, the floors of both chambers are set at 48 °C to evaluate any analgesic effect induced by the expectation of pain relief. The performance of mice was recorded for 3 min using a camera (acA1300, Basler) controlled by MATLAB (R2019b, MathWorks). The recorded videos were analysed using the machine-learning-based algorithm DeepLabCut67 or Ethovision XT15 (Noldus). We quantified and compared the latency of border crossings, time spent in each chamber and nocifensive behaviours (licking, rearing, jumping) of conditioned and unconditioned mice (Fig. 1).
Naloxone injection
To investigate whether endogenous opioid activity is necessary for PAC-induced placebo analgesia, we injected mice with saline or naloxone (N7758, Sigma-Aldrich) intraperitoneally (5 mg per kg) during the conditioning phase (day 4 to 6; Extended Data Fig. 1g–i) or before the post-conditioning test on day 7 (Extended Data Fig. 1d–f). After injection, the mice were returned to their home cage for at least 30 min to reduce injection-induced stress. Saline-injected mice were used as controls.
TRAP of rACC neurons during PAC
Two weeks after virus injection, TRAP2 mice were subjected to an adjusted PAC assay (30 min conditioning phase on days 4–6 instead of 3 min) to label the rACC neurons encoding expectation of pain relief. TRAP2 mice were injected with 4-hydroxytamoxifen (50 mg per kg, subcutaneous) on the last day of the conditioning phase (day 6) immediately before conducting the PAC trial. After injection, the mice were allowed to remain in the PAC apparatus for 30 min, and then returned to their home cages. Then, 2 weeks later, we perfused the mice and dissected the brains to determine synaptophysin–mRuby expression in the rACC and other brain areas. Mice that underwent the same procedure but with both chambers set at 30 °C were used as controls.
Pin prick
To examine the Ca2+ activity of rACC→Pn neurons during noxious mechanical stimulation (Extended Data Fig. 7), we gently touched the plantar surface of the hindpaw with a 25 G needle 10 times at an interval of around 30 s (Extended Data Fig. 7a). As a control, a needle with a blunt end was used to measure the Ca2+ activity of rACC→Pn neurons during innocuous mechanical stimulation. The entire procedure was recorded using a camera (acA1300, Basler) controlled by MATLAB (R2019b, MathWorks) and synchronized with the miniscope.
Hindpaw radiant heat (Hargreaves) test
To examine the Ca2+ activity of rACC→Pn neurons during noxious thermal stimulation (Extended Data Fig. 5), we used the Hargreaves test. Mice were placed in plastic chambers on a glass surface heated to 25 °C, through which a radiant heat source (Department of Anesthesiology, UC San Diego) could be focused onto the hindpaw. We recorded the performance of mice using a camera (acA1300, Basler) controlled by MATLAB (R2019b, MathWorks) and synchronized with the miniscope.
Von Frey withdrawal threshold test
Eight von Frey filaments (Stoelting), ranging from 0.007 to 6.0 g were used to assess mechanical withdrawal thresholds. Filaments were applied perpendicular to the ventral–medial hindpaw surface with sufficient force to cause a slight bending of the filament. A positive response was characterized by a rapid withdrawal of the paw away from the stimulus fibre within 4 s. The up–down method was used to determine the mechanical threshold (50% withdrawal threshold)68.
Von Frey withdraw frequency test
To evaluate mechanical sensitivity, we used six von Frey filaments (0.07, 0.16, 0.4, 1.0, 1.4 and 6.0 g). Filaments were applied perpendicular to the ventral–medial hindpaw surface with sufficient force to cause a slight bending of the filament. Each filament was applied for 1 s. A positive response was characterized by a rapid and immediate withdrawal of the paw away from the filament. Each filament was applied five times. The frequency of reflexive withdrawal responses was calculated.
Hotplate test
Mice were acclimatized to the testing environment as described above. The plate temperature was set at 48 °C or 52 °C to measure thermal pain threshold. The mouse was placed onto the plate and the latency preceding licking and/or biting of a hindpaw was scored. To prevent tissue damage, a cut-off of 3 min or 1 min was set for the 48 °C and 52 °C plates, respectively.
Formalin test
An intraplantar injection (20 µl) of 2.5% formalin was performed in the left hindpaw of mice after the conditioning phase of PAC. The mouse behaviour was recorded for 30 min within the PAC apparatus or using a four-camera set-up enabling synchronized capture of each lateral angle. The time spent licking the injected hindpaw was scored using Ethovision XT15 (Noldus) or automatically scored using DeepEthogram, an unbiased, pixel-based machine learning algorithm69.
scRNA-seq and snRNA-seq
Sample preparation, library generation and sequencing
For low-throughput, high-depth scRNA-seq, we used the SMART-seq v4 Ultra Low Input RNA Kit for Sequencing (SSv4; TakaraBio) as described previously70. To focus our high-depth analysis on neurons, we used Snap25-IRES2-cre;Ai14 (Snap25-tdT) mice, which express the fluorescent reporter tdTomato in neurons. The Pn was microdissected from two 8-week-old Snap25-tdT mice (one male and one female). The mice were anaesthetized with isoflurane and perfused with artificial cerebrospinal fluid comprising CaCl2 (0.5 mM), glucose (25 mM), HCl (96 mM), HEPES (20 mM), MgSO4 (10 mM), NaH2PO4 (1.25 mM), myo-inositol (3 mM), N-acetylcysteine (12 mM), NMDG (96 mM), KCl (2.5 mM), NaHCO3 (25 mM), sodium l-ascorbate (5 mM), sodium pyruvate (3 mM), taurine (0.01 mM) and thiourea (2 mM), bubbled with carbogen (95% O2 and 5% CO2). The Pn was microdissected and embedded in 2% agarose, sliced into 250 µm sections with a vibratome, then subjected to enzymatic digestion with pronase (1 mg ml−1) for 70 min at room temperature and triturated using fire-polished Pasteur pipettes to generate single-cell suspensions. Live single Pn neurons were isolated into eight-well strips containing SSv4 lysis buffer based on DAPI−tdTomato+ using fluorescence-activated cell sorting, then stored at −80 °C. To prepare single-cell transcriptome libraries, polyadenylated RNAs were reverse transcribed into full-length cDNA and subjected to 18 PCR amplification cycles according to the SSv4 protocol. Single-cell libraries were indexed and prepared for Illumina sequencing using the Nextera XT DNA Library Preparation Kit. Multiplexed libraries were sequenced on the HiSeq 2500 sequencers to generate 100 bp paired-end reads at a depth of 2.5 million reads per cell. Single-cell FastQ files were aligned to the mm10 mouse genome (GRCm38) using STAR (v.2.7.3a)71.
For high-throughput, low-depth single-nucleus RNA-seq (snRNA-seq) we used the 10x Chromium 3′ V3 System (10x Genomics). We microdissected the Pn from two 8-week-old female C57BL/6J mice. Pn tissues were pooled and flash-frozen on dry ice. Single-nucleus isolation was performed as described previously72. Tissue was placed into a prechilled Dounce homogenizer (Kimble) containing 500 µl chilled detergent lysis buffer (0.10% Triton X-100, 0.32 M sucrose, 10 mM HEPES (pH 8.0), 5 mM CaCl2, 3 mM MgAc, 0.1 mM EDTA, 1 mM dithiothreitol (DTT)). Tissue was homogenized by five strokes with the ‘loose’ pestle followed by ten strokes with the ‘tight’ pestle (Kimble). Then, 1 ml of sucrose buffer (0.32 M sucrose, 10 mM HEPES (pH 8.0), 5 mM CaCl2, 3 mM MgAc, 0.1 mM EDTA, 1 mM DTT) was added to the Dounce homogenizer and the combined solution was passed through a 40 μm cell strainer into a fresh tube containing 1 ml of 0.32 M sucrose buffer. An additional 1 ml of 0.32 M sucrose buffer was passed through the filter and the resulting 3.5 ml solution was centrifuged at 3,200g for 10 min at 4 °C. The pellet was resuspended with 3 ml of 0.32 M sucrose buffer and homogenized for 30 s (Ultra-Turrax disperser, setting 1). Next, 12.5 ml of 1 M sucrose buffer (1 M sucrose, 10 mM HEPES (pH 8.0), 3 mM MgAc, 1 mM DTT) was pipetted beneath the homogenate and the tube was centrifuged at 3,200g for 20 min at 4 °C. After decanting the supernatant, the pellet was resuspended in 1 ml of resuspension solution (0.4 mg ml−1 BSA, 0.2 U μl−1 RNase inhibitor (Lucigen) in 1× PBS), filtered through a 35 µm cell strainer and diluted to a final concentration of 225 cells per µl.
Single-nucleus suspensions were loaded onto two 10x Genomics chips (Chromium v3). snRNA-seq libraries were constructed according to the protocol provided by the manufacturer. Multiplexed snRNA-seq libraries were spiked with a PhiX control library (5%) and sequenced across two NextSeq 550 high-output flow-cell runs. Raw sequencing files were aligned to the mm10 mouse genome (GRCm38) and converted to gene expression matrices using the Cell Ranger pipeline (Cell Ranger v.5.0.1, default parameters). Intronic reads were included to increase assay sensitivity.
Normalization, clustering and differential gene expression
scRNA-seq data were analysed using Seurat (v.4.0)73. For 10x Genomics datasets, nuclei expressing fewer than 200 genes and genes expressed in fewer than 5 nuclei were removed. For SSv4 data, cells expressing fewer than 1,000 genes and genes expressed in fewer than 5 cells were removed. To focus our analysis on neurons, we performed broad preliminary clustering to define principal cell types and remove cells and nuclei that lacked expression of neuronal genes (Snap25 and Rbfox3) or expressed conventional glial cell markers (Mbp, Pdgfra, Gfap, Csf1r and Pecam1). The final datasets comprised 4,720 neuronal nuclei from 10x experiments (8,669 median transcripts per cell; 3,816 median genes per cell) and 212 neuronal cells from SSv4 experiments (481,098 median transcripts per cell; 9,956 median genes per cell). Each scRNA-seq dataset was normalized and transformed to a common scale separately using SCTransform with the following parameters: ncells = half the total number of cells; variable.features = median number of genes expressed per cell. The resulting datasets were integrated by SCT-Pearson residuals usinng Seurat’s FindIntegrationAnchors and IntegrateData functions using the default parameters.
We determined which principal components to use in subsequent clustering analyses by manually evaluating which principal components contributed to substantial variation (ElbowPlot function in Seurat). To increase cluster robustness, the optimal nearest neighbour parameter (k) was identified by iterating through nearest-neighbour values (FindNeighbors function in Seurat) and calculating the average silhouette score74. The k-nearest-neighbour value yielding the highest average silhouette score was used for Louvain clustering. Pairs of clusters that could not be reliably distinguished by a single gene using a binomial test (q < 0.01; log-effect size > 2.0) were dissolved and cells reassigned to the nearest cluster based on Euclidean distance in principal component (PC) space74. An initial round of clustering using this method was performed to detect principal cell types (for example, neurons, microglia, astrocytes). A subsequent round of clustering was performed on neuronal principal cell types based on enrichment of neuron-specific genes (Snap25, Rbfox3) and neurotransmitter vesicular transporters (Slc17a6, Slc17a7, Slc32a1).
Cell-type-specific marker genes were identified using a binomial test to determine which genes are expressed in cells within a given cluster compared to all other cells75. The expression frequency of a given gene (g) expressed in a specific cell population (N) was compared to the expression frequency in the remaining population (M). Thus, the P value for this test was calculated as follows: \({p}_{g}={\sum }_{k={N}_{g}}^{N}C(N,k)\gamma \wedge (k)(1-\gamma )\wedge (N-k)\), where γ is the proportional frequency of cells expressing the gene of interest (Mg/M). A complete list of cluster-specific marker genes is provided in Supplementary Table 1.
Statistics and reproducibility
Statistical analysis was performed using R v.4.0.3 (The R Project for Statistical Computing). All values are reported as mean ± s.e.m. Statistical significance was tested using two-sided Wilcoxon rank-sum tests, two-sided Wilcoxon matched-pairs signed-rank tests or one- or two-way ANOVA with Tukey post hoc test. P < 0.05 was considered to be significant. P values between 0.05 and 0.1 were noted in the figures. In experiments involving electrical fibre stimulation, stimulation artifacts were blanked for display purposes. In Figs. 1h,j and 4e, two mice were examined in each group, and similar results were generated. In Extended Data Figs. 8e and 9a–c, three independent repeats were performed with similar results and representative images were shown. In Extended Data Figs. 2a–e and 8g, two independent repeats were performed with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
scRNA-seq data are available through the Gene Expression Omnibus under accession numbers GSE267264 for SMART-seq and GSE267265 for 10x Genomics data. All raw data for behavioural, electrophysiological and immunohistochemical analyses are provided in the Article and its Supplementary Information. Processed Ca2+ imaging data are available at GitHub (https://github.com/ScherrerLab/PontineNucleus2024/). Raw Ca2+ imaging data are available on request due to the large file size. Source data are provided with this paper.
Code availability
We used the open-source software TurboReg62 for image registration (http://bigwww.epfl.ch/thevenaz/turboreg/) and EXTRACT63 for extracting individual neurons and their calcium activity traces from videos (https://github.com/schnitzer-lab/EXTRACT-public). We used R v.4.0.3 (The R Project for Statistical Computing) to write custom code for the following data processing: code for generating clusters and visualizations from raw count matrices and for processing Ca2+ imaging, behavioural and electrophysiology data. The R code is available at GitHub (https://github.com/ScherrerLab/PontineNucleus2024/).
References
Harrington, A. The Cure Within: A History of Mind-Body Medicine (W. W. Norton & Company, 2008).
Putnam, H. The Threefold Cord: Mind, Body, and World (Columbia Univ. Press, 1999).
Bingel, U. et al. The effect of treatment expectation on drug efficacy: imaging the analgesic benefit of the opioid remifentanil. Sci. Transl. Med. 3, 70ra14 (2011).
Colloca, L. & Benedetti, F. Placebo analgesia induced by social observational learning. Pain 144, 28–34 (2009).
Fields, H. L. How expectations influence pain. Pain 159, S3–S10 (2018).
Wager, T. D. & Atlas, L. Y. The neuroscience of placebo effects: connecting context, learning and health. Nat. Rev. Neurosci. 16, 403–418 (2015).
Finniss, D. G., Kaptchuk, T. J., Miller, F. & Benedetti, F. Biological, clinical, and ethical advances of placebo effects. Lancet 375, 686–695 (2010).
Bingel, U. Placebo 2.0: the impact of expectations on analgesic treatment outcome. Pain 161, S48–S56 (2020).
Gupta, U. & Verma, M. Placebo in clinical trials. Perspect. Clin. Res. 4, 49–52 (2013).
Ploghaus, A. et al. Dissociating pain from its anticipation in the human brain. Science 284, 1979–1981 (1999).
Petrovic, P., Kalso, E., Petersson, K. M. & Ingvar, M. Placebo and opioid analgesia—imaging a shared neuronal network. Science 295, 1737–1740 (2002).
Wager, T. D. et al. Placebo-induced changes in FMRI in the anticipation and experience of pain. Science 303, 1162–1167 (2004).
Bingel, U., Lorenz, J., Schoell, E., Weiller, C. & Büchel, C. Mechanisms of placebo analgesia: rACC recruitment of a subcortical antinociceptive network. Pain 120, 8–15 (2006).
Eippert, F. et al. Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron 63, 533–543 (2009).
Zunhammer, M., Spisák, T., Wager, T. D., Bingel, U. & Placebo Imaging Consortium. Meta-analysis of neural systems underlying placebo analgesia from individual participant fMRI data. Nat. Commun. 12, 1391 (2021).
Grevert, P., Albert, L. H. & Goldstein, A. Partial antagonism of placebo analgesia by naloxone. Pain 16, 129–143 (1983).
Enck, P. & Klosterhalfen, S. Does sex/gender play a role in placebo and nocebo effects? Conflicting evidence from clinical trials and experimental studies. Front. Neurosci. 13, 160 (2019).
Baker, A. et al. Specialized subpopulations of deep-layer pyramidal neurons in the neocortex: bridging cellular properties to functional consequences. J. Neurosci. 38, 5441–5455 (2018).
Craig, A. D., Bushnell, M. C., Zhang, E. T. & Blomqvist, A. A thalamic nucleus specific for pain and temperature sensation. Nature 372, 770–773 (1994).
Huang, T. et al. Identifying the pathways required for coping behaviours associated with sustained pain. Nature 565, 86–90 (2019).
Barceló, A. C., Filippini, B. & Pazo, J. H. The striatum and pain modulation. Cell. Mol. Neurobiol. 32, 1–12 (2012).
Sclocco, R. et al. Neuroimaging brainstem circuitry supporting cardiovagal response to pain: a combined heart rate variability/ultrahigh-field (7 T) functional magnetic resonance imaging study. Philos. Trans. A Math. Phys. Eng. Sci. 374, 20150189 (2016).
Hubbard, C. S. et al. Estrogen-dependent visceral hypersensitivity following stress in rats: An fMRI study. Mol. Pain 12, 1744806916654145 (2016).
Ruscheweyh, R. et al. Altered experimental pain perception after cerebellar infarction. Pain 155, 1303–1312 (2014).
Sallet, J. et al. Expectations, gains, and losses in the anterior cingulate cortex. Cogn. Affect. Behav. Neurosci. 7, 327–336 (2007).
Colloca, L. & Miller, F. G. How placebo responses are formed: a learning perspective. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 1859–1869 (2011).
Malenka, R. C. The long-term potential of LTP. Nat. Rev. Neurosci. 4, 923–926 (2003).
Owen, S. F., Berke, J. D. & Kreitzer, A. C. Fast-spiking interneurons supply feedforward control of bursting, calcium, and plasticity for efficient learning. Cell 172, 683–695 (2018).
Hu, H., Gan, J. & Jonas, P. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 345, 1255263 (2014).
Rancz, E. A. et al. High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature 450, 1245–1248 (2007).
Stoodley, C. J. & Schmahmann, J. D. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 46, 831–844 (2010).
Ghosh, K. K. et al. Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878 (2011).
Wang, S. S., Denk, W. & Häusser, M. Coincidence detection in single dendritic spines mediated by calcium release. Nat. Neurosci. 3, 1266–1273 (2000).
Roome, C. J. & Kuhn, B. Simultaneous dendritic voltage and calcium imaging and somatic recording from Purkinje neurons in awake mice. Nat. Commun. 9, 3388 (2018).
Roome, C. J. & Kuhn, B. Dendritic coincidence detection in Purkinje neurons of awake mice. eLife 9, e59619 (2020).
Mukamel, E. A., Nimmerjahn, A. & Schnitzer, M. J. Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760 (2009).
De Gruijl, J. R., Hoogland, T. M. & De Zeeuw, C. I. Behavioral correlates of complex spike synchrony in cerebellar microzones. J. Neurosci. 34, 8937–8947 (2014).
Melzack, R. & Casey, K. L. The Skin Senses (ed. Kenshalo, D. R.) 423–443 (Charles C. Thomas, 1968).
Keller, A., Akintola, T. & Colloca, L. Placebo analgesia in rodents: current and future research. Int. Rev. Neurobiol. 138, 1–15 (2018).
Popa, L. S. & Ebner, T. J. Cerebellum, predictions and errors. Front. Cell. Neurosci. 12, 524 (2018).
Timmann, D. et al. The human cerebellum contributes to motor, emotional and cognitive associative learning. A review. Cortex 46, 845–857 (2010).
Wagner, M. J., Kim, T. H., Savall, J., Schnitzer, M. J. & Luo, L. Cerebellar granule cells encode the expectation of reward. Nature 544, 96–100 (2017).
Wagner, M. J. et al. Shared cortex-cerebellum dynamics in the execution and learning of a motor task. Cell 177, 669–682 (2019).
Chen, C. H. et al. A Purkinje cell to parabrachial nucleus pathway enables broad cerebellar influence over the forebrain. Nat. Neurosci. 26, 1929–1941 (2023).
Frontera, J. L. et al. Bidirectional control of fear memories by cerebellar neurons projecting to the ventrolateral periaqueductal grey. Nat. Commun. 11, 5207 (2020).
Vaaga, C. E., Brown, S. T. & Raman, I. M. Cerebellar modulation of synaptic input to freezing-related neurons in the periaqueductal gray. eLife 9, e54302 (2020).
Economo, M. N. et al. Distinct descending motor cortex pathways and their roles in movement. Nature 563, 79–84 (2018).
Rojas-Piloni, G. et al. Relationships between structure, in vivo function and long-range axonal target of cortical pyramidal tract neurons. Nat. Commun. 8, 870 (2017).
Bissière, S., Humeau, Y. & Lüthi, A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat. Neurosci. 6, 587–592 (2003).
Gaffield, M. A., Rowan, M. J. M., Amat, S. B., Hirai, H. & Christie, J. M. Inhibition gates supralinear Ca2+ signaling in Purkinje cell dendrites during practiced movements. eLife 7, e36246 (2018).
Dhaka, A., Viswanath, V. & Patapoutian, A. Trp ion channels and temperature sensation. Annu. Rev. Neurosci. 29, 135–161 (2006).
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009).
Han, S., Soleiman, M. T., Soden, M. E., Zweifel, L. S. & Palmiter, R. D. Elucidating an affective pain circuit that creates a threat memory. Cell 162, 363–374 (2015).
Rodriguez, E. et al. A craniofacial-specific monosynaptic circuit enables heightened affective pain. Nat. Neurosci. 20, 1734–1743 (2017).
Liu, Y. et al. Touch and tactile neuropathic pain sensitivity are set by corticospinal projections. Nature 561, 547–550 (2018).
Corder, G. et al. An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281 (2019).
Choi, S. et al. Parallel ascending spinal pathways for affective touch and pain. Nature 587, 258–263 (2020).
Mercer Lindsay, N., Chen, C., Gilam, G., Mackey, S. & Scherrer, G. Brain circuits for pain and its treatment. Sci. Transl. Med. 13, eabj7360 (2021).
Gan, Z. et al. Layer-specific pain relief pathways originating from primary motor cortex. Science 378, 1336–1343 (2022).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657 (2019).
Zingg, B. et al. AAV-mediated anterograde transsynaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93, 33–47 (2017).
Thévenaz, P., Ruttimann, U. E. & Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998).
Inan, H. et al. Fast and statistically robust cell extraction from large-scale neural calcium imaging datasets. Preprint at bioRxiv https://doi.org/10.1101/2021.03.24.436279 (2021).
Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987).
Chen, C., Arai, I., Satterfield, R., Young, S. M. Jr & Jonas, P. Synaptotagmin 2 is the fast Ca2+ sensor at a central inhibitory synapse. Cell Rep. 18, 723–736 (2017).
Pernía-Andrade, A. J. et al. A deconvolution-based method with high sensitivity and temporal resolution for detection of spontaneous synaptic currents in vitro and in vivo. Biophys. J. 103, 1429–1439 (2012).
Mathis, A. et al. DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat. Neurosci. 21, 1281–1289 (2018).
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 (1994).
Bohnslav, J. P. et al. DeepEthogram, a machine learning pipeline for supervised behavior classification from raw pixels. eLife 10, e63377 (2021).
Yao, Z. et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Matson, K. J. E. et al. Isolation of adult spinal cord nuclei for massively parallel single-nucleus RNA sequencing. J. Vis. Exp. https://doi.org/10.3791/58413 (2018).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Loo, L. et al. Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun. 10, 134 (2019).
Shekhar, K. et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 166, 1308–1323 (2016).
Acknowledgements
This project was supported by the National Institutes of Health grants R01NS106301 (G.S.) and R01DA054583 (G.S., M.J.S. and H.Z.), the New York Stem Cell Foundation (G.S.), the Stanford School of Medicine Dean’s Fellowship (C.C.), seed funds from Biogen (G.S.), the Stanford Mind, Brain, Computation and Technology Program (F.D. and M.J.S.), the Vannevar Bush Faculty Fellowship (M.J.S.). We thank B. Ahanonu for training C.C. in miniscope cannula preparation and implantation, and related data acquisition and analysis at the beginning of this project; K. Kolar for discussions about Ca2+ imaging analysis; W. McCallum for technical assistance; J. Blair for manuscript discussions and editing; and M. Ehlers and the members of his former group at Biogen for providing seed funding and for engaging discussions at the beginning of this project.
Author information
Authors and Affiliations
Contributions
C.C. and G.S. conceptualized and designed the project. G.S., M.J.S., H.Z. and A.W.H. acquired funding, critical equipment and resources for the project and supervised work at UNC, Stanford, the Allen Institute for Brain Science, Janelia Research Campus/UNC, respectively. C.C. performed the Ca2+ imaging experiments, and F.D. and C.C. conducted the analysis. C.C. performed patch-clamp recording, viral tracing, behavioural experiments and data analysis. A.W.H., L.W., A.L., V.M. and H.Z. collected and provided the Pn scRNA-seq data, and J.K.N. performed the analysis. K.L.H. performed the in situ hybridization. A.L.B. conducted the rotarod test. C.C., K.L.H. and A.T. conducted the formalin test. S.A.S. generated a DNA targeting construct and screened ES cells to generate Oprd1cre mice. K.R. produced rabies and helper viruses for tracing studies. C.C. and G.S. wrote the manuscript with input from all of the authors.
Corresponding author
Ethics declarations
Competing interests
During part of this project, M.J.S. was a consultant for and had a financial interest in Inscopix, the company that makes the miniature microscope used for Ca2+ imaging in this study.
Peer review
Peer review information
Nature thanks Kirsty Bannister, Clifford Woolf and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Validation of an animal model for placebo analgesia investigation.
a, Experimental timeline to examine whether PAC-induced preference for chamber 2 is due to pain relief expectation. b, Latency for mice to cross back to chamber 1 for the first time (left; P = 1) and the proportion of time mice spent in chamber 2 (right; P = 0.91) while setting both chambers at 30 °C on the post-test day. n = 10 mice in each group. c, Experimental timeline to examine the timescale of PAC-induced analgesia (top). Comparison of the chamber preference and nociceptive behaviours of mice 1 (Day 7), 3 (Day 10), and 7 (Day 14) days after the conditioning phase of PAC (bottom). n = 10 in each group. d, Experimental timeline for the PAC assay (left) with naloxone injection during the post-test to test the dependency of PAC-induced pain relief on the endogenous opioid system. e, Latency for mice to cross back to chamber 1 for the first time (left; F(1,37) = 1.54, P = 0.66) and the proportion of time mice spent in chamber 2 (right; P = 0.22) after saline (red) or naloxone (purple) injection on the post-test day. n = 10 mice in each group. f, Latency preceding first paw licking (left; P = 0.007), rearing (middle; P = 0.07), and jumping (right; P = 0.007) behaviour after reaching chamber 2 on the post-test day for saline- or naloxone-injected mice. n = 10 mice in each group. g, Experimental timeline for the PAC assay (left) with naloxone injection during the conditioning phase to test the dependency of PAC-associated learning on the endogenous opioid system. h, Latency for mice to cross back to chamber 1 for the first time (left; P = 0.07) and the proportion of time mice spent in chamber 2 (right; P = 0.02) after saline (yellow) or naloxone (blue) injection on the post-test day. n = 10 mice in each group. i, Latency preceding first paw licking (left; P = 0.91), rearing (middle; P = 0.01), and jumping (right; P = 0.14) behaviour after reaching chamber 2 on the post-test day for saline- or naloxone-injected mice. n= 10 mice in each group. j, Experimental timeline to examine chemical pain sensitivity of mice during the post-test. k, Boxplots of the proportion of time mice spent in chamber 2 (left) and the duration of attending behaviours during the whole formalin test (right) in Ctrl and Cond groups. n = 4, 6 mice in Ctrl and Cond. l, Similar to (k), but for the first 3 min after formalin injection (P = 0.005 for time spent in chamber 2 and P = 0.03 for attending duration). n = 4, 6 mice in Ctrl and Cond. m, Experimental timeline to examine mechanical, thermal, and chemical pain sensitivity of mice after the conditioning phase of PAC. n, Boxplots of the mechanical (left), thermal (middle), and chemical (right) pain sensitivity after the conditioning phase of PAC, measured with von Frey, Hargreaves, and formalin injection, respectively. n = 10 mice in each group. o, Experimental timeline and strategy to examine the nocifensive behaviours of mice after confining them in either chamber 1 or chamber 2 by blocking the opening between chambers. p, Latency preceding first paw licking (left), rearing (middle), and jumping (right) behaviours of mice confined in chamber 1 (purple) and chamber 2 (red) on the post-test day. n = 5 mice in each group. q, Experimental timeline for the PAC with female mice. r, Boxplots of the latency for female mice to cross back to chamber 1 for the first time (left) and the proportion of time mice spent in chamber 2 (right) in Ctrl and Cond groups. n = 10 mice in each group. s, Latency preceding first paw licking (left; P = 0.01), rearing (middle), and jumping (right; P = 0.07) behaviour after reaching chamber 2 on the post-test day for female mice in Ctrl and Cond groups. n = 10 mice in each group. Two-sided Wilcoxon rank-sum test was used in (b), (f), (i), (k), (l), and (s); two-way ANOVA, Tukey post-hoc test in (e) and (h). In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual cells or mice.
Extended Data Fig. 2 Monosynaptic connection between rACC and Pn.
a, Schematic illustration of the viral strategy to trace projections from the rACC and motor cortex to the Pn. b, Injection sites in the motor cortex (top), rACC (middle), and the overlay (bottom). Scale bars, 500 μm. c, Projections from the rACC and motor cortex to the Pn. Note that the infected neurons in the motor cortex and rACC do not overlap and that the rACC only targets the rostral part of the Pn. Scale bars, 100 μm. d, Schematic illustration of the viral strategy to identify the brain areas that project to the Pn. SL1, stem-loop 1. e, Representative photomicrographs of labelled Pn projection neurons in several brain areas. M1, primary motor cortex; ACC, anterior cingulate cortex; S1, primary somatosensory cortex; S2, secondary somatosensory cortex; V1, primary visual cortex; CeA, central nucleus of the amygdala; Crus I, crus I of the ansiform lobule; Sp5I, interpolar nucleus. Scale bars, 100 μm. f, Schematic illustration of the strategy to test the synaptic connections between ACC projection neurons and neurons in the Pn. g, Confocal micrograph of two Pn neurons filled with biocytin during recording and labelled using a fluorescent conjugate of streptavidin. Scale bar, 50 μm. h, Average EPSC (black) of a Pn neuron holding at −70 mV evoked by photostimulation of rACC fibres. Adding CNQX (10 μM) in the perfusion solution abolished the EPSCs (red; left). EPSCs in a Pn neuron evoked by photostimulation of rACC fibres (top). TTX (1 μM) abolishes this response (middle), while 4-AP (1 mM) restores the response blocked by TTX (red, bottom).
Extended Data Fig. 3 PAC increases the activity of rACC→Pn neurons.
a, Cross-day alignment of rACC→Pn neurons across Pre, Cond, and Post phases of PAC using TRACKER. Neurons matched across 3 days are outlined in blue. Scale bar, 50 μm. b, Boxplot of the number of detected rACC→Pn neurons during Pre (grey), Cond (green), and Post (blue) phases, and the number of cross-day-aligned neurons (red). n = 6 mice. c, Z-scored activity traces (coloured traces) for the 10 neurons in (a). Raster traces show the binarized patterns of activity for each neuron. d, Mice with intracranial virus injection, GRIN lens implantation, and miniature microscope mounting (Mini) showed no difference in total walking distance and average movement speed versus mice without these manipulations (Ctrl) during PAC. n = 17 and 6 mice in Ctrl and Mini groups. e, Average Ca2+ activity of rACC→Pn neurons during first border crossing as a function of time in Pre (red), day 5 (green), and day 6 (blue). Note that their activity increased progressively during the conditioning phase of PAC. f, Line graph showing the progressively increased activity of rACC→Pn neurons during the conditioning phase of PAC. n = 6 mice in each group. g, Venn diagram showing cross-day-aligned rACC→Pn neurons that show increased activity in pairwise comparisons between Pre, Cond, and Post. h, Boxplots of the proportion of cross-day-aligned rACC→Pn neurons that show increased activity in pairwise comparisons between Pre, Cond, and Post. n = 6 mice. i, Discriminability index (d’) calculated between first crossing and crossing back for Ca2+ traces, averaged for individual neurons (left; P = 2 × 10−7; n = 233) and individual mice (right; P = 0.03; n = 6). j, Average firing rate of rACC→Pn neurons during first border crossing in Pre (cyan), Cond (green) and Post (blue; one-way ANOVA, Tukey post-hoc test, F(2,15) = 5, P = 0.022; n = 6 mice). k, Average firing rate as a function of time of rACC→Pn neurons during first border crossing in Pre (cyan), Cond (green) and Post (blue). l, Average firing rate of rACC→Pn neurons during first border crossing (red), first crossing back (green) and last border crossing (blue) on post-test day (one-way ANOVA, Tukey post-hoc test, F(2,15) = 16.8, P = 0.001; n = 6 mice). m, Discriminability index (d’) calculated between first crossing and crossing back for firing rate, averaged for individual neurons (left; two-sided Wilcoxon matched-pairs signed-rank test, P = 0.004; n = 233 neurons) and for individual mice (right; two-sided Wilcoxon matched-pairs signed-rank test, P = 0.07; n = 6). n, Control for (i, left) with randomized border crossing time. o, Control for Fig. 2f with randomized border crossing time. p, Control for (i, right) with randomized border crossing time. q, Control for (j) with randomized border crossing time. r, Control for (m, left) with randomized border crossing time. s, Control for (m, right) with randomized border crossing time. t, Boxplots of the correlation coefficient between the activity of rACC→Pn neurons and the velocity of mice during PAC with real neuronal activity trace (red) or randomly shuffled traces (green). n = 6 mice in each group. u, Scatterplot depicting the relationship between the Ca2+ activity of rACC→Pn neurons and the velocity of mice, specifically during periods of high neuronal activity in the pre-test. Shaded area represents the 95% confidence interval. v, Similar to (u), but specifically for periods during which mice display high moving velocity in the pre-test. Shaded area represents the 95% confidence interval. w, Average Ca2+ activity of rACC→Pn neurons over time during the first border crossing, illustrated in a longer time scale for Pre (cyan), Cond (green), and Post (blue). Note that the activity of rACC→Pn neurons decreases after reaching chamber 2 in Cond and Post. n = 6 in each group. Shaded area represents mean ± SEM. Pearson's two-sided correlation test in (u, v). In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual cells or mice. Data in (f), and shaded area in (k) and (w) are mean ± SEM.
Extended Data Fig. 4 rACC IT neurons are not activated during pain relief expectation.
a, Strategy and experimental timeline to record IT neuron activity in the rACC during PAC using in vivo Ca2+ imaging. b, Maximal projection of a Ca2+ video with 37 IT neurons. Scale bar, 50 μm. c, Z-scored activity traces (coloured traces) of the 10 example IT neurons outlined in (b). d, Boxplot of the number of detected IT neurons during Pre (grey), Cond (green), and Post (blue) phases, and the number of cross-day-aligned neurons (red; n = 3 mice). e, Activity of cross-day-aligned IT neurons (n = 133 cells from 3 mice) during first border crossing in Pre (left), Cond (middle) and Post (right) phases of PAC. Neurons are ordered according to their mean Ca2+ activity for each day. f, Averaged activity of IT neurons during first border crossing at the level of individual neurons in Pre (grey), Cond (green) and Post (blue) phases of PAC (one-way ANOVA, Tukey post-hoc test, F(2,396) = 2.19, P = 0.113; n = 133 neurons). g, Similar to (f), averaged for individual mice. h, Fisher information calculated between first crossing and crossing back for Ca2+ traces, averaged for individual neurons (two-sided Wilcoxon matched-pairs signed-rank test, P = 0.03; n = 133 neurons). i, Similar to (h), averaged for individual mice. n = 3 mice. j, Activity of cross-day-aligned IT neurons (n = 218 from 3 mice) during first border crossing (left), first crossing back (middle), and last border crossing (right) on post-test day. Neurons are ordered according to their mean Ca2+ activity for each condition. k, Averaged activity of IT neurons during first border crossing (red), first crossing back (green), and last border crossing (blue) at the level of individual cells on the post-test day (one-way ANOVA, Tukey post-hoc test, F(2,651) = 5.41, P = 0.004; n = 218 neurons). l, Similar to (k), for individual mice. n = 3 mice. m, Average firing rate of IT neurons during first border crossing in Pre (cyan), Cond (green), and Post (blue) phases of PAC. n = 3 mice. n, Average firing rate of rACC→Pn neurons during first border crossing (red), first crossing back (green), and last crossing (blue) on the post-test day. n = 3 mice. In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual neurons or mice.
Extended Data Fig. 5 Ca2+ dynamics of rACC→Pn neurons during thermal and mechanical pain.
a, Strategy to measure innocuous mechanical, noxious mechanical, or noxious heat responses while monitoring the Ca2+ activity of rACC→Pn neurons. b, Average silhouette width plotted for cluster numbers k from 1 to 10. Dashed line indicates the number of clusters corresponding to the maximal silhouette width. c, Averaged response (10 trials) of individual neurons upon each stimulation. Dashed lines (red) indicate the time point of stimulation (n = 335, 238, and 284 neurons for innocuous mechanical, noxious mechanical and noxious heat, respectively). Red and blue bars represent the cluster to which each neuron belongs (cluster 1 or cluster 2, respectively). d, Average activity of neurons in response to innocuous mechanical, noxious mechanical and noxious heat for cluster 1 (left) and 2 (right). e, Average activity of individual neurons in response to innocuous mechanical, noxious mechanical and noxious heat in cluster 1 (left; one-way ANOVA, Tukey post-hoc test, F(2,286) = 20.26, P = 1.1 × 10−8; n = 124, 92, and 73 neurons for innocuous mechanical, noxious mechanical and noxious heat, respectively) and 2 (right; one-way ANOVA, Tukey post-hoc test, F(2,565) = 16.73, P = 5.1 × 10−6; n = 211, 146, and 211 neurons for innocuous mechanical, noxious mechanical and noxious heat, respectively). Note that innocuous mechanical stimuli induced an increase in the activity of neurons in cluster 1, but a decrease in the activity of neurons in cluster 2. f, Average Ca2+ activity of rACC→Pn neurons during innocuous mechanical (red), noxious mechanical (green), and noxious heat (blue; n = 5 mice). g, Similar to (c), with cross-session-aligned neurons (n = 118 neurons). h, Similar to (d), with cross-session-aligned neurons. i, Similar to (e), with cross-session-aligned neurons (cluster 1; left; one-way ANOVA, Tukey post-hoc test, F(2,67) = 3.9, P = 0.02; n = 25, 20, and 25 neurons for innocuous mechanical, noxious mechanical and noxious heat; cluster 2; right; one-way ANOVA, Tukey post-hoc test, F(2,281) = 3.2, P = 0.04; n = 93, 98, and 93 neurons for innocuous mechanical, noxious mechanical and noxious heat). j, Similar to (f), with cross-session-aligned neurons. k, Heatmap of rACC→Pn neuron activity relative to licking (left) and rearing (right) behaviours in the post-test of PAC. l, Average Ca2+ activity of rACC→Pn neurons during licking (red) and rearing (green). n = 6 mice. In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual cells or mice. Shaded area in (d, f, h, j, l) represents mean ± SEM.
Extended Data Fig. 6 PAC alters the excitation and inhibition balance of rACC→Pn neurons.
a, Strategy to label Pn-projecting rACC neurons for electrophysiological recording. b, Time mice spent in chamber 2 during days 3–6 of PAC. n = 7, 8 in Ctrl and Cond groups. c, Boxplots of the resting membrane potential (RMP; left; P = 0.37) and the input resistance (right; P = 0.44) of rACC→Pn neurons from Ctrl and Cond mice. n = 14, 16 neurons in Ctrl and Cond groups, respectively. d, Boxplots of the peak amplitude (left; P = 0.85) and half-duration of the action potentials (right; P = 0.39). n = 14, 16 neurons in Ctrl and Cond groups, respectively. e, Action potential firing pattern of rACC→Pn neurons from littermate control (cyan) and PAC-conditioned mice (red). f, Action potential firing frequency evoked by different levels of injected current. n = 14 neurons/group. g, Percentage of rACC→Pn neurons displaying different numbers of spikelets in the first action potential. n = 14 neurons in Ctrl and 16 in Cond. h, Traces of the action potential firing pattern evoked by 1-s current injection (black, bottom) in tdTomato-negative (non rACC→Pn) neurons from Ctrl (green, top) and Cond (purple, middle) mice. i, Plot of the action potential firing frequency evoked by different levels of injected current (two-way ANOVA, Tukey post-hoc test, F(1,255) = 10.61, P = 0.001; n = 11, 22 neurons in Ctrl and Cond groups, respectively). j, Example traces of sEPSCs in a rACC→Pn neuron holding at −70 mV from a Ctrl mouse (cyan, top) and a Cond mouse (red, bottom). k, Cumulative histograms of sEPSC frequency from Ctrl (cyan) and Cond (red) mice. Inset: boxplot of sEPSC frequency (P = 0.56; n = 11 neurons in each group). l, Cumulative histograms of sEPSC amplitude from Ctrl (cyan) and Cond (red) mice. Inset: boxplot of the sEPSC peak amplitude (P = 0.028; n = 11 neurons in each group). m, Example traces of two EPSCs evoked at 50-ms intervals from Ctrl (cyan, top) and Cond (red, bottom) mice. n, Paired-pulse ratio as a function of Δt (20, 50, 100, 200, 500 ms) between two stimuli (two-way ANOVA, Tukey post-hoc test, F(1,87) = 0.37, P = 0.54) of the inputs to rACC→Pn neurons from Ctrl (cyan) and Cond (red) mice. n = 8, 11 in Ctrl and Cond groups. o, Recordings of mixed EPSCs and IPSCs while holding rACC→Pn neurons at −30 mV from Ctrl (cyan, top) and Cond (red, bottom) mice. Because the holding potential is between the reversal potentials for excitatory and inhibitory events, EPSCs are inwardly directed and IPSCs outwardly directed. p, Strategy to express excitatory opsin (ChR2) in PV+ interneurons and label the Pn-projecting rACC neurons for electrophysiological recording. q, Example trace of the action potential firing from a PV+ interneuron in the rACC (top) evoked by current injection (bottom). r, Whole-cell recording configuration to analyse the feedforward inhibition from PV+ interneurons to rACC→Pn neurons. A blue light (494 nm, 1 ms) was given to evoke neurotransmitter release from PV+ interneurons. s, Example trace showing a light-evoked IPSC (blocked by 10 μM SR-95531) in one rACC→Pn neuron from a control mouse. Blue bar indicates the time point of light stimulation. t, Boxplots of 20–80% rise time (left; P = 0.96) and half-duration (right; P = 0.65) of IPSCs from Ctrl (cyan) and Cond (red) mice. n = 8 neurons in each group. u, Paired-pulse ratio as a function of Δt between two light stimulations (20, 50, 100, 200, 500 ms) of the inhibitory inputs from PV+ interneurons to rACC→Pn neurons (two-way ANOVA, Tukey post-hoc test, F(1,65) = 0.018, P = 0.89) from Ctrl (cyan) and Cond (red) mice. n = 7 for each group. v, Light-evoked individual IPSCs (grey), and average IPSC (cyan or red) from Ctrl (top) and Cond (bottom) mice. Blue bar indicates the time point of light stimulation and dashed line indicates the IPSC onset of the neuron from the Ctrl group. Inset: average IPSCs at expanded time scale. w, Boxplots of the amplitude (left; P = 0.02) and latency (right; onset to onset; P = 0.01) of light-evoked IPSCs from Ctrl (cyan) and Cond (red) mice. n = 8 cells in each group. Two-sided Wilcoxon rank-sum test was used in (c), (d), (i), (j), (f) and (w). In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual cells. Data in (b, f, i, n, u) are mean ± SEM.
Extended Data Fig. 7 Photoexcitation of the rACC→Pn pathway enhances PAC-induced analgesia.
a, Strategy (top, left) and timeline (bottom, left) to optogenetically activate the rACC→Pn pathway during PAC. Illustration of bilateral implantation of cannula in the Pn (right). Dashed lines indicate cannula location. Scale bar, 500 μm. b, Timeline of the PAC assay, integrating optogenetics. c, Boxplots of the latency preceding first paw licking (two-sided Wilcoxon rank-sum test, P = 0.03; left), rearing (two-sided Wilcoxon rank-sum test, P = 0.24; middle), and jumping (two-sided Wilcoxon rank-sum test, P = 0.51; right). n = 10 in eYFP and 9 in ChR2 groups. d, Boxplot of the change in falling latency of mice during the rotarod test with and without photomanipulation of the rACC→Pn pathway (one-way ANOVA, Tukey post-hoc test, F(2,26) = 1.43, P = 0.25). n = 10 in eYFP, 10 in NpHR, and 9 in ChR2 groups. e, Quantification of the change in paw withdrawal frequency in response to 6 different von Frey filaments during optogenetic manipulation of the rACC→Pn pathway (two-way ANOVA, Tukey post-hoc test, F(2,156) = 55.18, P = 2 × 10−16). n = 10 in eYFP control, 10 in NpHR, and 9 in ChR2 groups. *P < 0.05 and ***P < 0.001. In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual cells or mice. Data in (d) are mean ± SEM.
Extended Data Fig. 8 Oprd1+ Pn neurons mediate PAC-induced analgesia.
a, UMAP plot of Pn neuron clusters with data from two single-cell RNA-seq platforms. 10x: 10x Chromium 3′ v3; SS: SMART-seq v4. b, UMAPs illustrating single-cell expression patterns of neuronal genes. c, Euler diagram showing coexpression of excitatory neuron marker genes and Oprd1 in Pn neurons. d, Strategy to confirm the connection between rACC projection neurons and Oprd1+ Pn neurons using an AAV1-Cre virus that exhibits anterograde transsynaptic spread properties. e, Fluorescence in situ hybridization (FISH) experiments show that the eGFP+ neurons are also Oprd1+. Scale bar, 50 μm. f, Boxplot of the overlay ratio of eGFP and DAPI signals. g, Strategy to label neurons that have a monosynaptic connection with Oprd1+ neurons in the Pn in a retrograde manner using G-deleted mutant rabies virus (RVdG). h, Left top: Coronal section from the Pn of Oprd1Cre mice injected with AAV helpers (red) and RVdG (green). Arrows indicate examples of co-infected starter cells (yellow). Left bottom panels show a magnified view of the dashed box shown in the left top panel. Scale bars, 50 μm (left top) and 20 μm (left bottom). Right: Representative images of GFP-labelled neurons in the ACC. Cg1, cingulate area 1; Cg2, cingulate area 2. Scale bar, 50 μm. i, Strategy and experimental timeline to optogenetically manipulate the activity of Oprd1+ neurons in the Pn during the conditioning phase of PAC. j, Boxplot of the latency preceding first border crossing during the post-test after photoinhibition of eYFP control (green) versus NpHR (blue) mice during the conditioning phase (P = 0.04; n = 10 for each group). k, Boxplots of the latency preceding first paw licking (left; P = 0.06), rearing (middle), and jumping (right; P = 0.05) on the post-test day after photoinhibition of eYFP control (green) versus NpHR (blue) mice during the conditioning phase of PAC. n = 10 for each group. l, Strategy to express inhibitory opsin specifically in the Oprd1+ Pn neurons that receive rACC inputs using an AAV1 virus. m, Representative coronal section of the Pn showing Oprd1+ neurons that express the inhibitory opsin eNpHR. Scale bar: 100 µm. n, Boxplot of the latency for mice to cross back to chamber 1 for the first time (left), and the proportion of time mice spent in chamber 2 (right) with photoinhibition of eYFP control (green) versus NpHR (red) mice on the post-test day. n = 8 in eYFP and 6 in NpHR groups. o, Boxplots of the latency preceding first paw licking (left; P = 0.02), rearing (middle), and jumping (right) on the post-test day with photoinhibition of eYFP control (green) versus NpHR (blue) mice. n = 8 in eYFP and 6 in NpHR groups. p, Experimental timeline for the PAC assay (left) with both µ- and δ-opioid receptor antagonists injection during the post-test to examine which opioid receptor contributes to PAC-induced pain relief. q, Boxplots of the latency preceding first paw licking (left), rearing (middle), and jumping (right) on the post-test day for mice injected with saline, naltrindole (δ-opioid receptor antagonist), or β-funaltrexamine (µ-opioid receptor antagonist). Note that injection of either naltrindole or β-funaltrexamine abolished PAC-induced analgesia. n = 10 for each group. r, Boxplot of the pain threshold of Oprd1Cre mice with eYFP (cyan) or NpHR (brown) injection in the von Frey assay with or without light stimulation (F(1,30) = 7.988, P = 0.008; n = 8 in eYFP control and 9 in NpHR groups). s, Boxplot of the latency preceding paw withdrawal on a 48 °C plate (P = 0.003; left) or 52 °C plate (P = 0.04; right). n = 8 in eYFP control and 9 in NpHR groups. t, Photoinhibition of Oprd1+ Pn neurons does not significantly affect locomotion in the open-field test (F(1,28) = 0.356, P = 0.55; n = 8 in eYFP and 9 in NpHR groups). u, Photoinhibition of Oprd1+ Pn neurons does not affect the latency of mice to fall during the rotarod test (F(1,30) = 0.03, P = 0.984; n = 8 in eYFP and 9 in NpHR groups). Two-sided Wilcoxon rank-sum test was used in (j), (k), (o) and (s); two-way ANOVA with Tukey post-hoc test in (n), (r), (t), and (u). In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual mice. Data in (t) are mean ± SEM.
Extended Data Fig. 9 Classification of Purkinje cell subpopulations during PAC.
a, Strategy to label Oprd1+ Pn neuron outputs. b, Cerebellar sections showing projections from Oprd1+ Pn neurons. Scale bars: 50 μm (top), 20 μm (bottom). c, Cerebellar sections showing projections in lobule VI, Crus I, and Crus II. Scale bars, 200 μm. d, Boxplot of the number of Purkinje cells detected in each mouse during the Pre (grey), Cond (green), and Post (blue) phases of PAC, and the number of cross-day-aligned neurons (red). n = 6 mice. e, Activity of cross-day-aligned Purkinje cells in cluster 1 (n = 58 neurons from 6 mice) and cluster 2 (n = 218 neurons from 6 mice) during the first border crossing, first crossing back, and the last border crossing on the post-test day. Neurons are ordered according to their cluster and mean Ca2+ activity during the first border crossing. f, Averaged activity of Purkinje cells within each cluster during first border crossing (red), first crossing back (green), and last border crossing (blue) on the post-test day at the level of individual neurons. g, Similar to (f), for individual mice. h, Averaged activity of Purkinje cells in cluster 2 during first border crossing at the level of individual neurons in the Pre (grey), Cond (green), and Post (blue) phases of PAC (F(2,651) = 10.28, P = 4 × 10−5; n = 218 neurons). i, Similar to (h), but at the level of individual mice (F(2,15) = 7.13, P = 0.006; n = 6 mice). j, Ca2+ spike frequency of cluster 1 Purkinje cells during first border crossing (F(2,171) = 13.43, P = 3.8 × 10−6). k, similar to (j), for cluster 2 Purkinje cells (two-sided Wilcoxon signed-rank test; n = 218 neurons). l, Cumulative histograms of Ca2+ spike amplitudes from cluster 1 Purkinje cells. Inset: Average amplitude of spikes >3 z-score (P = 0.0001, n = 75, 93, 178 in Pre, Cond, Post). m, similar to (l), but for cluster 2 Purkinje cells. n, Averaged waveforms of extracted Ca2+ spikes with amplitudes exceeding 3 z-scored ΔF/F for Purkinje cells in cluster 1 (left) and cluster 2 (right) during the Pre, Cond, and Post phases of PAC. Traces are aligned by the time point of their peak. o, Scatterplot of the latency preceding first border crossing against averaged signal of Purkinje cells in cluster 1 during day 3 (Pre), day 5, day 6 (Cond), and day 7 (Post) of PAC for each mouse. Data points were fit by linear regression. p, Similar to (o), for Purkinje cells in cluster 2. q, Purkinje cell activity during first border crossing (left), first crossing back (middle), and last border crossing (right) during the post-test. Neurons are ordered by mean activity during the first border crossing. r, s, Activity of Purkinje cells averaged for individual neurons (r) (F(2,2079) = 11.91, P = 1.3 × 10−6; n = 694) and individual mice (s) (F(2,15) = 13.1, P = 0.0004; n = 6). t, Control for Fig. 4l and (r) with randomized border crossing time. One-way ANOVA with Tukey post-hoc test was used in (f-m) and (r-t). In boxplots, horizontal lines represent median; boxes, quartiles; whiskers, most extreme data points ≤ interquartile range from box edges; and single points, data from individual cells or mice. Shaded area in (n) represents mean ± SEM.
Supplementary information
Supplementary Table 1
Marker genes for each Pn neuron cluster (related to Fig. 5). The spreadsheet contains 10 tabs, each corresponding to a cluster of Pn neurons classified in Fig. 5b. In each tab, genes with a log-transformed effect size greater than 0.5 are listed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, C., Niehaus, J.K., Dinc, F. et al. Neural circuit basis of placebo pain relief. Nature 632, 1092–1100 (2024). https://doi.org/10.1038/s41586-024-07816-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07816-z
This article is cited by
-
How do placebos ease pain? Mouse brain study offers clues
Nature (2024)
-
Placebo effect involves unexpected brain regions
Nature (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
