
Abstract
Parkinson disease (PD) is a progressive neurodegenerative disease that affects peripheral organs as well as the central nervous system and involves a fundamental role of neuroinflammation in its pathophysiology. Neurohistological and neuroimaging studies support the presence of ongoing and end-stage neuroinflammatory processes in PD. Moreover, numerous studies of peripheral blood and cerebrospinal fluid from patients with PD suggest alterations in markers of inflammation and immune cell populations that could initiate or exacerbate neuroinflammation and perpetuate the neurodegenerative process. A number of disease genes and risk factors have been identified as modulators of immune function in PD and evidence is mounting for a role of viral or bacterial exposure, pesticides and alterations in gut microbiota in disease pathogenesis. This has led to the hypothesis that complex gene-by-environment interactions combine with an ageing immune system to create the ‘perfect storm’ that enables the development and progression of PD. We discuss the evidence for this hypothesis and opportunities to harness the emerging immunological knowledge from patients with PD to create better preclinical models with the long-term goal of enabling earlier identification of at-risk individuals to prevent, delay and more effectively treat the disease.
Similar content being viewed by others

Introduction
Bidirectional communication between the brain and other organ systems is essential for brain health and the overall health of the organism. Once thought to be immune privileged, the brain is now acknowledged as a highly immune-specialized organ with its own brain-resident immune cells. These cells shape neuronal circuitry1 and lymphatic and glymphatic systems that regulate the complex efflux of immune cells and fluids exchanging from the cerebrospinal space with the rest of the circulation2. The study of the crosstalk between the central and peripheral immune systems has intensified in recent decades.
Parkinson disease (PD) is a progressive neurodegenerative disorder pathologically characterized by the loss of dopaminergic neurons in the substantia nigra and the presence of protein inclusions termed Lewy bodies. Previously, the disease was largely considered to be a movement disorder, classically characterized by a tetrad of motor deficits, including resting tremor, bradykinesia, postural instability, and rigidity of the neck, trunk and limbs. However, PD is now understood to be a multi-system disorder with notable neuroinflammation and immune dysfunction that has been implicated in the development of various non-motor symptoms such as sleep and gastrointestinal dysfunction, which can precede the disease diagnosis by decades3,4,5.
Most cases of PD are considered idiopathic; however, with the advancement of technology and accessibility to patient populations, the genetic architecture of PD is becoming further defined. Mutations in more than 20 genes have been identified that cause monogenic forms of PD6, yet they are rare and account for ~30% of familial PD and only ~3–5% of sporadic cases7. Interestingly, patients exhibiting the same genetic mutation typically do not have similar clinical presentations, suggesting that the aetiology of the disease is compounded by a complex interaction among environmental, age-associated and genetic factors (Fig. 1). Genome-wide association studies (GWAS) have identified over 90 risk variants, and the variants explain ~22% of the heritability of the disease8. Of the significant single-nucleotide polymorphisms (SNPs), several are found in genes associated with the function of the immune system8,9. Furthermore, previous studies have found common genetic variants between patients with PD and other autoimmune and inflammatory diseases10, including Crohn’s disease11, further suggesting the influence of the immune system in PD pathogenesis. Additionally, exposure to environmental insecticides heightened immune responses in HLA-DR variant carriers and increased disease risk 2.48-fold12. Together, these findings place the immune system at the intersection between genetic and environment interactions that confer risk for PD. In this Review, we discuss clinical evidence from genetic, pharmacological, immunological, neuroimaging and epidemiological studies and mention animal studies that have served to interrogate and validate pathways that could be targeted for therapeutic intervention to delay, prevent or treat PD.

Although primarily considered a motor-related disorder, Parkinson disease (PD) affects multiple systems, and patients commonly present with accompanying non-motor symptoms, which often start in the prodromal phase. The concept of prodromal PD is supported by the Braak theory (blue box), in which Lewy body pathologies begin in the periphery and olfactory bulb and advance to the brainstem and towards higher brain centres following a predictable caudal-rostral pattern204. During the prodromal stage, when neuronal dysfunction begins, a combination of factors, from an ageing immune system, genes and environment, can create the perfect storm to enable the development and progression of PD pathogenesis. Age-associated alterations in the immune system include immunosenescence and inflammageing as well as an impaired adaptive immune system defined by a decline in naive T cells and B cells and memory cell accumulation and a reduction in T cell receptor and B cell receptor diversity and sensitivity to stimuli13,14,15. These deficiencies contribute to an increase in susceptibility to infection and a type of age-acquired autoimmunity where autoantibodies may begin to appear. There are now multiple lines of evidence that suggest a relationship between environmental stressors, including viral and bacterial exposures, pesticides, diet, and alterations in gut microbiota, and the increased risk of developing PD. Treg cell, regulatory T cell.
Inflammation in PD pathogenesis and progression
Considering age is the greatest risk factor for many neurodegenerative diseases, the ageing of the immune system is the most underappreciated and understudied contributing factor in the neurodegeneration field. Immunosenescence is characterized by two primary features, namely an age-acquired immunodeficiency and inflammageing. Inflammageing is characterized by excess low-level production of circulating inflammatory mediators, or cytokines — most notably C-reactive protein (CRP), IL-6 and tumour necrosis factor (TNF) — from chronically stimulated innate and adaptive immune cells13,14,15. Both the innate and adaptive immune systems lose competence with ageing and are also notably altered in PD16,17 (Fig. 2).

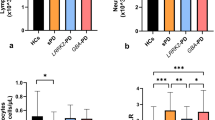
The figure highlights inflammatory manifestations that have been identified in patients with Parkinson disease (PD). Intestinal dysbiosis and inflammation (step 1), increases in levels of circulating pro-inflammatory cytokines (step 2), innate and adaptive immune cell activation and changes in frequency (step 3), blood–brain barrier permeability and peripheral immune cell infiltration of the central nervous system (step 4) and neuroinflammation (step 5) are hallmarks of a pro-inflammatory immune phenotype in PD. ROS, reactive oxygen species.
Innate immunity
Microglia are densely populated in the substantia nigra pars compacta and striatum of the brain — areas that are both affected in PD. One of the first pieces of evidence linking neuroinflammation to the pathogenesis of PD came in 1988, when McGeer et al. showed HLA-DR+ reactive microglia in post mortem tissue from patients who had PD18. The number of HLA-DR+ microglia increases with neuronal degeneration throughout the nigrostriatal pathway19. Activated microglia are partially responsible for the elevated levels of TNF, IL-1β, TGFβ, IL-6, reactive oxygen species (ROS), nitric oxide species and pro-apoptotic proteins found in the substantia nigra pars compacta, striatum, and cerebrospinal fluid (CSF) of patients with PD20,21. In vivo evaluation of microglial activity has been performed using positron emission tomography (PET) ligands to measure and trace neuroinflammation in the brains of patients with PD. The use of ligands such as [11C](R)-PK11195, which binds translocator protein (TSPO, formerly known as the peripheral benzodiazepine receptor), a receptor which was considered selectively expressed on activated microglia, showed increased microglial activation in the brains of patients with PD but the levels of microglial activation did not correlate with clinical severity22,23. Usage of this first-generation PET ligand allowed researchers to conclude that microglia are activated early in the disease process, leaving them to promote neuroinflammation in vulnerable PD-associated brain regions. However, the accuracy and interpretation of TSPO radioligand binding may be influenced by a number of issues such as TSPO polymorphisms found with second-generation ligands, low TSPO density in the healthy brain and multicellular expression, including infiltrating cells from the periphery24. New, more accurate targets are necessary to ensure microglia specificity and function.
Historically, microglia in the areas of neurodegeneration have been termed ‘activated’ due to their ameboid morphology, a description often perceived as a damaging inflammatory state. However, evidence suggests that microglia present along a spectrum of phenotypes and play a number of distinct roles in PD pathogenesis. For example, microglia can contribute to neuronal death through the production of inflammatory factors, they may interact with α-synuclein to contribute to α-synuclein propagation and aggregation or can alternatively have protective functions through production of neurotrophic factors25,26. Dysfunctional phagocytosis in glial cells due to lysosomal defects imparted by PD-related mutations may be one mechanism that contributes to microgliosis and neuroinflammation27. Extracellular α-synuclein can directly activate microglia in a conformation-specific and mutation-specific context, such that α-synuclein fibrils and mutations associated with early-onset PD induce the most robust immune responses in BV2 microglial-like cells28,29. The NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome signalling in microglia is a multiprotein complex involved in the activation of a pro-inflammatory state30. In PD models, NLRP3 inflammasome signalling in microglia is reportedly triggered by α-synuclein31 and different α-synuclein species lead to specific NLRP3 inflammasome microglial responses, which can comprise α-synuclein degradation32, indicating its potential role in PD.
In addition to microglia, monocytes may contribute to disease pathogenesis. Within the total monocyte population, frequencies of classical CD14+CD16– monocytes are elevated in patients with PD, and these cells display an altered transcriptome33. CC-chemokine ligand 2 (CCL2) is upregulated in patients with PD, suggesting an increase in monocyte recruitment and inflammation. Leucine-rich repeat kinase 2 (LRRK2) levels are elevated in monocyte populations from patients with PD and contribute to monocyte dysregulation34,35. Distinct gene expression patterns of monocytes have been noted in patients in early stages of the disorder, including genes involved in immune activation such as HLA-DQB1, MYD88, REL and TNF36. More recently, a transcriptome-wide association study positively identified gene associations between lysosomal pathways and innate immune function in dorsolateral prefrontal cortex and peripheral monocytes as risk factors for PD37.
Adaptive immunity
Ample evidence suggests a role for adaptive immunity in disease pathogenesis. In the same incipient study that identified HLA-DR+ microglia in the brains of patients with PD, McGeer et al. showed that CD3+ T cells infiltrate the brains of patients with PD, a finding which has been replicated in other studies and in animal models18,38. Subsequent studies have focused on T cell subsets in the brain and in the periphery to understand their role in the inflammatory pathogenesis associated with PD. In the brain, CD4+ and CD8+ T cells were present in the substantia nigra pars compacta of patients with PD at greater levels than in control patients38. Peripherally, multiple studies, including a meta-analysis of 943 cases of PD, have identified a reduction in circulating CD4+ T cells in patients39,40. More specifically, HLA-DR+ T cells and CD45RO+ memory T cells have been shown to be increased in patients with PD relative to healthy controls, while naive CD4+ T cells are reduced, and mixed results have been reported on the frequency of CD25+ regulatory T (Treg) cells40,41,42. CD4+FOXP3+ Treg cells have increased suppressive activity in patients with PD43. This correlates with the finding that dopamine, which is deficient in patients with PD, lowers Treg cell function40,44. Other functional studies have not demonstrated a difference between patients treated with dopamine-replacement medication compared to untreated patients, suggesting that dopaminergic drugs may not alter T cell activity42,43. Interestingly, the expression of specific dopamine receptors found on T cell subsets correlates with disease severity in patients with PD, highlighting a potential role of immune cell dopamine receptors in the development or progression of PD45,46. T cell dysregulation in PD has been suggested as T cells exhibit increased TNF receptor expression47 along with increased production of IFNγ and TNF by effector T cells, even in the presence of Treg cells42. In 2017, a ground-breaking study by Sulzer et al. was the first to suggest that, in patients with PD, specific T cell subsets — mainly CD4+ T cells — recognized certain α-synuclein peptides48, further supporting a role of adaptive immunity in PD pathogenesis. A more recent study has identified that α-synuclein T cell immunoreactivity in peripheral blood mononuclear cells is associated with preclinical and early motor PD, suggesting that monitoring this in at-risk populations could potentially enable earlier detection of disease49. Although there are discrepancies between findings in T cell populations concerning their dysregulation and their roles in PD pathogenesis, some of the variability may be explained by the heterogenous nature of the patient populations that were evaluated in these studies. It remains clear that dysregulation in immune cell trafficking can promote a pro-inflammatory environment that can contribute to the neuronal cell death associated with PD.
The role of B cells in PD is less well understood and is being actively explored. Reports suggest that B cells are reduced in number in the blood of patients with PD relative to controls50,51 but these findings are not consistent across studies52. IgG deposits have been found on dopaminergic neurons in the brain, and the IgG receptor FcγRI was found on activated microglia53, suggesting that humoral immunity may play a role in neuroinflammation and neurodegeneration. Autoantibodies against α-synuclein, dopamine and melanin are present in the sera and CSF of patients with PD54,55. The levels of α-synuclein autoantibodies in CSF and plasma in patients with mild or moderate PD have been correlated with disease activity, supporting the hypothesis that α-synuclein autoantibodies could serve as a potential biomarker for PD56. Previous infections may be linked to the production of autoantibodies through molecular mimicry as has been suggested for infections with herpes simplex virus 1 (HSV1) and Helicobacter pylori57,58.
Cytokines in PD
Collectively, data from the innate and adaptive immune systems provide evidence that immune dysregulation in both the periphery and brain can cause upregulation of inflammatory cytokines that initiate a cascade of pro-inflammatory signalling events that ultimately result in the neurotoxicity that is associated with PD. In a similar manner to what has been observed in the brain, levels of the pro-inflammatory cytokines TNF, IFNγ, IL-1β, IL-6, IL-2, CXC-chemokine ligand 8 (CXCL8) and CCL2 are elevated in the serum of patients with PD and correlate with disease severity and disability59,60. This may be a consequence of altered lymphocyte populations that contribute to immune cell dysregulation as higher levels of IFNγ-producing T cells relative to IL-4-producing T cells have been identified, which is in accordance with a reduced CD4 to CD8 T cell ratio in patients with PD61.
While a number of different signalling pathways have been implicated in PD pathogenesis, two of the ones that have received increasing attention are the IFNγ (also called type II interferon) and TNF pathways62. Multiple studies have reported TNF to be elevated in the sera, CSF and brains of patients with PD19,20,63,64,65,66, and consistent with a role for TNF in nigral degeneration, selective neutralization of soluble TNF signalling significantly attenuates dopaminergic (DA) neuron death in rodent models67,68,69,70. As described in more detail below, epidemiological evidence suggests that TNF signalling in individuals with autoimmune diseases is linked to increased risk of developing PD, whereas anti-TNF therapy in patients with autoimmunity has been associated with a decreased risk for PD71. Notably, as TNF is produced by intestinal epithelial cells72, it may alter the gut environment, impacting inflammation and α-synuclein accumulation.
A significant amount of evidence implicates IFNγ signalling in PD pathophysiology. Near the end of World War I in 1918, an influenza pandemic gave rise to a high incidence of postencephalitic parkinsonism, suggesting vulnerability of basal ganglia DA neurons to pathogen-driven immune response73, and a type II interferon response was directly implicated by transcriptomic analysis in the selective vulnerability of DA neurons74. In the midbrain regions of patients with PD, higher levels of IFNγ have been reported along with significant co-expression of α-synuclein75. Preclinical rodent models of PD-like degeneration have also implicated IFNγ signalling in progressive DA neuron death76,77. Two recent studies have demonstrated synergistic neurodegenerative effects between lipopolysaccharide (LPS)-induced inflammation involving IFNγ pathways and mutant LRRK2 in mice78 and in metabolic reprogramming of neurotoxic microglia from patient-derived induced pluripotent stem cells79. Interestingly, LRRK2 is an IFNγ target gene regulated in immune cells in response to pathogens80,81,82; genetic polymorphisms in LRRK2 (discussed below) have been associated with Crohn’s disease and leprosy, suggesting that pathogenic mechanisms may be common to certain chronic inflammatory conditions, infections and PD.
Genetic evidence of immune involvement
Although age remains the greatest risk factor for developing sporadic PD, mutations in several genes cause autosomal dominant and autosomal recessive monogenic forms of PD. Several genetic variants that modulate the risk of idiopathic disease have been identified; some of these genes, including LRRK2, SNCA (which encodes α-synuclein), GBA (which encodes glucocerebrosidase (GBA); also known as lysosomal acid glucosylceramidase), PRKN (encoding E3 ubiquitin-protein ligase parkin) and PINK1 (encoding PTEN-induced kinase 1 (PINK1)), encode proteins that also modulate immune function. The fundamentals of these genetic findings in PD are described in Box 1.

LRRK2 variants in PD
Mutations in the gene encoding LRRK2 are the most common cause of familial PD and explain ~1% of sporadic cases83,84,85. LRRK2 expression is tightly regulated in peripheral immune cells and increases in response to microbial pathogens in human B cells, T cells, macrophages and non-classical monocytes34,81,86,87. Furthermore, LRRK2 is a member of the receptor interacting protein (RIP) kinase family, which is a group of proteins that detect and respond to cellular stress by regulating cell death and activation of the immune system88.
To date, eight pathogenic mutations have been identified in LRRK2 in patients with PD, with the G2019S mutation being the most common89, as well as two non-coding variants that confer a twofold increase in the risk for disease90. The G2019S mutation has consistently been reported to increase LRRK2 kinase activity91,92,93, which is associated with neuronal toxicity94,95. Peripheral pro-inflammatory cytokine levels are higher in a subset of asymptomatic individuals carrying the G2019S mutation96. Taken together with the fact that overall LRRK2 levels are increased in immune cells of patients with sporadic PD34, it is suggested that inflammation plays an early role in the disease and may be driven by increased LRRK2 kinase activity. This pathological mechanism is further supported by reports that LRRK2 toxicity, both in vitro and in vivo, is ameliorated following pharmacological LRRK2 kinase inhibition97.
Interestingly, LRRK2 expression is also associated with a number of bacterial infections and infectious diseases. Meta-analysis of human cell gene expression in response to a Mycobacterium tuberculosis infection identified LRRK2 as a highly significant differentially enriched gene98. Furthermore, Lrrk2-knockout mice showed limited bacterial burdens and enhanced inflammatory profiles, suggesting that LRRK2 may control innate immune pathways following M. tuberculosis infection99. LRRK2 has also been implicated in the regulation of the enteric pathogens Salmonella typhimurium100 and Listeria monocytogenes101. Knock-in mice harbouring the G2019S mutation showed improved control of S. typhimurium infection with reduced bacterial growth and longer survival during sepsis. However, animals with the G2019S mutation that were subjected to reovirus-induced encephalitis exhibited increased mortality and had elevated ROS production and concentrations of α-synuclein in the brain102, potentially suggesting antagonistic pleiotropic effects of the G2019S mutation. Similar effects are seen with the gain-of-kinase function R1628P mutation, which is a risk variant for PD but protective for leprosy, and associated with excessive inflammatory type 1 reactions103,104. These data highlight the versatility of LRRK2 in regulating immune responses and imply potential opposing effects of LRRK2 kinase-mediated inflammation in the central nervous system (CNS) versus the periphery that may also be dependent on additional complex genetic interactions (Box 2).

GBA mutations in PD
The neurodegenerative manifestations of GBA-associated PD may arise from the toxic effects of accumulated lipids, autophagic perturbations and endoplasmic reticulum stress in neurons105. However, it is becoming more apparent that inflammation plays a key role in the pathology of GBA-associated PD. For example, selective deletion of GBA in neurons, oligodendrocytes and astrocytes of mice increased inflammatory cytokine production, oxidative stress and morphologically active microglia despite normal microglial GBA expression106,107. Furthermore, deletion of the Ripk3 gene (which encodes for the receptor-interacting kinase 3 that is essential for programmed cell death in response to TNF) attenuates microglial cell activation and motor impairment upon glucocerebrosidase (GCase) inhibition in mice108. Additionally, mice expressing the L444P point mutation in the Gba gene exhibit multisystem inflammation, including evidence of B cell hyperproliferation109.
Induced pluripotent stem cell-derived macrophages from individuals with a GBA mutation exhibit increased upregulation of TNF mRNA expression upon LPS stimulation110 and secrete higher levels of the pro-inflammatory cytokines TNF, IL-6 and IL-1β111. Patients with GBA-associated PD have higher plasma levels of CXCL8, CCL2 and CCL3 (also known as MIP1α) than patients with idiopathic PD112, suggesting GBA may alter the immune environment. Additionally, enzymatic GBA activity is significantly reduced in monocytes from both patients with idiopathic PD and patients with GBA-associated PD compared to controls113, suggesting that GBA dysfunction in immune cells may be instrumental in both idiopathic and GBA-associated PD.
PRKN and PINK1 mutations in PD
Seventy-nine different PRKN mutations have been reported in familial and sporadic PD, and these mutations are the most common cause of autosomal recessive early-onset cases114. The second most common cause are mutations in the PINK1 gene115, which are typically missense mutations, though copy number variants and exonic rearrangements, such as the homozygous deletion of exon 7, have been described116,117,118. Parkin, a cytosolic protein, mediates mitophagy in conjunction with the ubiquitin kinase PINK1. PINK1 accumulates on the outer mitochondrial membrane when mitochondria are damaged or depolarized. It assists in the recruitment of parkin to mitochondria, where parkin then ubiquitinates proteins found on the outer mitochondrial membrane with K63-linked polyubiquitin chains, targeting these mitochondria for lysosomal degradation. A loss of Pink1 or Prkn dysregulates mitophagy and increases mitochondria stress, mitochondrial ROS and mitochondrial DNA (mtDNA)119. Mito-inflammation has recently been directly connected to PD-like pathology in mice deficient in Pink1 or Prkn that are subjected to acute (exhaustive exercise induced) or chronic (mtDNA mutation induced) mitochondrial stress120. This mitochondrial stress induced dopaminergic neurodegeneration in the substantia nigra pars compacta in the absence of Prkn or Pink1 and increased pro-inflammatory cytokines in the serum. These phenotypes were successfully rescued by the genetic inactivation of Sting1, a component of the DNA-sensing cyclic GMP–AMP synthase (cGAS)–STING innate immune pathway that can be activated by mtDNA from damaged mitochondria121. STING signalling also exacerbates progression in other models of chronic neurodegenerative disease such as the ME7 prion disease model122. Nazmi et al. confirmed STING as a critical driver of type I interferon-mediated neurodegeneration and, in this model, mice deficient in STING or IFNAR1 displayed attenuated neuroinflammation122. Interestingly, Lrrk2-/- macrophages exhibit chronic cGAS engagement caused by mtDNA leakage into the cytosol, leading to altered innate immune gene expression123. These reports highlight a convergence between PD-associated genes and emphasize an important connection between mitochondrial stress and inflammation in the context of PD.
As in mito-inflammation, both PINK1 and parkin have been implicated in adaptive immunity via presentation of mitochondrial antigens. Although antigen presentation can be mediated by autophagy, mitochondrial antigen presentation relies on the generation and trafficking of mitochondrially derived vesicles124. With the knockdown of Pink1, the amount of glycoprotein B of HSV1 targeted to the mitochondrial matrix is upregulated in a murine macrophage cell line, with the overexpression of parkin eliciting the opposite phenotype124. Furthermore, when CD11c+ dendritic cells isolated from Pink1 or Prkn-knockout mice were treated with a single dose of LPS, they exhibited increased presentation of an endogenous mitochondrial antigen, the mitochondrial matrix protein 2-oxoglutarate de-hydrogenase, relative to wild type controls. Such data provide support for a non-cell-autonomous model in which autoimmune mechanisms mediated by mitochondria-specific cytotoxic T cell activity may contribute to pathology in PRKN-associated and PINK1-associated PD. In addition to this role of PINK1 and parkin in mitochondrial quality control, the loss of their activity during PD may increase mitochondrial antigen presentation during inflammation. In support of this, intestinal infection with Gram-negative bacteria in Pink1−/− mice engages mitochondrial antigen presentation and autoimmune mechanisms that elicit the establishment of cytotoxic mitochondria-specific CD8+ T cells in the periphery and in the brain124. Furthermore, these mice show a decrease in the density of dopaminergic axons in the striatum, increased infiltrating monocytes and T cells into the CNS, and are affected by motor impairment. Collectively, these data support the idea that PINK1 is a repressor of the immune system and provide a pathophysiological model in which intestinal infection acts as a triggering event in PD, highlighting the relevance of the gut–brain axis in the disease.
SNCA in PD
Due to the earlier onset and more rapid disease progression observed in patients with SNCA gene triplications relative to those with duplications, a dosage effect of the SNCA gene is expected125. Such observations indicate a neurotoxic effect of increased α-synuclein, even when point mutations are not present, and have prompted the development of various animal models of overexpression. Overexpression of α-synuclein in dopaminergic neurons of the substantia nigra pars compacta in an adeno-associated virus mouse model of PD robustly induces neuroinflammation126,127,128. This model also results in substantial infiltration of pro-inflammatory CC-chemokine receptor 2 (CCR2)+ peripheral monocytes into the CNS, and genetic knockout of Ccr2 prevents α-synuclein-induced inflammation and neuronal degeneration129. Furthermore, when in contact with α-synuclein fibrils or oligomers, both astrocytes and microglia exhibit increased neurotoxicity26,130,131,132.
Increased expression of α-synuclein also affects immune cells outside the CNS. For example, α-synuclein peptides can stimulate the secretion of TNF in lymphocytes from patients with PD but not from controls133 and can trigger helper and cytotoxic T cells to secrete cytokines, including IFNγ, IL-2 and IL-5 (ref.48). Fibrillar α-synuclein can act via Toll-like receptor (TLR) and inflammasome pathways in monocytes, leading to IL-1β production134. Similarly, human peripheral blood mononuclear cells increase inflammasome-related cytokines in response to treatment with α-synuclein monomers or fibrils135. Such data imply that increased α-synuclein expression, in the form of monomers or fibrils, may stimulate cytokine production in the periphery, which may be a major contributor to immune activation in PD.
Multiple lines of evidence indicate that inflammation associated with infection triggers increased α-synuclein expression in vivo, with examples including norovirus infection in the human gastrointestinal tract136 and West Nile virus in mice137. In preclinical models, LPS can trigger increased α-synuclein expression138,139, which may be a structurally distinct fibril strain that induces specific pathological patterns of synucleinopathies in mice140. These reports suggest that α-synuclein may have a bidirectional relationship with inflammation or that an inflamed environment may affect α-synuclein processing.
VPS35 mutations in PD
RAB10, a member of the small RAB GTPase family, is a known substrate of LRRK2 (ref.141) and the D620N mutation in Vps35 leads to LRRK2-dependent increases in RAB10 phosphorylation in the spleens of Vps35D620N knock-in mice relative to non-transgenic littermates142. Furthermore, neutrophils and monocytes from patients with PD who carry the D620N mutation exhibited this same increase in LRRK2-mediated RAB10 phosphorylation relative to idiopathic PD and healthy controls142. Such data support the idea that the D620N mutation results in a toxic gain-of-function in an upstream regulator of the LRRK2 kinase pathway. How this interaction and PD-associated mutations affect neutrophil and monocyte function has yet to be elucidated.
PARK7 in PD
DJ1, which is encoded by PARK7, regulates TLR signalling in mouse primary astrocytes, suggesting that DJ1 may play a role in innate immunity143. Indeed, DJ1-deficient Caenorhabditis elegans exposed to pathogenic Pseudomonas aeruginosa exhibit increased p38 phosphorylation and hyper-induction of PMK1 target genes144, which are genes implicated in innate immunity in C. elegans145. More recently, it has been revealed that DJ1-deficient mice exhibit decreased generation of thymus-derived Treg cells and generate increased levels of ROS146. Interestingly, DJ1-deficient Treg cells had higher levels of AKT–mTOR signalling and were less sensitive to TGFβ and IL-2. Treg cells are thought to be neuroprotective in PD via their role in suppressing the inflammatory activity of microglial cells exposed to oxidative stress and inflammation147. Coupled with the observation that loss of DJ1 leads to constitutively active BV2 microglia148, this suggests a mechanism behind the neuroprotective effects of DJ1 in PD.
PD risk factor SNPs and genes are associated with the immune system
In addition to the genetic mutations discussed above, 91 genes and/or risk loci for PD have been identified from GWAS9,149,150, with several of the candidate genes associated with the immune system (Box 1).
Although the association between PD and the HLA region is complex, the hits at HLA-DRB6 and HLA-DQA1 may imply regulation of antigen presentation as a potential mechanism by which the immune response links environmental factors to genetic susceptibility in conferring risk for PD12. Indeed, α-synuclein-derived fragments may act as antigenic epitopes displayed by MHC molecules48. Several SNPs around the GPNMB gene (encoding glycoprotein NMB (GPNMB)) have been linked to risk for developing PD150,151. It has been suggested that GPNMB may regulate systemic immune responses, including inhibition of T cell activation and reducing macrophage inflammatory responses to LPS152,153. Although the function of transmembrane protein 175 (TMEM175) has been recently characterized in neurons, with TMEM175 deficiency decreasing GCase activity and impairing lysosome-dependent clearance of α-synuclein fibrils154, TMEM175 function in glial and immune cells is not well understood. Given that TMEM175 is heavily implicated in the lysosomal pathway, and that lysosomal function is critical for immune cell behaviour, it is highly likely that TMEM175 dysfunction may also affect immune cells. Mutations in the GRN gene that lead to haploinsufficiency of the progranulin (PGRN) protein have been reported to cause frontotemporal dementia155, with the rs5848 SNP increasing PD risk156. Additionally, reduced serum levels of PGRN have been reported in PD157. Although a number of receptors, such as Ephrin 2A receptors158, are regulated by PGRN, current data suggest that PGRN also fulfils receptor-independent intracellular functions, which appear to converge on the lysosome149.
Interestingly, several histological markers and cytokines upregulated in patients with PD are risk factors for PD. An interaction between IL-6 and oestrogen receptor polymorphisms increases the risk for early-onset PD159. Polymorphisms in the TNF gene also increase the risk of PD and earlier disease onset160,161. Similarly, IL1B polymorphisms are more abundant in patients with PD relative to healthy controls and may affect the onset of disease162,163. Despite not being a risk factor for PD, allelic distribution of IFNG is significantly different between early-onset and late-onset PD164, implicating a role of T cells in PD pathogenesis as T cells are major producers of IFNγ. In addition, pathway analysis has implicated genes involved in the ‘regulation of leukocyte/lymphocyte activity’ as conferring an increased susceptibility to PD165.
Four loci associated with PD relate to the transcription factor nuclear factor-κB (NF-κB) that regulates a number of immune genes in response to different stimuli. These loci include DDRGK1 (ref.149) and SCARB2 (encoding lysosomal integral membrane protein 2 (LIMP2); also known as SCARB2) as reported in an earlier GWAS meta-analysis150. Interestingly, SCARB2 is a receptor for GCase and regulates the production of type I interferon through TLR9, another known PD risk locus149.
Epidemiological evidence
Given the heterogeneity of manifestations and sporadic incidence of PD, it is no surprise that disease incidence has been associated with numerous environmental factors that either increase or decrease the risk of its development166. These include links between caffeine consumption167, exercise168, use of nonsteroidal anti-inflammatory drugs (NSAIDs), and smoking169 and reduced incidence of PD and increased incidence after pesticide exposure170, traumatic brain injury171, consumption of dairy products172, and exposure to certain viral or bacterial infections173,174. These findings suggest that inflammation or immunological challenges may synergize with genetic predisposition to trigger PD pathogenesis and progression (Fig. 1). For an in-depth discussion of how gene-by-environment interactions converge on immune and inflammatory pathways and contribute to lifetime risk of PD, we refer the reader to a recent review175.
Pesticides and PD risk
Pesticides and occupational exposure to chemicals can increase the risk of PD176,177. Paraquat is the chemical most linked to risk, and even rotenone, a ‘natural’ plant-derived pesticide used by home gardeners, has been associated with disease178. Both compounds inhibit mitochondrial respiration, which could impact an already dysfunctional endolysosomal pathway in individuals with particular genetic mutations179,180. In addition to paraquat and rotenone, exposure to the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), after its accidental discovery as an agent that caused dopaminergic neuronal loss in humans during recreational drug use, has become one of the main methods to induce dopaminergic neuronal loss and degeneration in animal models181,182,183. Studies have shown that paraquat, rotenone and MPTP all induce both central and peripheral inflammation and oxidative stress184,185,186,187,188.
Links to autoimmune conditions
With overwhelming evidence for a role of inflammation in PD pathogenesis, epidemiological studies have focused on identifying genetic overlap or pleiotropic loci between PD and autoimmune disorders to identify potential common genetic pathways. PD has been epidemiologically linked to several organ-specific and multi-organ autoimmune disorders. Patients with multiple sclerosis, Graves disease (hyperthyroidism), Hashimoto disease (hypothyroidism), pernicious anaemia or polymyalgia rheumatica have a 33% increased risk of developing PD, and this risk increases further after hospitalization with an autoimmune disorder189. A recent GWAS identified 17 shared loci between PD and 7 autoimmune diseases, including type 1 diabetes, Crohn’s disease, ulcerative colitis, rheumatoid arthritis, coeliac disease, psoriasis and multiple sclerosis10. In addition, it is well established that mutations in the LRRK2 gene increase risk of developing PD in patients with Crohn’s disease10 or ulcerative colitis190. Furthermore, LRRK2 has been identified by GWAS as a major susceptibility gene for Crohn’s disease190, and the PD-associated G2019S mutation contributed genetic risk for Crohn’s disease11,191. Collectively, autoimmune diseases represent a group of disorders in which the peripheral immune system is chronically activated and producing inflammatory mediators that may stimulate neuroinflammation and thereby promote PD pathogenesis.
NSAIDs and PD
Given that NSAIDs may reduce the risk of other neurodegenerative diseases192, it is unsurprising that this association has been observed in PD. One of the first reports linking NSAIDs and PD showed that ibuprofen, acetaminophen and aspirin protect dopaminergic neurons in vitro, thus promoting dopaminergic neuronal integrity193. Furthermore, the NSAIDs sodium salicylate, aspirin and meloxicam protect against MPTP-induced dopaminergic neurotoxicity in animal models194,195,196. Following these initial reports, an epidemiological study found that patients with regular use of non-aspirin NSAIDs (two or more tablets per day) had a lower risk of developing PD relative to non-regular users of non-aspirin NSAIDs197. This was further supported by a follow-up study showing that ibuprofen users but not acetaminophen or aspirin users had a 35% lower risk of developing PD than non-users198. This was corroborated by a study which found that, while NSAIDs as a class did not modify the risk, ibuprofen had a slightly protective effect on risk for PD, indicating that certain NSAIDs may provide protective benefits199. These protective properties may be based on the well-known role of NSAIDs in inhibiting cyclooxygenase 1 (COX1) and COX2, thereby reducing the generation of nitric oxide radicals and oxidative stress, to which dopaminergic neurons are particularly susceptible200. In 2006, a study reported that non-aspirin NSAIDs reduced the risk for PD by 20% in men but increased the incidence in women by 20%, being one of the first studies to indicate sex differences with NSAID use and PD incidence201. Some studies have not been able to replicate the link between incidence of PD and NSAID use but have suggested that patients with PD have a higher rate of immediate-type hypersensitivity (asthma, hay fever or allergic rhinitis), providing further evidence for an inflammatory link in the pathogenesis of PD and the need to effectively evaluate the impact of NSAID use prior to the development of PD202,203.
Gut dysbiosis and inflammatory bowel disease
In 2003, Braak et al. introduced a hypothesis that PD pathogenesis originates in the gut204 and that the associated gastrointestinal dysfunction, including a history of constipation, preceded motor symptoms and a PD diagnosis in the clinic by decades3,205. Since then, the hypothesis has been further developed to incorporate contributions from the gut microbiota and intestinal inflammation as a mechanism driving pathology206,207. Differences in gut microbial composition between patients with PD and controls have been extensively reported, indicating that the gastrointestinal environment and its microbial inhabitants are impacted in PD208. Although several studies have identified changes in relative abundance of certain bacteria, including Prevotellaceae, Bifidobacterium, Akkermansia and Lactobacillus, in patients with PD, results often vary given differences in study design and methodology, patient populations, and choice of controls209. While it remains unknown how specific taxa of gut bacteria could contribute to or trigger PD, several studies have shown associations between motor symptoms or disease progression as well as conditions associated with early, pre-motor stages of PD and the relative abundance of certain bacterial families within faecal samples from patients210,211,212. Furthermore, changes in gut bacterial composition have been associated with intestinal inflammation in PD206,213. Higher levels of numerous inflammatory mediators have been found in stool of patients with PD compared to controls, including IL-1α, IL-1β, CXCL8, CRP and calprotectin213,214,215,216, and the levels of some of these molecules are inversely associated with the age of PD symptom onset, suggesting that they could contribute to the development of the disorder213. Levels of Bacteroides and Verrucomicrobia also correlate with plasma levels of TNF and IFNγ217, respectively. These findings are consistent with the hypothesis that gut dysbiosis is linked to an inflammatory environment that may contribute to the initiation of PD pathology. How to effectively target the gut microbiome to delay or mitigate the development of PD remains an area of active research.
Several epidemiological studies have associated risk for PD with inflammatory bowel disease (IBD)11,218,219. A meta-analysis suggests that patients with IBD have a 28–30% increased risk of developing PD220. A systematic review and meta-analysis also found that patients with IBD who are on chronic anti-inflammatory therapy with anti-TNF biologics had 78% lower odds of developing PD than patients with IBD not on anti-TNF drugs, further supporting the hypothesis that chronic inflammation contributes to PD pathogenesis11,221. Additional research is needed to identify therapeutic windows, time and duration of therapy, and whether it should be prophylactic or prior to clinical (motor) symptoms. More importantly, the current FDA-approved anti-TNF biologics may not be the most suitable drug candidates for long-term chronic use because they immunosuppress the patient due to their ability to block both membrane-bound and soluble forms of TNF. In addition, they have very limited brain penetrance. Pegipanermin — a second-generation biologic that is a non-immunosuppressive soluble TNF-selective drug due to its unique dominant-negative mechanism of action222 — crosses the blood–brain barrier and has been shown to have neuroprotective activity in multiple preclinical models of ageing, neuronal dysfunction and neurodegeneration223,224,225,226. This drug is currently in clinical trials for the treatment of pulmonary complications in COVID-19 under the name QUELLOR (ClinicalTrials.gov identifier NCT04370236) and may be worth exploring in PD and other chronic neuroinflammatory brain disorders.
Therapeutic landscape
Given the overwhelming evidence that immune activation and inflammation are hallmarks of PD, it is unsurprising that anti-inflammatory drugs and interventions targeting the immune system have moved forward in the PD clinic as they have in other neurodegenerative diseases like amyotrophic lateral sclerosis and Alzheimer disease. Table 1 lists clinical trials that involve immunotherapies against α-synuclein and immunomodulatory or anti-inflammatory approaches that have been completed or are in progress. Most of these trials have clinical (motor) and not immunological end points, and some include measures of target engagement, which is deemed critical for hypothesis-driven decision-making. As in other neurodegenerative diseases, the outcome of anti-inflammatory drug trials in PD has been terribly disappointing thus far, and changes in the approach to such trials are warranted. The addition of immune-related end points may be an important clinical consideration that could impact the design of future, potentially more successful studies using combination therapies that target multiple processes, including immunological responses. Additionally, the selection of patient cohorts and enrolment criteria for any clinical study is critical to the outcome as is the point at which any given therapy or drug is initiated. Specifically, for inflammation in PD pathogenesis, if the inflammatory process or immune dysfunction begins early in the disease process, an anti-inflammatory intervention that is started after a clinical (motor) diagnosis is made in a neurologist’s office is unlikely to have a disease-modifying effect. Based on the abundant data supporting a prodromal, preclinical stage of PD that includes sleep, olfactory and gastrointestinal dysfunction that begins decades before motor symptoms develop, biofluids and immune cells should be collected from individuals with these features and interrogated for dysfunction and inflammation in order to find inflammatory endophenotypes for enrolment into these trials in order to directly test hypotheses about inflammation and target engagement. This could position the neurodegeneration field for greater success at the level of the FDA by proposing the performance of shorter biomarker-directed clinical trials with a clear target engagement study design that will inform better decision-making going forward into phase III trials.
Challenges and future directions
As the field of neurodegeneration moves forward to critically interrogate the role of central and peripheral inflammation in the pathogenesis and progression of PD, one major challenge is the development of new techniques, such as single-cell multi-omics, machine learning or advanced patient-derived models227, that will enable the field to rigorously test whether or not the immune system and inflammation are critical components of the disease, how immune cells protect or predispose neurons to injury, and at which stages immune processes play critical roles and are predictive of disease trajectory. To do this, we need to have a better understanding of how genes and the environment impact immune function over time. To this end, the field will need to engage in longitudinal collection of immune cells and tissues from individuals at genetic and environmental risk for PD, and not just from those with clinical motor features, to perform deep genetic, transcriptional and immune profiling. This will enable researchers to look for meaningful changes in responses to immune challenges as opposed to comparing baseline differences between disease cases and healthy controls — specifically, the detection of an immune dysfunctional trait rather than an immune dysfunctional state. A second challenge will be the development of non-invasive tools to identify individuals exhibiting neuroinflammation. Currently, PET imaging is the best technique available to the field. However, high costs, low signal-to-noise ratio and genetic polymorphisms in the TSPO gene in the human population significantly curtail its widespread use228,229. Perhaps the use of non-invasive breath volatile organic compound mass-spectrometry technology230 will enable monitoring of treatment responsiveness and measurement of target engagement simultaneously. Another promising technology is free-water measurement in diffusion-tensor magnetic resonance imaging (DT-MRI), which is a sensitive method widely used in patients with Alzheimer disease and PD to identify clinical pathology. This technology could be leveraged to also identify neuroinflammatory changes in the vicinity of white-matter tracts in the CNS231,232,233,234.
While genetic research is informative, obtaining clinical samples from patients with monogenic forms of PD or specific risk SNPs is not trivial because these individuals are relatively rare. As an example, heterozygote carriers of the LRRK2G2019S mutation are prevalent in the Ashkenazi Jewish ancestry community; however, when trials with LRRK2 kinase inhibitors begin in the clinic, their participation will be highly sought by a large number of pharmaceutical companies, and the interpretation of results from individuals participating in multiple trials will be complex. Enrolling specific clinical populations for assessment of immunomodulatory therapies, including patients of genetic risk tied to the immune system, will be crucial to determine the neuroprotective effects as a response to a genetic contribution. Additionally, while the field has made significant strides in identifying genes and gene variants associated with PD risk in individuals with European ancestry, better efforts are needed to perform similar studies in diverse populations. These efforts have been severely lagging, a serious problem since immune-related genes are expected to vary widely based on ethnicity and geography. Immune-based therapies are therefore unlikely to have equivalent efficacy in all populations and should be tested in cohorts with diverse ethnic backgrounds.
Perhaps the most pressing challenge in the field is the need to develop tools enabling earlier diagnosis to deliver effective therapies to prevent, delay or arrest disease. The overall sentiment in the field from preclinical data is that, if PD begins outside the brain and in peripheral organs and then progresses into the brain, diagnosis in the pre-motor stages of the disease must be achievable. A deeper understanding and accurate detection of the immunological mechanisms underlying the earliest signs of parkinsonism will lead to new therapies and may one day enable clinicians to intervene effectively with novel or repurposed anti-inflammatory and immunomodulatory therapies to slow or delay the progression of disease from the periphery to the CNS.
References
Czirr, E. & Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Invest. 122, 1156–1163 (2012). A review underscoring the importance of bidirectional communication between the peripheral and central immune and nervous systems for brain health and potential role of intrinsic innate molecules in neurodegeneration.
Papadopoulos, Z., Herz, J. & Kipnis, J. Meningeal lymphatics: from anatomy to central nervous system immune surveillance. J. Immunol. 204, 286–293 (2020). The role of meningeal lymphatics in communication between the brain and the peripheral immune system is discussed within the context of CNS-related immune responses.
Savica, R. et al. Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology 73, 1752–1758 (2009).
Okun, M., Malaty, I. & Deeb, W. Living with Parkinson’s Disease: A Complete Guide for Patients and Caregivers (Robert Rose, 2020).
Lindqvist, D. et al. Non-motor symptoms in patients with Parkinson’s disease - correlations with inflammatory cytokines in serum. PLoS ONE 7, e47387 (2012).
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
Klein, C. & Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a008888 (2012).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Pierce, S. & Coetzee, G. A. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS ONE 12, e0175882 (2017).
Witoelar, A. et al. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 74, 780 (2017).
Hui, K. Y. et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl Med. 10, eaai7795 (2018). Exosome sequencing studies revealed shared LRRK2 alleles in PD and Crohn’s disease providing insight into potential underlying disease mechanisms.
Kannarkat, G. T. et al. Common genetic variant association with altered HLA expression, synergy with pyrethroid exposure, and risk for Parkinson’s disease: an observational and case–control study. NPJ Parkinsons Dis. 1, 15002 (2015). Immunophenotyping studies of human peripheral blood mononuclear cells support functional consequences of gene-by-environment interplay between specific pesticide exposure and an HLA-DRA polymorphism associated with risk for late-onset PD.
Franceschi, C., Valensin, S., Fagnoni, F., Barbi, C. & Bonafe, M. Biomarkers of immunosenescence within an evolutionary perspective: the challenge of heterogeneity and the role of antigenic load. Exp. Gerontol. 34, 911–921 (1999).
Franceschi, C., Bonafe, M. & Valensin, S. Human immunosenescence: the prevailing of innate immunity, the failing of clonotypic immunity, and the filling of immunological space. Vaccine 18, 1717–1720 (2000).
Goronzy, J. J., Li, G., Yang, Z. & Weyand, C. M. The janus head of T cell aging - autoimmunity and immunodeficiency. Front. Immunol. 4, 131 (2013). A critical review of the age-acquired autoimmunity and immunodeficiency displayed by the human adaptive immune system.
Calabrese, V. et al. Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 115, 80–91 (2018).
Meszaros, A. et al. Neurovascular inflammaging in health and disease. Cells 9, 1614 (2020).
McGeer, P. L., Itagaki, S., Boyes, B. E. & McGeer, E. G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291 (1988). Neuropathological studies of autopsy brains documenting the presence of activated innate immune cells in midbrains of patients with PD and AD.
Imamura, K. et al. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 106, 518–526 (2003).
Nagatsu, T., Mogi, M., Ichinose, H. & Togari, A. Cytokines in Parkinson’s disease. J. Neural Transm. Suppl. 58, 143–151 (2000).
Harms, A. S., Ferreira, S. A. & Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 141, 527–545 (2021).
Gerhard, A. et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 21, 404–412 (2006).
Bartels, A. L. et al. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat. Disord. 16, 57–59 (2010).
Tronel, C. et al. Molecular targets for PET imaging of activated microglia: the current situation and future expectations. Int. J. Mol. Sci. 18, 802 (2017).
Joers, V., Tansey, M. G., Mulas, G. & Carta, A. R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 155, 57–75 (2017).
Sanchez-Guajardo, V., Barnum, C. J., Tansey, M. G. & Romero-Ramos, M. Neuroimmunological processes in Parkinson’s disease and their relation to alpha-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 5, 113–139 (2013).
Tremblay, M. E., Cookson, M. R. & Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 14, 16 (2019).
Schapansky, J., Nardozzi, J. D. & LaVoie, M. J. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience 302, 74–88 (2015).
Grozdanov, V. et al. Increased immune activation by pathologic alpha-synuclein in Parkinson’s disease. Ann. Neurol. 86, 593–606 (2019).
Govindarajan, V., de Rivero Vaccari, J. P. & Keane, R. W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J. Neuroinflammation 17, 260 (2020).
Gordon, D. et al. Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol. Dis. 121, 148–162 (2019).
Scheiblich, H. et al. Microglial NLRP3 inflammasome activation upon TLR2 and TLR5 ligation by distinct alpha-synuclein assemblies. J. Immunol. 207, 2143–2154 (2021).
Grozdanov, V. et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol. 128, 651–663 (2014).
Cook, D. A. et al. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis. 3, 11 (2017).
Bliederhaeuser, C. et al. LRRK2 contributes to monocyte dysregulation in Parkinson’s disease. Acta Neuropathol. Commun. 4, 123 (2016).
Schlachetzki, J. C. M. et al. A monocyte gene expression signature in the early clinical course of Parkinson’s disease. Sci. Rep. 8, 10757 (2018).
Li, J. et al. Parkin impairs antiviral immunity by suppressing the mitochondrial reactive oxygen species-Nlrp3 axis and antiviral inflammation. iScience 16, 468–484 (2019).
Brochard, V. et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 119, 182–192 (2009). One of the earliest studies supporting a role for peripheral immune cell infiltration in experimental parkinsonism.
Jiang, S., Gao, H., Luo, Q., Wang, P. & Yang, X. The correlation of lymphocyte subsets, natural killer cell, and Parkinson’s disease: a meta-analysis. Neurol. Sci. 38, 1373–1380 (2017).
Saunders, J. A. H. et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharmacol. 7, 927–938 (2012).
Fiszer, U., Mix, E., Fredrikson, S., Kostulas, V. & Link, H. Parkinson’s disease and immunological abnormalities: increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol. Scand. 90, 160–166 (1994).
Kustrimovic, N. et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflammation 15, 205 (2018).
Rosenkranz, D. et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J. Neuroimmunol. 188, 117–127 (2007).
Kipnis, J. et al. Dopamine, through the extracellular signal-regulated kinase pathway, downregulates CD4+CD25+ regulatory T-cell activity: implications for neurodegeneration. J. Neurosci. 24, 6133–6143 (2004).
Elgueta, D. et al. Dopamine receptor D3 expression is altered in CD4+ T-cells from Parkinson’s disease patients and its pharmacologic inhibition attenuates the motor impairment in a mouse model. Front. Immunol. 10, 981 (2019).
Kustrimovic, N. et al. Dopaminergic receptors on CD4+ T naive and memory lymphocytes correlate with motor impairment in patients with Parkinson’s disease. Sci. Rep. 6, 33738 (2016).
Bongioanni, P., Castagna, M., Maltinti, S., Boccardi, B. & Dadone, F. T-lymphocyte tumor necrosis factor-alpha receptor binding in patients with Parkinson’s disease. J. Neurol. Sci. 149, 41–45 (1997).
Sulzer, D. et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661 (2017). One of the first studies to report the presence of α-synuclein-specific T cells in peripheral blood of patients with PD.
Lindestam Arlehamn, C. S. et al. -Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 11, 1875 (2020).
Stevens, C. H. et al. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 252, 95–99 (2012).
Bas, J. et al. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J. Neuroimmunol. 113, 146–152 (2001).
Lee, C. et al. The lysosomal potassium channel TMEM175 adopts a novel tetrameric architecture. Nature 547, 472–475 (2017).
Orr, C. F., Rowe, D. B., Mizuno, Y., Mori, H. & Halliday, G. M. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 128, 2665–2674 (2005).
Yanamandra, K. et al. alpha-synuclein reactive antibodies as diagnostic biomarkers in blood sera of Parkinson’s disease patients. PLoS ONE 6, e18513 (2011).
Double, K. L. et al. Anti-melanin antibodies are increased in sera in Parkinson’s disease. Exp. Neurol. 217, 297–301 (2009).
Horvath, I., Iashchishyn, I. A., Forsgren, L. & Morozova-Roche, L. A. Immunochemical detection of alpha-synuclein autoantibodies in Parkinson’s disease: correlation between plasma and cerebrospinal fluid levels. ACS Chem. Neurosci. 8, 1170–1176 (2017).
Suwarnalata, G. et al. Augmentation of Autoantibodies by Helicobacter pylori in Parkinson’s disease patients may be linked to greater severity. PLoS ONE 11, e0153725 (2016).
Caggiu, E. et al. Humoral cross reactivity between alpha-synuclein and herpes simplex-1 epitope in Parkinson’s disease, a triggering role in the disease? J. Neuroimmunol. 291, 110–114 (2016).
Reale, M. et al. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 23, 55–63 (2009).
Brodacki, B. et al. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 441, 158–162 (2008).
Baba, Y., Kuroiwa, A., Uitti, R. J., Wszolek, Z. K. & Yamada, T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat. Disord. 11, 493–498 (2005).
McCoy, M. K., Ruhn, K. A., Blesch, A. & Tansey, M. G. TNF: a key neuroinflammatory mediator of neurotoxicity and neurodegeneration in models of Parkinson’s disease. Adv. Exp. Med. Biol. 691, 539–540 (2011).
Mogi, M. et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 180, 147–150 (1994).
Mogi, M. et al. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 165, 208–210 (1994).
Blum-Degen, D. et al. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20 (1995).
Eidson, L. N. et al. Candidate inflammatory biomarkers display unique relationships with alpha-synuclein and correlate with measures of disease severity in subjects with Parkinson’s disease. J. Neuroinflammation 14, 164 (2017). Twenty-four-hour qualification study in CSF and serum of patients with PD and healthy controls documenting fluctuations in inflammatory analytes, correlation between central and peripheral compartments, differences between PD and controls, and relationship to neurodegeneration peptides and clinical symptoms.
Barnum, C. J. et al. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro(R)1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Parkinsons Dis. 4, 349–360 (2014).
McCoy, M. K. et al. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol. Ther. 16, 1572–1579 (2008).
McCoy, M. K. et al. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 26, 9365–9375 (2006).
Harms, A. S. et al. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson’s disease. Mol. Ther. 19, 46–52 (2011).
Peter, I. et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 75, 939–946 (2018). Epidemiological studies supporting an association between IBD and PD risk and the potential therapeutic utility of anti-TNF therapy to mitigate risk for development of PD.
Roulis, M., Armaka, M., Manoloukos, M., Apostolaki, M. & Kollias, G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl Acad. Sci. USA 108, 5396–5401 (2011).
Dale, R. C. et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain 127, 21–33 (2004).
Keo, A. et al. Transcriptomic signatures of brain regional vulnerability to Parkinson’s disease. Commun. Biol. 3, 101 (2020).
Liscovitch, N. & French, L. Differential co-expression between α-synuclein and IFN-γ signaling genes across development and in Parkinson’s disease. PLoS ONE 9, e115029 (2014).
Chakrabarty, P. et al. Interferon-gamma induces progressive nigrostriatal degeneration and basal ganglia calcification. Nat. Neurosci. 14, 694–696 (2011).
Mount, M. P. et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337 (2007).
Kozina, E. et al. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain 141, 1753–1769 (2018). Studies of immune challenge in bone-marrow chimeric mice with wild-type LRRK2 in the brain and mutant LRRK2 in the periphery support the primacy of the peripheral immune system genotype in the neurodegeneration outcome.
Panagiotakopoulou, V. et al. Interferon-gamma signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat. Commun. 11, 5163 (2020).
Gardet, A. et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 185, 5577–5585 (2010).
Thevenet, J., Pescini Gobert, R., Hooft van Huijsduijnen, R., Wiessner, C. & Sagot, Y. J. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS ONE 6, e21519 (2011).
Moehle, M. S. et al. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 32, 1602–1611 (2012).
Paisan-Ruiz, C. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004).
Zimprich, A. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004).
Hernandez, D. G., Reed, X. & Singleton, A. B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 139, 59–74 (2016).
Gardet, A. et al. LRRK2 is involved in the IFN-γ response and host response to pathogens. J. Immunol. 185, 5577–5585 (2010).
Kuss, M., Adamopoulou, E. & Kahle, P. J. Interferon-gamma induces leucine-rich repeat kinase LRRK2 via extracellular signal-regulated kinase ERK5 in macrophages. J. Neurochem. 129, 980–987 (2014).
Rideout, H. J. & Re, D. B. LRRK2 and the “LRRKtosome” at the crossroads of programmed cell death: clues from RIP kinase relatives. Adv. Neurobiol. 14, 193–208 (2017).
Di Fonzo, A. et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365, 412–415 (2005).
Monfrini, E. & Di Fonzo, A. Leucine-rich repeat kinase (LRRK2) genetics and Parkinson’s Disease. Adv. Neurobiol. 14, 3–30 (2017).
Smith, W. W. et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 9, 1231–1233 (2006).
Anand, V. S. & Braithwaite, S. P. LRRK2 pathobiology in Parkinson’s disease. J. Neurochem. 276, 6428–6435 (2009).
Anand, V. S. et al. Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 276, 466–478 (2009).
Greggio, E. et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Nuerobiol. Dis. 23, 329–341 (2006).
West, A. B. et al. From the cover: Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA 102, 16842–16847 (2005).
Dzamko, N., Rowe, D. B. & Halliday, G. M. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov. Disord. 31, 889–897 (2016).
Lee, B. D. et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 16, 998–1000 (2010).
Wang, Z., Arat, S., Magid-Slav, M. & Brown, J. R. Meta-analysis of human gene expression in response to Mycobacterium tuberculosis infection reveals potential therapeutic targets. BMC Syst. Biol. 12, 3 (2018).
Hartlova, A. et al. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J. 37, e98694 (2018).
Liu, W. et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 214, 3051–3066 (2017).
Zhang, Q. et al. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 16, 918–926 (2015).
Shutinoski, B. et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl Med. 11, eaas9292 (2019). LRRK2 alleles modulate the outcome of Salmonella and reovirus-induced encephalitic microbial infections in mice.
Ross, O. A. et al. Analysis of Lrrk2 R1628P as a risk factor for Parkinson’s disease. Ann. Neurol. 64, 88–92 (2008).
Fava, V. M. et al. Pleiotropic effects for Parkin and LRRK2 in leprosy type-1 reactions and Parkinson’s disease. Proc. Natl Acad. Sci. USA 116, 15616–15624 (2019).
Fernandes, H. et al. ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 6, 342–356 (2016).
Farfel-Becker, T. et al. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 23, 843–854 (2014).
Vitner, E. B., Farfel-Becker, T., Eilam, R., Biton, I. & Futerman, A. H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 135, 1724–1735 (2012).
Vitner, E. B. et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 20, 204–208 (2014).
Mizukami, H. et al. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Invest. 109, 1215–1221 (2002).
Panicker, L. M. et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl Acad. Sci. USA 109, 18054–18059 (2012). Inflammatory phenotypes and glucocerebrosidase deficits in human islet-derived precursor cells from patients with Gaucher’s disease.
Panicker, L. M. et al. Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cell 32, 2338–2349 (2014).
Chahine, L. M. et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 70, 852–858 (2013).
Atashrazm, F. et al. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov. Disord. 34, 406–415 (2019).
Ferreira, M. & Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 135, 273–284 (2017).
Kumazawa, R. et al. Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch. Neurol. 65, 802–808 (2008).
Samaranch, L. et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133, 1128–1142 (2010).
Camargos, S. T. et al. Familial parkinsonism and early onset Parkinson’s disease in a Brazilian movement disorders clinic: phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov. Disord. 24, 662–666 (2009).
Marongiu, R. et al. Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum. Mutat. 28, 98 (2007).
Scarffe, L. A., Stevens, D. A., Dawson, V. L. & Dawson, T. M. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37, 315–324 (2014).
Sliter, D. A. et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018).
West, A. P. & Shadel, G. S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375 (2017).
Nazmi, A. et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 67, 1254–1276 (2019).
Weindel, C. G. et al. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. eLife 9, e51071 (2020).
Matheoud, D. et al. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 166, 314–327 (2016). PINK1 and parkin identified as suppressors of immune-related inflammatory responses by inhibiting formation and trafficking of vesicles involved in mitochondrial antigen presentation.
Chartier-Harlin, M. C. et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
Harms, A. S. et al. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. 33, 9592–9600 (2013).
Volpicelli-Daley, L. A., Kirik, D., Stoyka, L. E., Standaert, D. G. & Harms, A. S. How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J. Neurochem. 139, 131–155 (2016).
Earls, R. H. et al. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflammation 16, 250 (2019).
Harms, A. S. et al. Peripheral monocyte entry is required for alpha-synuclein induced inflammation and neurodegeneration in a model of Parkinson disease. Exp. Neurol. 300, 179–187 (2018).
Kim, C. et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562 (2013).
Zhang, W. et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542 (2005).
Rocha, E. M., De Miranda, B. & Sanders, L. H. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257 (2018).
Caggiu, E. et al. Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson’s disease. J. Neuroimmunol. 310, 26–31 (2017).
Codolo, G. et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375 (2013).
White, A. J. et al. The peripheral inflammatory response to alpha-synuclein and endotoxin in Parkinson’s disease. Front. Neurol. 9, 946 (2018).
Stolzenberg, E. et al. A role for neuronal alpha-synuclein in gastrointestinal immunity. J. Innate Immun. 9, 456–463 (2017).
Beatman, E. L. et al. Alpha-synuclein expression restricts RNA viral infections in the brain. J. Virol. 90, 2767–2782 (2015).
Gao, H. M. et al. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J. Neurosci. 28, 7687–7698 (2008).
La Vitola, P. et al. Peripheral inflammation exacerbates alpha-synuclein toxicity and neuropathology in Parkinson’s models. Neuropathol. Appl. Neurobiol. 47, 43–60 (2021).
Kim, C. et al. Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci. Rep. 6, 30891 (2016).
Steger, M. et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 6, e31012 (2017).
Mir, R. et al. The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochemical J. 475, 1861–1883 (2018).
Waak, J. et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 23, 2478–2489 (2009).
Cornejo Castro, E. M. et al. Parkinson’s disease-associated DJ-1 modulates innate immunity signaling in Caenorhabditis elegans. J. Neural Transm. 117, 599–604 (2010).
Bolz, D. D., Tenor, J. L. & Aballay, A. A conserved PMK-1/p38 MAPK is required in caenorhabditis elegans tissue-specific immune response to yersinia pestis infection. J. Biol. Chem. 285, 10832–10840 (2010).
Singh, Y. et al. Differential effect of DJ-1/PARK7 on development of natural and induced regulatory T cells. Sci. Rep. 5, 17723 (2015).
Reynolds, A. D., Banerjee, R., Liu, J., Gendelman, H. E. & Mosley, R. L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 82, 1083–1094 (2007).
Meiser, J. et al. Loss of DJ-1 impairs antioxidant response by altered glutamine and serine metabolism. Neurobiol. Dis. 89, 112–125 (2016).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516 (2017).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993 (2014).
Murthy, M. N. et al. Increased brain expression of GPNMB is associated with genome wide significant risk for Parkinson’s disease on chromosome 7p15.3. Neurogenetics 18, 121–133 (2017).
Chung, J. S. et al. DC-HIL is a negative regulator of T lymphocyte activation. Blood 109, 4320–4327 (2007).
Ripoll, V. M., Irvine, K. M., Ravasi, T., Sweet, M. J. & Hume, D. A. Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J. Immunol. 178, 6557–6566 (2007).
Jinn, S. et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl Acad. Sci. USA 114, 2389–2394 (2017).
Baker, M. et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 (2006).
Chang, K.-H. et al. Association between GRN rs5848 polymorphism and Parkinson’s disease in Taiwanese population. PLoS ONE 8, e54448 (2013).
Mateo, I. et al. Reduced serum progranulin level might be associated with Parkinson’s disease risk. Eur. J. Neurol. 20, 1571–1573 (2013).
Neill, T. et al. EphA2 is a functional receptor for the growth factor progranulin. J. Cell Biol. 215, 687–703 (2016).
Hakansson, A. et al. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am. J. Med. Genet. B 133B, 88–92 (2005).
Nishimura, M. et al. Tumor necrosis factor gene polymorphisms in patients with sporadic Parkinson’s disease. Neurosci. Lett. 311, 1–4 (2001).
Chu, K., Zhou, X. & Luo, B. Y. Cytokine gene polymorphisms and Parkinson’s disease: a meta-analysis. Can. J. Neurol. Sci. 39, 58–64 (2012).
Schulte, T. et al. Polymorphisms in the interleukin-1 alpha and beta genes and the risk for Parkinson’s disease. Neurosci. Lett. 326, 70–72 (2002).
Nishimura, M., Kuno, S., Kaji, R., Yasuno, K. & Kawakami, H. Glutathione-S-transferase-1 and interleukin-1beta gene polymorphisms in Japanese patients with Parkinson’s disease. Mov. Disord. 20, 901–902 (2005).
Mizuta, I. et al. Relation between the high production related allele of the interferon-gamma (IFN-gamma) gene and age at onset of idiopathic Parkinson’s disease in Japan. J. Neurol. Neurosurg. Psychiatry 71, 818–819 (2001).
Holmans, P. et al. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Hum. Mol. Genet. 22, 1039–1049 (2013).
Simon, D. K., Tanner, C. M. & Brundin, P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin. Geriatr. Med. 36, 1–12 (2020).
Hernan, M. A., Takkouche, B., Caamano-Isorna, F. & Gestal-Otero, J. J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 52, 276–284 (2002).
Yang, F. et al. Physical activity and risk of Parkinson’s disease in the Swedish National March Cohort. Brain 138, 269–275 (2015).
Ritz, B. et al. Pooled analysis of tobacco use and risk of Parkinson disease. Arch. Neurol. 64, 990–997 (2007).
Priyadarshi, A., Khuder, S. A., Schaub, E. A. & Shrivastava, S. A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology 21, 435–440 (2000).
Kenborg, L. et al. Head injury and risk for Parkinson disease: results from a Danish case-control study. Neurology 84, 1098–1103 (2015).
Park, M. et al. Consumption of milk and calcium in midlife and the future risk of Parkinson disease. Neurology 64, 1047–1051 (2005).
Brugger, F. et al. Why is there motor deterioration in Parkinson’s disease during systemic infections-a hypothetical view. NPJ Parkinsons Dis. 1, 15014 (2015).
Costa Sa, A. C., Madsen, H. & Brown, J. R. Shared molecular signatures across neurodegenerative diseases and herpes virus infections highlights potential mechanisms for maladaptive innate immune responses. Sci. Rep. 9, 8795 (2019).
Kline, E. M. et al. Genetic and environmental factors in Parkinson’s disease converge on immune function and inflammation. Mov. Disord. 36, 25–36 (2021). Environmental factors (pesticides and infections) increase the risk for idiopathic PD via the immune system. This review highlights the major PD-relevant genes expressed in immune cells and key environmental factors that activate immune cells and that, alone or in combination with other factors, may contribute to idiopathic PD pathogenesis.
Steenland, K. et al. Polychlorinated biphenyls and neurodegenerative disease mortality in an occupational cohort. Epidemiology 17, 8–13 (2006).
Goldman, S. M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 54, 141–164 (2014).
Tanner, C. M. et al. Rotenone, paraquat, and Parkinson’s disease. Env. Health Perspect. 119, 866–872 (2011).
Plotegher, N. & Duchen, M. R. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front. Cell Dev. Biol. 5, 110 (2017).
Kett, L. R. & Dauer, W. T. Endolysosomal dysfunction in Parkinson’s disease: recent developments and future challenges. Mov. Disord. 31, 1433–1443 (2016).
Langston, J. W. MPTP neurotoxicity: an overview and characterization of phases of toxicity. Life Sci. 36, 201–206 (1985).
Langston, J. W. & Ballard, P. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): implications for treatment and the pathogenesis of Parkinson’s disease. Can. J. Neurol. Sci. 11, 160–165 (1984).
Do, C. B. et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 7, e1002141 (2011).
Lofrumento, D. D. et al. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation 18, 79–88 (2011).
Joers, V. et al. Microglia, inflammation and gut microbiota responses in a progressive monkey model of Parkinson’s disease: a case series. Neurobiol. Dis. 144, 105027 (2020).
Maitra, U., Scaglione, M. N., Chtarbanova, S. & O’Donnell, J. M. Innate immune responses to paraquat exposure in a Drosophila model of Parkinson’s disease. Sci. Rep. 9, 12714 (2019).
Thakur, P. & Nehru, B. Inhibition of neuroinflammation and mitochondrial dysfunctions by carbenoxolone in the rotenone model of Parkinson’s disease. Mol. Neurobiol. 51, 209–219 (2015).
Morais, L. H., Hara, D. B., Bicca, M. A., Poli, A. & Takahashi, R. N. Early signs of colonic inflammation, intestinal dysfunction, and olfactory impairments in the rotenone-induced mouse model of Parkinson’s disease. Behav. Pharmacol. 29, 199–210 (2018).
Li, X., Sundquist, J. & Sundquist, K. Subsequent risks of Parkinson disease in patients with autoimmune and related disorders: a nationwide epidemiological study from Sweden. Neurodegener. Dis. 10, 277–284 (2012).
Villumsen, M., Aznar, S., Pakkenberg, B., Jess, T. & Brudek, T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut 68, 18–24 (2019).
Rivas, M. A. et al. Correction: Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet. 15, e1008190 (2019).
Zhang, C., Wang, Y., Wang, D., Zhang, J. & Zhang, F. NSAID exposure and risk of Alzheimer’s disease: an updated meta-analysis from cohort studies. Front. Aging Neurosci. 10, 83 (2018).
Casper, D., Yaparpalvi, U., Rempel, N. & Werner, P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 289, 201–204 (2000).
Sairam, K., Saravanan, K. S., Banerjee, R. & Mohanakumar, K. P. Non-steroidal anti-inflammatory drug sodium salicylate, but not diclofenac or celecoxib, protects against 1-methyl-4-phenyl pyridinium-induced dopaminergic neurotoxicity in rats. Brain Res. 966, 245–252 (2003).
Aubin, N., Curet, O., Deffois, A. & Carter, C. Aspirin and salicylate protect against MPTP-induced dopamine depletion in mice. J. Neurochem. 71, 1635–1642 (1998).
Ferger, B., Teismann, P., Earl, C. D., Kuschinsky, K. & Oertel, W. H. Salicylate protects against MPTP-induced impairments in dopaminergic neurotransmission at the striatal and nigral level in mice. Naunyn Schmiedebergs Arch. Pharmacol. 360, 256–261 (1999).
Chen, H. et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 60, 1059–1064 (2003).
Chen, H. et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 58, 963–967 (2005).
Samii, A., Etminan, M., Wiens, M. O. & Jafari, S. NSAID use and the risk of Parkinson’s disease: systematic review and meta-analysis of observational studies. Drugs Aging 26, 769–779 (2009).
Asanuma, M., Nishibayashi-Asanuma, S., Miyazaki, I., Kohno, M. & Ogawa, N. Neuroprotective effects of non-steroidal anti-inflammatory drugs by direct scavenging of nitric oxide radicals. J. Neurochem. 76, 1895–1904 (2001).
Hernan, M. A., Logroscino, G. & Garcia Rodriguez, L. A. Nonsteroidal anti-inflammatory drugs and the incidence of Parkinson disease. Neurology 66, 1097–1099 (2006).
Ton, T. G. et al. Nonsteroidal anti-inflammatory drugs and risk of Parkinson’s disease. Mov. Disord. 21, 964–969 (2006).
Poly, T. N., Islam, M. M. R., Yang, H. C. & Li, Y. J. Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: a meta-analysis. Eur. J. Clin. Pharmacol. 75, 99–108 (2019).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Noyce, A. J. et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 72, 893–901 (2012).
Romano, S. et al. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 7, 27 (2021).
Houser, M. C. & Tansey, M. G. The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis. 3, 3 (2017). Evidence that intestinal dysfunction is present in PD and that it may reflect the earliest manifestations of disease pathology, and the link to dysregulated immune activity is reviewed along with potential mechanisms associated to neuropathology.
Bhattarai, Y. & Kashyap, P. C. Parkinson’s disease: are gut microbes involved? Am. J. Physiol. Gastrointest. Liver Physiol. 319, G529–G540 (2020).
Boertien, J. M., Pereira, P. A. B., Aho, V. T. E. & Scheperjans, F. Increasing comparability and utility of gut microbiome studies in Parkinson’s disease: a systematic review. J. Parkinsons Dis. 9 (Suppl. 2), S297–S312 (2019).
Aho, V. T. E. et al. Gut microbiota in Parkinson’s disease: temporal stability and relations to disease progression. EBioMedicine 44, 691–707 (2019).
Scheperjans, F. et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015).
Heinzel, S. et al. Gut microbiome signatures of risk and prodromal markers of Parkinson disease. Ann. Neurol. 88, 320–331 (2020).
Aho, V. T. E. et al. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol. Neurodegener. 16, 6 (2021).
Houser, M. C. et al. Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov. Disord. 33, 793–804 (2018).
Mulak, A., Koszewicz, M., Panek-Jeziorna, M., Koziorowska-Gawron, E. & Budrewicz, S. Fecal calprotectin as a marker of the gut immune system activation is elevated in Parkinson’s disease. Front. Neurosci. 13, 992 (2019).
Schwiertz, A. et al. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat. Disord. 50, 104–107 (2018).
Lin, C. H. et al. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflammation 16, 129 (2019).
Lin, J. C., Lin, C. S., Hsu, C. W., Lin, C. L. & Kao, C. H. Association between Parkinson’s disease and inflammatory bowel disease: a nationwide Taiwanese retrospective cohort study. Inflamm. Bowel Dis. 22, 1049–1055 (2016).
Weimers, P. et al. Inflammatory bowel disease and Parkinson’s disease: a nationwide Swedish cohort study. Inflamm. Bowel Dis. 25, 111–123 (2019).
Zhu, F. et al. The risk of Parkinson’s disease in inflammatory bowel disease: a systematic review and meta-analysis. Dig. Liver Dis. 51, 38–42 (2019).
Park, S. et al. Patients with inflammatory bowel disease are at an increased risk of Parkinson’s disease: a south Korean nationwide population-based study. J. Clin. Med. 8, 1191 (2019).
Steed, P. M. et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 301, 1895–1898 (2003).
Cavanagh, C. et al. Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 47, 41–49 (2016).
MacPherson, K. P. et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 102, 81–95 (2017).
McAlpine, F. E. et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 34, 163–177 (2009).
Sama, D. M. et al. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS ONE 7, e38170 (2012).
Rajewsky, N. et al. LifeTime and improving European healthcare through cell-based interceptive medicine. Nature 587, 377–386 (2020).
Milenkovic, V. M. et al. Effects of genetic variants in the TSPO gene on protein structure and stability. PLoS ONE 13, e0195627 (2018).
Owen, D. R. et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 32, 1–5 (2012).
Patterson, S. G. et al. Breath analysis by mass spectrometry: a new tool for breast cancer detection? Am. Surg. 77, 747–751 (2011).
Dumont, M. et al. Free water in white matter differentiates MCI and AD from control subjects. Front. Aging Neurosci. 11, 270 (2019).
Ofori, E. et al. Free-water imaging of the hippocampus is a sensitive marker of Alzheimer’s disease. Neuroimage Clin. 24, 101985 (2019).
Ofori, E. et al. Free water improves detection of changes in the substantia nigra in parkinsonism: a multisite study. Mov. Disord. 32, 1457–1464 (2017).
Yang, J. et al. Multimodal dopaminergic and free-water imaging in Parkinson’s disease. Parkinsonism Relat. Disord. 62, 10–15 (2019).
Gendelman, H. E. et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Parkinsons Dis. 3, 10 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03790670 (2020).
Athauda, D. et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675 (2017).
. Pioglitazone in early Parkinson’s disease: a phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 14, 795–803 (2015).
Peball, M. et al. Non-motor symptoms in Parkinson’s disease are reduced by nabilone. Ann. Neurol. 88, 712–722 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03773796 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02818777 (2019).
Jankovic, J. et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. 75, 1206–1214 (2018).
Schenk, D. B. et al. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 32, 211–218 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03100149 (2021).
Brys, M. et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov. Disord. 34, 1154–1163 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03318523 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02216188 (2015).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02618941 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01885494 (2015).
McFarthing, K. & Simuni, T. Clinical trial highlights: phase III study in spotlight. J. Parkinsons Dis. 9, 3–4 (2019).
Braczynski, A. K., Schulz, J. B. & Bach, J. P. Vaccination strategies in tauopathies and synucleinopathies. J. Neurochem. 143, 467–488 (2017).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04075318 (2021).
Pagan, F. L. et al. Nilotinib effects on safety, tolerability, and potential biomarkers in parkinson disease: a phase 2 randomized clinical trial. JAMA Neurol. 77, 309–317 (2020).
Aharon-Peretz, J., Rosenbaum, H. & Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977 (2004).
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661 (2009).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998).
Valente, E. M. et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160 (2004).
Kruger, R. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108 (1998).
Zarranz, J. J. et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 (2004).
Appel-Cresswell, S. et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813 (2013).
Lesage, S. et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471 (2013).
Polymeropoulos, M. H. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997).
Ahn, T. B. et al. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70, 43–49 (2008).
Ibanez, P. et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171 (2004).
Kara, E. et al. A 6.4 Mb duplication of the alpha-synuclein locus causing frontotemporal dementia and Parkinsonism: phenotype-genotype correlations. JAMA Neurol. 71, 1162–1171 (2014).
Zimprich, A. et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175 (2011).
Vilariño-Güell, C. et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167 (2011).
Thirstrup, K. et al. Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci. Rep. 7, 10300 (2017).
Sharma, M. et al. Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology 79, 659–667 (2012).
Bonifati, V. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 (2003).
Canet-Aviles, R. M. et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl Acad. Sci. USA 101, 9103–9108 (2004).
Clements, C. M., McNally, R. S., Conti, B. J., Mak, T. W. & Ting, J. P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl Acad. Sci. USA 103, 15091–15096 (2006).
Fan, J. et al. DJ-1 decreases Bax expression through repressing p53 transcriptional activity. J. Biol. Chem. 283, 4022–4030 (2008).
Zhang, D. et al. Identification of potential target genes for RFX4_v3, a transcription factor critical for brain development. J. Neurochem. 98, 860–875 (2006).
Du, C. et al. Synaptotagmin-11 inhibits cytokine secretion and phagocytosis in microglia. Glia 65, 1656–1667 (2017).
Cang, C., Aranda, K., Seo, Y. J., Gasnier, B. & Ren, D. TMEM175 is an organelle K+ channel regulating lysosomal function. Cell 162, 1101–1112 (2015).
Robak, L. A. et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140, 3191–3203 (2017).
Gan-Or, Z., Dion, P. A. & Rouleau, G. A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 11, 1443–1457 (2015).
Wallings, R. L. & Tansey, M. G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 47, 1581–1595 (2019).
Wallings, R. L., Humble, S. W., Ward, M. E. & Wade-Martins, R. Lysosomal dysfunction at the centre of parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci. 42, 899–912 (2019).
Depboylu, C. et al. Brain-resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T-lymphocytes in the MPTP mouse model of Parkinson disease. Exp. Neurol. 238, 183–191 (2012).
Stefanova, N. et al. Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 179, 954–963 (2011).
Papadopoulos, V. E. et al. Modulation of beta-glucocerebrosidase increases alpha-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum. Mol. Genet. 27, 1696–1710 (2018).
Kim, K. S. et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease. Proc. Natl Acad. Sci. USA 115, E5164–E5173 (2018).
Nash, Y., Schmukler, E., Trudler, D., Pinkas-Kramarski, R. & Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 143, 584–594 (2017).
Haenseler, W. et al. Excess α-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci. Rep. 7, 9003 (2017).
Gardai, S. J. et al. Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS ONE 8, e71634 (2013).
Bonet-Ponce, L. et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 6, eabb2454 (2020).
Hipolito, V. E. B., Ospina-Escobar, E. & Botelho, R. J. Lysosome remodelling and adaptation during phagocyte activation. Cell. Microbiol. 20, e12824 (2018).
Mantegazza, A. R. et al. TLR-dependent phagosome tubulation in dendritic cells promotes phagosome cross-talk to optimize MHC-II antigen presentation. Proc. Natl Acad. Sci. USA 111, 15508–15513 (2014).
Sanyal, A. et al. Lysosome and inflammatory defects in GBA1-mutant astrocytes are normalized by LRRK2 inhibition. Mov. Disord. 35, 760–773 (2020).
Colombo, E. & Farina, C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 37, 608–620 (2016).
Healy, D. G. et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590 (2008).
Hentati, F. et al. LRRK2 parkinsonism in Tunisia and Norway: a comparative analysis of disease penetrance. Neurology 83, 568–569 (2014).
Marder, K. et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 85, 89–95 (2015).
Trinh, J. et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome-wide linkage and association study. Lancet Neurol. 15, 1248–1256 (2016).
Hoyer, M. J. et al. A novel class of ER membrane proteins regulates ER-associated endosome fission. Cell 175, 254–265.e14 (2018).
Lai, D. et al. Genomewide association studies of LRRK2 modifiers of Parkinson’s disease. Ann. Neurol. 90, 76–88 (2021).
Pellegrini, L. et al. Proteomic analysis reveals co-ordinated alterations in protein synthesis and degradation pathways in LRRK2 knockout mice. Hum. Mol. Genet. 27, 3257–3271 (2018).
Acknowledgements
The authors thank members of the Tansey lab for useful discussions. The authors’ research is supported by grants from the US National Institutes of Health (NS092122, AG051514, AG057247 to M.G.T.), the Michael J. Fox Foundation for Parkinson’s Research (16778.01, 18212, 18319, 18891 to M.G.T., V.J. and R.L.W.) and the Parkinson Foundation (PF-RCE-1945 to M.G.T.). The authors regret that several important studies could only be cited indirectly through comprehensive reviews, owing to space and reference number limitations.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
M.G.T. is an ex-employee of and co-inventor on the Xencor and INmune Bio patents describing the dominant-negative TNFs and is a consultant to and has stock ownership in Xencor and Inmune Bio, which has licensed Xpro1595 (pegipanermin) for neurological indications. All other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks C. Klein, P. Seibler and P. West for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Substantia nigra
-
The midbrain nucleus, which supplies the basal ganglia with dopamine, involved in reward, motivation and addiction.
- Bradykinesia
-
Slowness of movement.
- Inflammageing
-
Low-grade inflammation that occurs during ageing.
- Mitophagy
-
The selective degradation of mitochondria by autophagy.
- Mito-inflammation
-
Inflammation that arises through mitochondrial pathways.
- Substantia nigra pars compacta
-
An area of the substantia nigra that contains the majority of neuromelanin-containing dopaminergic neurons, as opposed to the pars reticulata, which is comprised of mainly GABAergic neurons.
- Synucleinopathies
-
A group of neurodegenerative disorders in which the protein α-synuclein accumulates abnormally to form inclusions in the cell bodies or axons of neurons or oligodendrocytes.
- Pernicious anaemia
-
A condition in which the body cannot make enough healthy red blood cells because it does not have enough vitamin B12.
- Polymyalgia rheumatica
-
An inflammatory disorder that causes muscle pain and stiffness, especially in the shoulders and hips.
Rights and permissions
About this article

Cite this article
Tansey, M.G., Wallings, R.L., Houser, M.C. et al. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22, 657–673 (2022). https://doi.org/10.1038/s41577-022-00684-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-022-00684-6
This article is cited by
-
Single cell analysis reveals the roles and regulatory mechanisms of type-I interferons in Parkinson’s disease
Cell Communication and Signaling (2024)
-
Immunotherapy with an antibody against CD1d modulates neuroinflammation in an α-synuclein transgenic model of Lewy body like disease
Journal of Neuroinflammation (2024)
-
Beyond genetics: including the environmental dimension in amyloidosis mouse models for Alzheimer’s disease
Acta Neuropathologica Communications (2024)
-
Integration of National Health Insurance claims data and animal models reveals fexofenadine as a promising repurposed drug for Parkinson’s disease
Journal of Neuroinflammation (2024)
-
Preclinical translational platform of neuroinflammatory disease biology relevant to neurodegenerative disease
Journal of Neuroinflammation (2024)
