
Abstract
Dyskinesia is one of the most disabling motor complications in Parkinson’s Disease (PD). Sleep is crucial to keep neural circuit homeostasis, and PD patients often suffer from sleep disturbance. However, few prospective studies have been conducted to investigate the association of sleep quality with dyskinesia in PD. The objective of the current study is to investigate the association between sleep quality and dyskinesia and build a prediction model for dyskinesia in PD. We prospectively followed a group of PD patients without dyskinesia at baseline for a maximum of 36 months. Univariable and multivariable Cox regression with stepwise variable selection was used to investigate risk factors for dyskinesia. The performance of the model was assessed by the time-dependent area under the receiver-operating characteristic curve (AUC). At the end of follow-up, 32.8% of patients developed dyskinesia. Patients with bad sleep quality had a significantly higher proportion of dyskinesia compared with those with good sleep quality (48.1% vs. 20.6%, p = 0.023). Multivariable Cox regression selected duration of PD, sleep quality, cognition, mood, and levodopa dose. Notably, high Pittsburgh sleep quality index (PSQI) score was independently associated with an increased risk of dyskinesia (HR = 2.96, 95% CI 1.05–8.35, p = 0.041). The model achieved a good discriminative ability, with the highest AUC being 0.83 at 35 months. Our results indicated that high PSQI score may increase the risk of developing dyskinesia in PD, implying that therapeutic intervention targeting improving sleep quality may be a promising approach to prevent or delay the development of dyskinesia in PD.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the second common aging-related central neurodegenerative disease with different types of motor and non-motor symptoms. Levodopa, which can improve patients’ clinical symptoms including bradykinesia, rigidity, and akinesia, is considered to be the gold standard of PD treatment1. However, as the disease progresses, a majority of patients suffer from motor complications, including dyskinesia2. In 2012, Manson et al.3 reviewed studies of the incidence of dyskinesia in PD patients. They concluded that 40–50% of patients developed dyskinesia within 5 years since initiation of antiparkinsonian treatment and the percentage increased to 50–75% after 10 years. As one of the most disabling motor complications in PD patients, dyskinesia imposes a huge burden on the patients, their family, and society4.
Dyskinesia can have a variety of clinical forms including dystonic and choreic movements. It appears initially on the more affected body side at different points of a levodopa drug cycle5. The clinical manifestations of dyskinesia can be divided into three main patterns, including peak-dose dyskinesia, “off” dyskinesia, and biphasic dyskinesia6, with peak-dose dyskinesia being the most common in PD. Dyskinesia is believed to be associated with dysfunction of neural circuit2,5,7. Good sleep quality is crucial to maintain neural circuit homeostasis8,9. Many studies10 showed that PD patients usually suffered from sleep disturbance, such as long periods spent in bed while not asleep and unable to remain asleep and excessive daytime sleepiness. In addition, there is a growing body of evidence supporting that sleep disturbance may precede the development of neurodegenerative diseases, including PD11,12,13. A recent study found that an animal model of PD dyskinesia experienced a change in cortical slow-wave activity, which is of great importance to homeostasis during non–rapid eye movement sleep14. An in vivo study also showed that sleep deprivation aggravated dyskinesia and led to an earlier onset of dyskinesia14. However, few prospective studies have been conducted to explore the relationship between sleep quality and dyskinesia in PD patients.
Here, we prospectively followed a group of PD patients without dyskinesia at baseline for a maximum of 36 months to investigate the association between sleep quality and dyskinesia and build a prediction model for the development of dyskinesia.
Results
Patients’ clinical characteristics at the baseline
A total of 61 participants were included in this study. Characteristics of the study participants are presented in Table 1. The mean baseline age of the study cohort was 66.6 ± 6.1 years and the mean age of onset of PD was 61.6 ± 7.0 years. There were 41 (67.2%) men. The median duration of PD was 4.5 years (interquartile range [IQR] 2.9–7.8 years). The median MDS-UPDRS III was 22 (IQR 16.5–30.0). There were 27 (44.3%) patients who had motor fluctuation and 19 (31.1%) patients who had FOG. The median LEDD was 375.0 (IQR 275.0–611.9) mg and the median levodopa equivalent daily dose (LEDD) per kilogram body weight (LEDD/Weight) was 9.6 (IQR 6.6–12.9) mg/kg. The median Pittsburgh sleep quality index (PSQI) was 5.0 (IQR 3.0–9.5).
Incidence of dyskinesia during follow-up
During a median follow-up of 3.0 years (mean 2.8 years), 20 (32.8%) patients developed dyskinesia. The cumulative incidence of dyskinesia was 8.2%, 11.5%, 21.3%, and 32.8% at the 24-, 28-, 32-, and 35-month follow-up, respectively (Fig. 1). At the end of follow-up, patients with bad sleep quality had a significantly higher proportion of dyskinesia compared with those with good sleep quality (48.1% vs. 20.6%, p = 0.023) (Fig. 2).

Kaplan–Meier estimates showing cumulative risk of dyskinesia in PD patients.

PSQI, Pittsburgh Sleep Quality Index.
Risk factors of dyskinesia
Kaplan–Meier curves comparing the cumulative incidence of dyskinesia with respect to NMSS, HAMA, LEDD, LEDD/weight, duration of PD, and PSQI were shown in Fig. 3a–f. Kaplan–Meier curves comparing the cumulative incidence of dyskinesia with respect to MOCA, H-Y, and PDSS were shown in Supplementary Fig. 1a–c. We found that patients who had bad sleep quality had a significantly higher risk to develop dyskinesia than those who had good sleep quality (p = 0.017, Fig. 3f).

a NMSS (“1” represents the first tertile, “2” represents the second tertile, and “3” represents the third tertile), b HAMA (“1” represents <7, and “2” represents ≥7), c LEDD (“1” represents ≤400 mg, “2” represents 400~600 mg, and “3” represents >600 mg), d LEDD/Weight (“1” represents ≤median, and “2” represents >median), e duration of PD (“1” represents ≤5 y, “2” represents 5~10 y, and “3” represents > 10 y), f PSQI (“1” represents <6, and “2” represents ≥6). NMSS Non-Motor Symptom Scale, HAMA Hamilton Anxiety Rating Scale, LEDD Levodopa equivalent daily dose, PSQI Pittsburgh Sleep Quality Index.
In addition, Cox proportional hazards analysis was conducted to examine risk factors for dyskinesia in PD (Table 2). In the univariable Cox analysis, long duration of PD was associated with a greater risk of developing dyskinesia (Hazard ratio; HR = 2.32, 95% confidence interval (CI) 1.38–3.92, p = 0.002). In addition, high LEDD (HR = 1.92, 95% CI 1.15–3.20, p = 0.012) and high LEDD/Weight (HR = 5.35, 95% CI 1.78–16.10, p = 0.003) also showed significant association with greater risk of dyskinesia. Furthermore, multiple non-motor clinical characteristics were associated with a greater risk of dyskinesia, including high NMSS score (HR = 2.29, 95% CI 1.27–4.16, p = 0.006), high HAMA score (HR = 2.64, 95% CI 1.05–6.62, p = 0.039) and high PSQI score (HR = 2.93, 95% CI 1.17–7.36, p = 0.022).
The results of the multivariable Cox regression analysis were shown in Table 2. Forest plot of the multivariable Cox regression analysis was displayed in Fig. 4. We found that high PSQI was independently associated with the risk of dyskinesia (HR = 2.96, 95% CI 1.05–8.35, p = 0.041). In addition, long duration of PD (HR = 2.09, 95% CI 1.05–4.17, p = 0.037), and low MOCA score (HR = 4.12, 95% CI 1.40–14.00, p = 0.011) also showed significant association with dyskinesia. However, the association of high LEDD/Weight (HR = 3.06, 95% CI 0.93–10.10, p = 0.064) and high HAMD score (HR = 0.38, 95% CI 0.12–1.17, p = 0.090) with dyskinesia were of borderline significance. To evaluate the performance of the Cox model, we generated time-dependent ROC curves (Fig. 5). The area under receiver-operating characteristic curve (AUC) was 0.78, 0.80, and 0.78 at 24 months, 28 months, and 32 months since baseline, respectively, with the highest AUC being 0.83, which was reached at 35 months since baseline.

LEDD Levodopa equivalent daily dose, MOCA Montreal Cognitive Assessment, PSQI Pittsburgh Sleep Quality Index, HAMD Hamilton Depression Rating Scale.

AUC Area under curve.
Discussion
In this study, we prospectively followed a group of patients without dyskinesia at baseline for a maximum of 36 months. We found that a high PSQI score was independently associated with the development of dyskinesia and the predictive model achieved a good discriminative ability. These results highlight the important effect of sleep quality on the risk of dyskinesia and imply that therapeutic intervention targeting improving sleep quality may be a promising approach to prevent or delay the development of dyskinesia in PD.
Disrupted slow-wave sleep could accelerate the progression of PD15,16. Prior research indicated that sleep disturbance might precede motor symptoms in PD13,17. It is widely believed that slow homeostatic adjustment of intrinsic excitability occurring during sleep is fundamental for the stabilization of the network by gradual modification of plasticity thresholds14,18. Galati et al.14 conducted an assessment on synaptic downscaling across sleep episodes in a parkinsonian rat model showing dyskinetic movements similar to levodopa-induced dyskinesia. They observed a synaptic homeostasis impairment during sleep in rats showing dyskinesia14. Moreover, sleep deprivation led to anticipation and aggravation of dyskinesia in rats, supporting an association between sleep disturbance and the development of dyskinesia14. In recent years, a bulk of evidence has shown that dyskinesia is associated with neurodegeneration in cortical and subcortical areas, including the prefrontal cortex, primary motor cortex, striatum, subthalamic nucleus, and cerebellum4. Cortical activity was observed to be dominated by spindles and slow waves during sleep19,20,21. Sleep could enable a downward firing rate to maintain homeostasis and overall sleep slow-wave activity potentiates the majority of cortical synaptic plasticity22,23, implying that sleep disturbance could disrupt synaptic plasticity or synaptic homeostasis24. As a result, bad sleep quality may increase the risk of developing dyskinesia in PD by interrupting neural circuit homeostasis25. These findings, together with ours, provide important clues for further investigation of potential treatments of dyskinesia.
Previous research reported that disease duration and dose of levodopa were important etiopathogenic factors for LID25. In this study, the duration of PD was confirmed as a predictor for dyskinesia, while the association of LEDD/Weight with dyskinesia was of borderline significance in the multivariable Cox regression analysis, probably due to limited statistical power resulting from the limited sample size of our study. Cumulative LEDD26 was found to be a risk factor for LID, indicating that levodopa use, especially in a day-night pulsatile manner, could be a cause of subsequently aggravated LID22. Nevertheless, levodopa is necessary but not enough to generate LID. Dyskinesia is caused by pre-synaptic nigro-striatal degeneration and deteriorates along with disease duration. It is also associated with a relatively spared post-synaptic nigro-striatal system27, as supported by a prior study28 showing that dyskinesia development was a function of disease duration. Moreover, previous studies supported that long-term levodopa treatment for PD resulted in dyskinesia, suggesting a sleep-dependent depotentiation impairment in the dorsal striatum29. Future therapeutic strategies for PD may benefit from targeting sleep-dependent depotentiation impairment. Interestingly, a case-control polysomnographic study showed that reduced total sleep time was associated with increased LEDD and levodopa use could be a cause of reduced total sleep time22. These results highlight the importance of further investigate the relationship between sleep disturbance and dyskinesia.
In our study, we found that cognitive impairment as assessed by the global MOCA score was also associated with dyskinesia. Indeed, the reduction of the connectivity between the left inferior frontal cortex and the right motor cortex has been reported in dyskinesia patients30. It was speculated that decreased inhibitory action of the prefrontal cortex could produce disinhibition in both motor and cognitive control loops in PD7. Further studies with functional magnetic resonance and scales of different part of cognition in larger samples are needed to explore the association between cognitive impairment and dyskinesia.
To assess the relationship between change of PSQI and change of other clinical characteristics, we also calculated their annual change and the corresponding correlation (Table 3). The annual change of PSQI was ~0 (IQR ~0–0.343). The annual change of PSQI was associated with worsened non-motor symptoms, greater anxiety, worsened cognition, and increased levodopa dose, suggesting that patients experienced more impaired mood disturbance and cognitive dysfunction as sleep quality deteriorated. These findings are consistent with previous studies which observed a strong association between sleep disturbance and subsequent progression of PD31,32 and rapid motor progression in patients with rapid eye movement sleep behavior disorder (RBD). The exact pathophysiology of the association of sleep disturbance with dyskinesia needs to be explored in future studies.
There are some limitations of this study. First, the assessments of dyskinesia and sleep were conducted with subjective scales, which might suffer from subjective assessment bias. However, these scales are convenient, reliable, and practicable for clinicians. In addition, to minimize biases, each patient was assessed by two raters in a randomized and assessor-masking way. Second, the sample size of our study is limited, preventing us to match important characteristics other than PQSI to estimate more precisely the effect of PSQI on the risk of dyskinesia. As a result, the findings of this study need to be validated in future studies of large sample sizes, especially studies that match important clinical characteristics that might affect the risk of dyskinesia. Despite these limitations, the present work was among the few prospective studies to examine the relationship between dyskinesia and sleep disturbance, with a relatively long follow-up.
In summary, the current study found that a high PSQI score was a risk factor for the development of dyskinesia in patients with PD, suggesting that therapeutic intervention targeting sleep quality may be a promising approach to prevent or delay the development of dyskinesia in PD. The findings of our study need to be validated and the pathophysiological mechanisms underlying the findings need to be explored. Indeed, we are planning a new cohort study with larger sample size and a new project utilizing animal models to validate our findings and explore the pathological mechanisms.
Methods
Participants
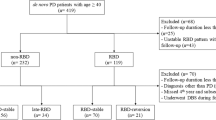
A total of 76 patients were diagnosed with PD by experienced experts in the Department of Neurology, Shanghai Tongji Hospital, School of Medicine, Tongji University and Department of Neurology, Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine based on the Unified Kingdom PD Society Brain Bank Clinical Diagnostic Criteria33. Exclusion criteria included: atypical and secondary parkinsonism, sleep apnea syndrome, severe mental diseases, never taken levodopa, a history of deep brain stimulation surgery, and unable to complete clinical evaluation due to cognitive impairment. Of the 76 patients, 15 patients were excluded and 61 patients who were without dyskinesia at the baseline were prospectively followed (Fig. 6). These subjects all received levodopa medications regularly for at least one month22. All patients were followed through outpatients visit in a blinded manner every three months until the onset of dyskinesia or the end of 36 months. This study was approved by the Ethics Committees of Shanghai Tongji Hospital, School of Medicine, Tongji University, and Xinhua Hospital Affiliated with Shanghai Jiao Tong University School of Medicine. Written informed consent was obtained from all patients.

DBS deep brain stimulation, PD Parkinson’s disease.
Assessments of demographic and clinical characteristics
Demographic and clinical characteristics include age, sex, age of PD onset, years of education, duration of PD, and weight. All patients’ motor symptoms were evaluated by the Movement Disorder Society–sponsored revision of the Unified Parkinson Disease Rating Scale III (MDS-UPDRS III) and H-Y stage. Levodopa dose was defined as the dose taken at the time of the first visit. Total LEDD was calculated based on the established algorithm34. Global non-motor symptoms were evaluated by the non-motor symptoms scale (NMSS). Cognition was evaluated by the Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MOCA, score ≥26 was used as the reference in Cox regression analysis in this study). Mood complaints were investigated by the Hamilton Anxiety Rating Scale (HAMA) and the Hamilton Depression Rating Scale (HAMD). Patients’ quality of life was assessed by the Parkinson’s Disease Questionnaire-8 (PDQ-8). Freezing of gait (FOG) was evaluated by the New Freezing of Gait Questionnaire35. Annual change of clinical characteristics was calculated. For example, the annual change in HAMD was calculated as (follow-up HAMD—the baseline HAMD)/time of follow-up (year).
Assessments of dyskinesia and sleep
Dyskinesia was assessed by two raters blinded to patients’ sleep based on the Movement Disorder Society-sponsored revision of the Unified Parkinson Disease Rating Scale IV22,36. If evidence for determining dyskinesia came from patients or their caregivers, they need to provide the raters with related videos. It is worth noting that the PD patients who developed dyskinesia during the follow-up were all peak-dose dyskinesia rather than biphasic dyskinesia or “off” dyskinesia. Four sleep scales were used to assess sleep functioning, including the PSQI37, the Epworth Sleepiness Scale (ESS)38, the Rapid Eye Movement Sleep Behavior Disorder Questionnaire—Hong Kong (RBDQ-HK)39,40, and the Parkinson’s Disease Sleep Scale (PDSS)41. PSQI, the most commonly used measurement tool for sleep quality in clinical application, was designed to assess general sleep quality over the past month in clinical populations37. It has 19 subitems categorized into seven components, and score was summed to obtain a global score. According to conventions in previous studies, patients were dichotomized as having bad (PSQI ≥ 6) and good (PSQI < 6) sleep quality42,43,44. Other continuous variables were converted to categorical ones when necessary, with cutoff being set based on previous conventions or the median value in the total sample (Fig. 3a–f and Supplementary Fig. 1a–c).
In our study, all patients were evaluated in the “on” state. All raters received homogeneity training at the start of the study to reduce inter-rater disagreement.
Statistical analysis
Continuous data were presented as mean ± standard deviation or median (IQR) and compared by using independent t test or Mann–Whitney U test, as appropriate. Categorical variables were presented as numbers and percentages and compared by Pearson’s chi-square or Fisher’s exact test, as appropriate. Cumulative incidence of dyskinesia was displayed using Kaplan–Meier curves with log-rank test to compare the differences. Cox proportional hazards regression was applied to explore risk factors for developing dyskinesia. We conducted univariable Cox models for all baseline variables, followed by stepwise variable selection to select variables for the multivariable Cox regression, with the significance level for entry and stay being 0.1 (the default setting) to avoid rejection of potentially important variables due to uncontrolled confounders. HR with 95% CI was calculated. This was done using the my.stepwise.coxph in the My.stepwise R package. Multicollinearity of the selected variables was assessed using the variance inflation factor (VIF) (Supplementary Table 1). The assumption of proportional hazard was tested using scaled Shoenfeld residuals. The performance of the multivariable Cox model was assessed using a time-dependent area under the receiver-operating characteristic (ROC) curve (AUC).
A two-sided p value < 0.05 was considered to be statistically significant. Statistical analysis was performed using R version 4.2.0 (Foundation for Statistical Computing, Vienna, Austria).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the Department of Neurology, Shanghai Tongji Hospital, School of Medicine, Tongji University, via the corresponding authors.
Code availability
R codes for data analysis are available upon reasonable request.
References
Connolly, B. S. & Lang, A. E. Pharmacological treatment of Parkinson disease: a review. JAMA 311, 1670–1683 (2014).
Voon, V. et al. Impulse control disorders and levodopa-induced dyskinesias in Parkinson’s disease: an update. Lancet Neurol. 16, 238–250 (2017).
Manson, A., Stirpe, P. & Schrag, A. Levodopa-induced-dyskinesias clinical features, incidence, risk factors, management and impact on quality of life. J. Parkinsons Dis. 2, 189–198 (2012).
Bastide, M. F. et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 132, 96–168 (2015).
Espay, A. J. et al. Levodopa-induced dyskinesia in Parkinson disease: current and evolving concepts. Ann. Neurol. 84, 797–811 (2018).
Tran, T. N., Vo, T. N. N., Frei, K. & Truong, D. D. Levodopa-induced dyskinesia: clinical features, incidence, and risk factors. J. Neural Transm. (Vienna) 125, 1109–1117 (2018).
Picazio, S., Ponzo, V., Caltagirone, C., Brusa, L. & Koch, G. Dysfunctional inhibitory control in Parkinson’s disease patients with levodopa-induced dyskinesias. J. Neurol. 265, 2088–2096 (2018).
Tononi, G. & Cirelli, C. Sleep and synaptic down-selection. Eur. J. Neurosci. 51, 413–421 (2020).
Kramer, M. A. et al. Focal sleep spindle deficits reveal focal thalamocortical dysfunction and predict cognitive deficits in sleep activated developmental epilepsy. J. Neurosci. 41, 1816–1829 (2021).
Lebrun, C. et al. Presleep cognitive arousal and insomnia comorbid to parkinson disease: evidence for a serial mediation model of sleep-related safety behaviors and dysfunctional beliefs about sleep. J. Clin. Sleep. Med. 15, 1217–1224 (2019).
Lysen, T. S., Darweesh, S. K. L., Ikram, M. K., Luik, A. I. & Ikram, M. A. Sleep and risk of parkinsonism and Parkinson’s disease: a population-based study. Brain 142, 2013–2022 (2019).
Videnovic, A. & Willis, G. L. Circadian system - a novel diagnostic and therapeutic target in Parkinson’s disease? Mov. Disord. 31, 260–269 (2016).
Tang, X. et al. Association of sleep disturbance and freezing of gait in Parkinson disease: prevention/delay implications. J. Clin. Sleep. Med. 17, 779–789 (2021).
Galati, S. et al. Evidence of an association between sleep and levodopa-induced dyskinesia in an animal model of Parkinson’s disease. Neurobiol. Aging 36, 1577–1589 (2015).
Schreiner, S. J. et al. Slow-wave sleep and motor progression in Parkinson disease. Ann. Neurol. 85, 765–770 (2019).
Bohnen, N. I. & Hu, M. T. M. Sleep disturbance as potential risk and progression factor for Parkinson’s disease. J. Parkinsons Dis. 9, 603–614 (2019).
Gan-Or, Z., Alcalay, R. N., Rouleau, G. A. & Postuma, R. B. Sleep disorders and Parkinson disease; lessons from genetics. Sleep. Med. Rev. 41, 101–112 (2018).
Tononi, G. & Cirelli, C. Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014).
Timofeev, I. & Chauvette, S. Sleep slow oscillation and plasticity. Curr. Opin. Neurobiol. 44, 116–126 (2017).
Torrado Pacheco, A., Bottorff, J., Gao, Y. & Turrigiano, G. G. Sleep promotes downward firing rate homeostasis. Neuron 109, 530–544 e536 (2021).
Ma, Y. et al. Association between sleep duration and cognitive decline. JAMA Netw. Open 3, e2013573 (2020).
Amato, N. et al. Levodopa-induced dyskinesia in Parkinson disease: sleep matters. Ann. Neurol. 84, 905–917 (2018).
de Vivo, L. et al. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355, 507–510 (2017).
Tamaki, M. et al. Complementary contributions of non-REM and REM sleep to visual learning. Nat. Neurosci. 23, 1150–1156 (2020).
Picconi, B., Hernandez, L. F., Obeso, J. A. & Calabresi, P. Motor complications in Parkinson’s disease: striatal molecular and electrophysiological mechanisms of dyskinesias. Mov. Disord. 33, 867–876 (2018).
Prange, S. et al. Age and time course of long-term motor and nonmotor complications in Parkinson disease. Neurology 92, e148–e160 (2019).
Luca, A. et al. Cognitive impairment and levodopa induced dyskinesia in Parkinson’s disease: a longitudinal study from the PACOS cohort. Sci. Rep. 11, 867 (2021).
Cilia, R. et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain 137, 2731–2742 (2014).
Poe, G. R. Sleep is for forgetting. J. Neurosci. 37, 464–473 (2017).
Cerasa, A. et al. A network centred on the inferior frontal cortex is critically involved in levodopa-induced dyskinesias. Brain 138, 414–427 (2015).
Sommerauer, M. et al. Revisiting the impact of REM sleep behavior disorder on motor progression in Parkinson’s disease. Parkinsonism Relat. Disord. 20, 460–462 (2014).
Liguori, C. et al. Sleep problems affect quality of life in Parkinson’s disease along disease progression. Sleep. Med. 81, 307–311 (2021).
Gibb, W. R. & Lees, A. J. The significance of the Lewy body in the diagnosis of idiopathic Parkinson’s disease. Neuropathol. Appl. Neurobiol. 15, 27–44 (1989).
Warren Olanow, C. et al. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov. Disord. 28, 1064–1071 (2013).
Amundsen Huffmaster, S. L., Lu, C., Tuite, P. J. & MacKinnon, C. D. The transition from standing to walking is affected in people with Parkinson’s disease and freezing of gait. J. Parkinsons Dis. 10, 233–243 (2020).
Hong, J. Y. et al. Presynaptic dopamine depletion predicts levodopa-induced dyskinesia in de novo Parkinson disease. Neurology 82, 1597–1604 (2014).
Mollayeva, T. et al. The Pittsburgh sleep quality index as a screening tool for sleep dysfunction in clinical and non-clinical samples: a systematic review and meta-analysis. Sleep. Med. Rev. 25, 52–73 (2016).
Hogl, B. et al. Scales to assess sleep impairment in Parkinson’s disease: critique and recommendations. Mov. Disord. 25, 2704–2716 (2010).
Telarovic, S., Mijatovic, D. & Telarovic, I. Effects of various factors on sleep disorders and quality of life in Parkinson’s disease. Acta Neurol. Belg. 115, 615–621 (2015).
Li, S. X. et al. Validation of a new REM sleep behavior disorder questionnaire (RBDQ-HK). Sleep. Med. 11, 43–48 (2010).
Chaudhuri, K. R. et al. The Parkinson’s disease sleep scale: a new instrument for assessing sleep and nocturnal disability in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 73, 629–635 (2002).
Buysse, D. J., Reynolds, C. F. 3rd, Monk, T. H., Berman, S. R. & Kupfer, D. J. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 28, 193–213 (1989).
Yamamoto, R. et al. Sleep quality and sleep duration with CKD are associated with progression to ESKD. Clin. J. Am. Soc. Nephrol. 13, 1825–1832 (2018).
Pilz, L. K., Keller, L. K., Lenssen, D. & Roenneberg, T. Time to rethink sleep quality: PSQI scores reflect sleep quality on workdays. Sleep 41, https://doi.org/10.1093/sleep/zsy029 (2018).
Acknowledgements
This study was supported by the Projects of National Science Foundation of China (82271227, 81671273, 81171204, and 30870874), National Key R&D Program of China (2022YFF1202800), the Project of Shanghai Tongji Hospital (RCQD2203 and ITJ(ZD)1810), the Project of Suzhou BenQ Medical Center (SZMJ2002) and NIH/NIA grants (P30AG10161, R01AG15819, R01AG17917, R01AG033678, R01AG36042, U01AG61356, and 1RF1AG064312– 01). We would like to thank all patients and their caregivers who collaborated in this study.
Author information
Authors and Affiliations
Contributions
Xijin Wang is the primary contact for this paper. Study concept and design: Xijin Wang, Xiaohui Tang, and Guozhao Ma; Literature search, data collection, and data analysis: Xiaohui Tang, Jingyun Yang, Yining Zhu, Haiyan Gong, Hui Sun, Fan Chen, Qiang Guan, Lijia Yu, Weijia Wang, Zengping Zhang, and Li Li; drafting of the manuscript: Xiaohui Tang, Yining Zhu, Guozhao Ma, and Xijin Wang; Review, critique, and supervision: Xijin Wang and Guozhao Ma. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, X., Yang, J., Zhu, Y. et al. High PSQI score is associated with the development of dyskinesia in Parkinson’s disease. npj Parkinsons Dis. 8, 124 (2022). https://doi.org/10.1038/s41531-022-00391-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-022-00391-y
